4.1.2. Experimental Procedures
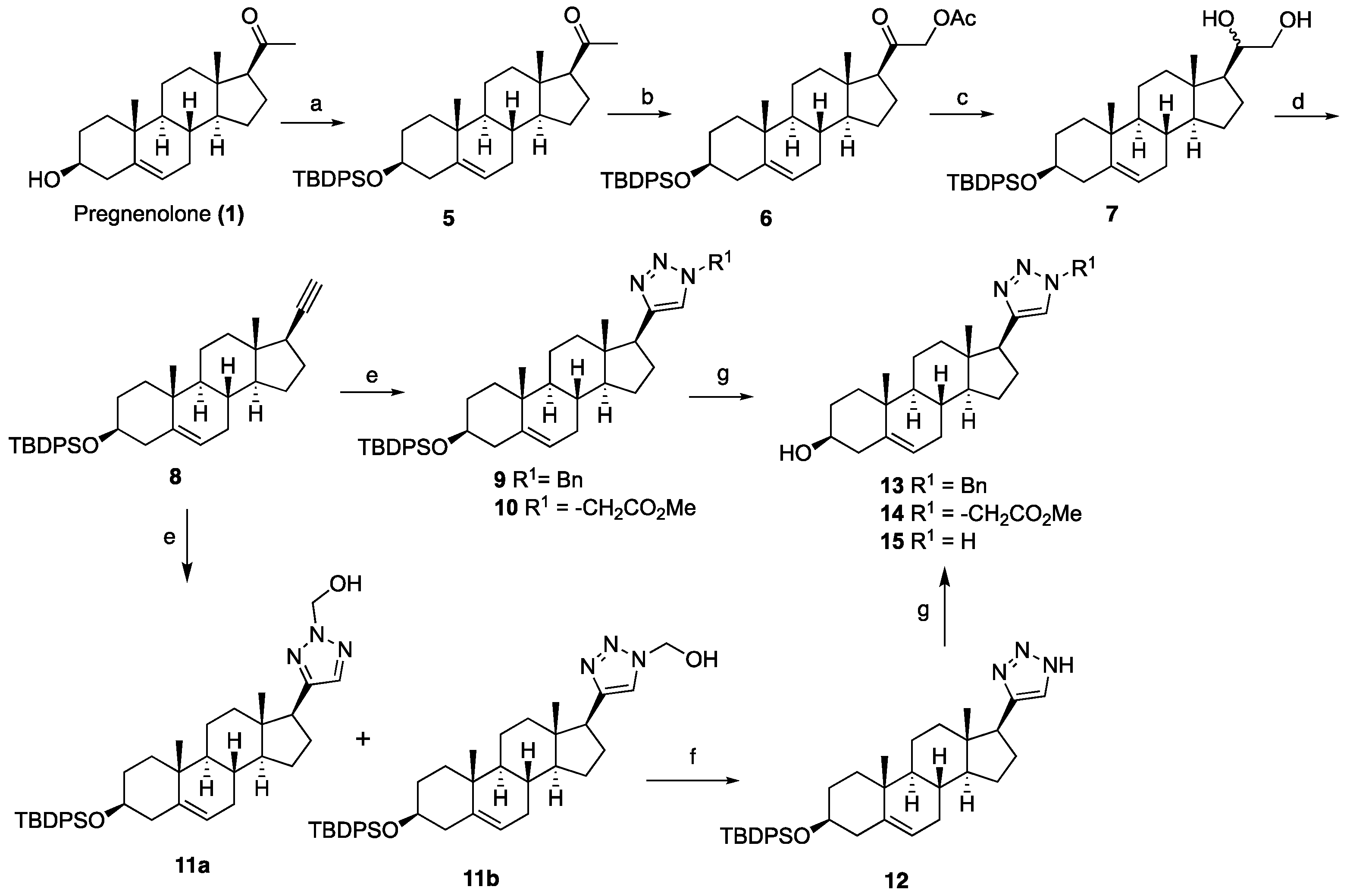
1-((3S,8S,9S,10R,13S,14S,17S)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethan-1-one (5): To a solution of pregnenolone (1, 4.00 g, 12.65 mmol) in DCM (100 mL) at 0 °C, imidazole (2.58 g, 38.0 mmol) and TBDPSCl (4.80 mL, 19.0 mmol) were added. After being stirred at room temperature for 4 h, the reaction mixture was quenched with saturated aqueous NaHCO3 (60 mL). The organic phase was separated and extracted with an additional DCM (2 × 100 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (silica gel, EtOAc/hexanes, 1:9) to obtain TBDPS-ether 5 (5.96 g, 85%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.75–7.65 (m, 4H), 7.46–7.32 (m, 6H), 5.17–5.09 (m, 1H), 3.66–3.42 (m, 1H), 2.59–2.44 (m, 1H), 2.43–2.26 (m, 1H), 2.22–2.08 (m, 5H), 2.07–1.85 (m, 2H), 1.80–1.32 (m, 10H), 1.31–1.15 (m, 1H), 1.14–1.03 (m, 10H), 0.99 (s, 3H), 0.87 (ddd, J = 13.9, 6.6, 3.6 Hz, 2H), 0.61 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 209.5, 141.2, 135.7 (2C), 134.8, 134.7, 129.4 (2C), 127.4 (2C), 120.8, 73.1, 63.7, 56.9, 49.9, 43.9, 42.4, 38.8, 37.2, 36.5, 31.8 (2C), 31.7, 31.5, 27.0, 24.7, 22.8, 21.0, 19.4, 19.1, 13.2.
2-((3S,8S,9S,10R,13S,14S,17S)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethyl acetate (6): To a stirred solution of compound 5 (1.00 g, 1.80 mmol) in toluene (28 mL), methanol (3.40 mL) containing BF3.OEt2 (3.37 mL, 27.0 mmol) and lead tetraacetate (0.870 g, 1.98 mmol) were added at room temperature. After being stirred at room temperature for 4 h, the mixture was poured into ice water and extracted with DCM (3 × 30 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give a residue, which was purified by column chromatography (silica gel, EtOAc/hexanes, 15:85) to obtain compound 6 (0.696 g, 63%) as a white powder: 1H NMR (400 MHz, CDCl3) δ 7.75–7.61 (m, 4H), 7.50–7.29 (m, 6H), 5.12 (d, J = 5.1 Hz, 1H), 4.70 (dd, J = 16.9, 6.6 Hz, 1H), 4.51 (dd, J = 16.8, 5.9 Hz, 1H), 3.63–3.44 (m, 1H), 2.55–2.25 (m, 2H), 2.24–2.08 (m, 5H), 2.07–1.81 (m, 2H), 1.74–1.52 (m, 6H), 1.50–1.37 (m, 3H), 1.35–1.18 (m, 2H), 1.15–0.92 (m, 13H), 0.85 (ddd, J = 22.5, 14.4, 8.3 Hz, 2H), 0.68–0.61 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 203.8, 170.2, 141.3, 135.7, 135.6, 134.7 (2C), 129.45 (2C), 127.4 (2C), 120.7, 73.1, 69.1, 59.2 (2C), 57.0, 49.8, 44.6, 42.4, 38.5, 37.1, 36.5, 31.8 (2C), 31.7, 24.5, 22.8, 20.9, 20.4, 19.3, 19.1, 13.0.
1-((3S,8S,9S,10R,13S,14S,17S)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethane-1,2-diol (7): A 100-mL, two-necked, round-bottomed flask was charged with LiAlH4 (0.860 g, 22.7 mmol) under nitrogen and cooled to 0 °C. Anhydrous THF (40 mL) followed by compound 6 (3.48 g, 5.68 mmol) in THF (10 mL) was added dropwise, and the resulting mixture was stirred at ambient temperature for 30 min. The reaction mixture was quenched with H2O (0.9 mL), NaOH solution (0.9 mL, 15%), and H2O (2.7 mL) and stirred for another 10 min. The precipitate was filtered and washed with Et2O. The filtrate was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give a residue, which was purified by column chromatography (silica gel, EtOAc/hexanes, 3:7) to obtain diol 7 (2.18 g, 67%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 7.72–7.63 (m, 4H), 7.47–7.31 (m, 6H), 5.12 (d, J = 5.0 Hz, 1H), 3.64 (d, J = 9.3 Hz, 2H), 3.60–3.46 (m, 1H), 3.36 (t, J = 9.2 Hz, 1H), 2.33 (t, J = 12.2 Hz, 1H), 2.22–1.98 (m, 2H), 1.99–1.79 (m, 3H), 1.77–1.53 (m, 4H (overlapped with H2O), 1.52–1.33 (m, 5H), 1.31–1.10 (m, 3H), 1.05 (s, 9H), 0.99 (s, 3H), 0.92–0.78 (m, 2H), 0.76 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.3, 135.7 (2C), 134.8 (2C), 129.4 (2C), 127.4 (2C), 120.8, 74.6, 73.2, 66.4, 55.9, 52.4, 50.0, 42.4 (2C), 39.7, 37.2, 36.5, 31.8 (2C), 31.7, 26.9, 24.6, 24.5, 20.8, 19.4, 19.1, 12.3.
tert-Butyl(((3S,8S,9S,10R,13S,14S,17R)-17-ethynyl-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)oxy)diphenylsilane (8): Sodium periodate (0.560 g, 2.62 mmol) was added to a solution of diol 7 (0.500 g, 0.872 mmol) in THF:H2O (8:2, 20 mL) and the solution was stirred for 1 h. The reaction mixture was then diluted with water (20 mL). The organic phase was separated and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (silica gel, EtOAc/hexanes, 3:7) to afford the corresponding aldehyde (0.500 g), which was used for the next step without further purification.
To a solution of tetrabromomethane (0.910 g, 2.77 mmol) in anhydrous DCM (20 mL) triphenylphosphine (1.45 g, 5.55 mmol) was added at 0 °C, and the resulting mixture was stirred for 10 min. Subsequently, a solution of the above aldehyde (0.500 g, 0.920 mmol) in DCM (5 mL) was added. After stirring for 20 min, the reaction mixture was diluted with DCM (30 mL). The organic phase was washed with water (30 mL) and saturated NaCl solution (30 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give dibromoalkene (0.700 g), which was also used for the next step without further purification.
A solution of n-BuLi (1.6 M) in hexanes (2.51 mL, 4.02 mmol) was added to a solution of dibromoalkene (0.700 g, 1.00 mmol) in anhydrous THF (20 mL) at –78 °C, and the resulting mixture was stirred at the same temperature for 1 h. The reaction mixture was quenched with saturated aqueous NH4Cl (20 mL), and the mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (silica gel, hexanes/EtOAc, 1:9) to afford the alkyne 8 (0.330 g, 70%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.76–7.61 (m, 4H), 7.51–7.28 (m, 6H), 5.12 (d, J = 5.2 Hz, 1H), 3.69–3.42 (m, 1H), 2.35 (dt, J = 13.2, 11.3 Hz, 1H), 2.21–1.78 (m, 6H), 1.75–1.57 (m, 5H), 1.53–1.35 (m, 4H), 1.35–1.12 (m, 1H), 1.06 (s, 9H), 1.03 (d, J = 4.5 Hz, 1H), 1.00 (s, 3H), 0.86 (m, 3H), 0.79 (s, 3H).
General procedure A for synthesizing silyl or Fmoc or Boc-protected triazole analogs (9, 10, and 18), simplified small molecules 33, and 39: A mixture of alkyne (1.0 equiv), azide (1.5 equiv), and DMF (6 mL) was combined with sodium ascorbate (0.40 equiv) in H2O (3 mL) and stirred for two min at ambient temperature. Next, CuSO4·5H2O (0.20 equiv) in H2O (3 mL) was added to the mixture. The mixture was stirred at room temperature for 12 h, and then water was added (6 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (silica gel, hexanes/EtOAc, 2:8) to yield the triazole analogs.
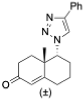
1-Benzyl-4-((3S,8S,9S,10R,13S,14S,17S)-3-((tert-butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1H-1,2,3-triazole (9): Compound 9 was prepared following by general procedure A, using alkyne 8 (0.300 g, 0.559 mmol), benzyl azide (0.112 g, 0.838 mmol), CuSO4·5H2O (27.0 mg, 0.011 mmol), and sodium ascorbate (44.3 mg, 0.022 mmol)) to yield 9 (0.300 g, 80%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 7.70–7.65 (m, 4H), 7.45–7.32 (m, 9H), 7.23–7.20 (m, 2H), 7.10 (s, 1H), 5.55–5.46 (m, 2H), 5.13 (d, J = 5.0 Hz, 1H), 3.55–3.44 (m, 1H), 2.75 (t, J = 9.8 Hz, 1H), 2.37–2.26 (m, 1H), 2.16–1.90 (m, 4H), 1.80–1.20 (m, 12H), 1.05 (s, 9H), 0.97 (s, 3H), 0.92–0.81 (m, 2H), 0.44 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.4, 141.3, 135.8 (2C), 135.12, 134.6, 129.5 (2C), 129.0, 128.5, 127.8, 127.5 (2C), 121.0, 120.9, 73.1, 55.9, 53.9, 50.0, 47.8, 43.5, 42.4, 37.5, 37.1, 36.5, 32.2, 31.8 (2C), 27.0, 26.6, 24.5, 20.6, 19.5, 19.1, 13.0. HRMS (ESI) calcd for C44H56N3OSi (M + H)+ 670.4193, found 670.4189.
Methyl 2-(4-((3S,8S,9S,10R,13S,14S,17S)-3-((tert-butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1H-1,2,3-triazol-1-yl)acetate (10): Compound 10 was prepared using general procedure A, using alkyne 8 (400 mg, 0.745 mmol), methyl 2-azidoacetate (257 mg, 2.23 mmol), CuSO4·5H2O (37.2 mg, 0.015 mmol), and sodium ascorbate (59.0 mg, 0.030 mmol) to yield 10 (0.330 g, 68% yield) as a white foam: 1H NMR (400 MHz, CDCl3) δ 7.70–7.65 (m, 4H), 7.44–7.34 (m, 7H), 5.15–5.11 (m, 3H), 3.79 (s, 3H), 3.60–3.46 (m, 1H), 2.80 (t, J = 9.8 Hz, 1H), 2.37–2.30 (m, 1H), 2.17–1.94 (m, 4H), 1.83–1.59 (m, 5H), 1.58–1.44 (m, 3H), 1.40–1.21 (m, 3H), 1.18–1.11 (m, 1H), 1.06 (s, 9H), 0.98 (s, 3H), 0.94–0.82 (m, 2H), 0.49 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.0, 149.3, 141.4, 135.7 (2C), 134.8, 129.4 (2C), 127.4 (2C), 122.2, 120.9, 73.2, 56.0, 52.9, 50.5, 50.1, 47.8, 43.5, 42.4, 37.6, 37.2, 36.6, 32.2, 31.8 (2C), 27.0, 26.6, 24.5, 20.6, 19.4, 19.1, 12.9; HRMS (ESI) calcd for C40H54N3O3Si (M + H)+ 652.3934, found 652.3919.
General procedure B for synthesizing hydroxymethyl triazole analogs (11 and 34): The mixture of HCHO (10 equiv, 37% aq), glacial AcOH (1.5 equiv), and 1,4- dioxane (1 mL) was stirred for 15 min, then NaN3 (1.5 equiv), followed by the alkyne (1 equiv), was added to the reaction. After 10 min of stirring, sodium ascorbate (0.2 equiv) and CuSO4.5H2O (0.4 equiv) were added. The mixture was stirred for 18 h at room temperature, then diluted with H2O (20 mL), extracted with CHCl3 (3 × 20 mL), dried over Na2SO4, filtered, and concentrated on a rotary evaporator to give a residue. The crude product was purified by column chromatography (acetone: hexanes, 3:7) to give a regio-isomeric mixture of hydroxy methyl triazoles, which was subjected to the next step without further purification.
(4-((3S,8S,9S,10R,13S,14S,17S)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1H-1,2,3-triazol-1-yl)methanol (11a+b): Compound 11a+b was prepared following general procedure B using alkyne 8 (0.400 g, 0.745 mmol), HCHO (0.600 mL, 7.45 mmol, 37% aq), and glacial AcOH (64.0 μL, 1.19 mol), then NaN3 (73.0 mg, 1.5 mmol), sodium ascorbate (59.0 mg, 0.030 mol), and CuSO4.5H2O (37.0 mg, 0.015 mmol). The crude product was purified by flash column chromatography (acetone: hexanes, 3:7) to give a regio-isomeric mixture of hydroxy methyl triazoles 11a and 11b as a white foam (0.309 g, 65% yield), which was used for the next step without further purification.
4-((3S,8S,9S,10R,13S,14S,17S)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1H-1,2,3-triazole (12): The regioisomeric mixture 11a+b (0.220g, 0.361 mmol) and active MnO2 (0.314 g, 3.61 mmol) in CHCl3 (20 mL) was stirred under reflux for 20 h. Then, the reaction was filtered through Celite, washed with CHCl3:MeOH, 1:1, and the solvent was removed under reduced pressure. The residue was purified by column chromatography to obtain 12 (0.172 g, 82%) as a white foam: 1H NMR (400 MHz, CDCl3) δ 7.72–7.64 (m, 4H), 7.50 (s, 1H), 7.45–7.32 (m, 6H), 5.14 (d, J = 5.2 Hz, 1H), 3.64–3.45 (m, 1H), 2.77 (t, J = 9.8 Hz, 1H), 2.33 (td, J = 13.4, 6.8 Hz, 1H), 2.23–1.87 (m, 4H), 1.86–1.10 (m, 13H), 1.06 (d, J = 5.7 Hz, 9H), 1.01–0.96 (m, 3H), 0.95–0.80 (m, 2H), 0.49 (s, 3H). HRMS (ESI) calcd for C37H50N3OSi (M + H)+ 580.3723, found 580.3740.
General procedure C for silyl deprotection: The silylated triazole (0.10 mmol HCl in MeOH (1 N, 5 mL) and the solution were stirred for 5 h at room temperature. The reaction mixture was concentrated in vacuo. The residue was E3N (1 mL) to obtain a solid, which was filtered off and purified by column chromatography (silica gel, acetone/hexanes, 4:6) or washing with DCM to give the triazole targets.
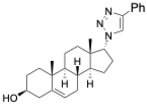
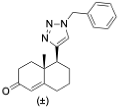
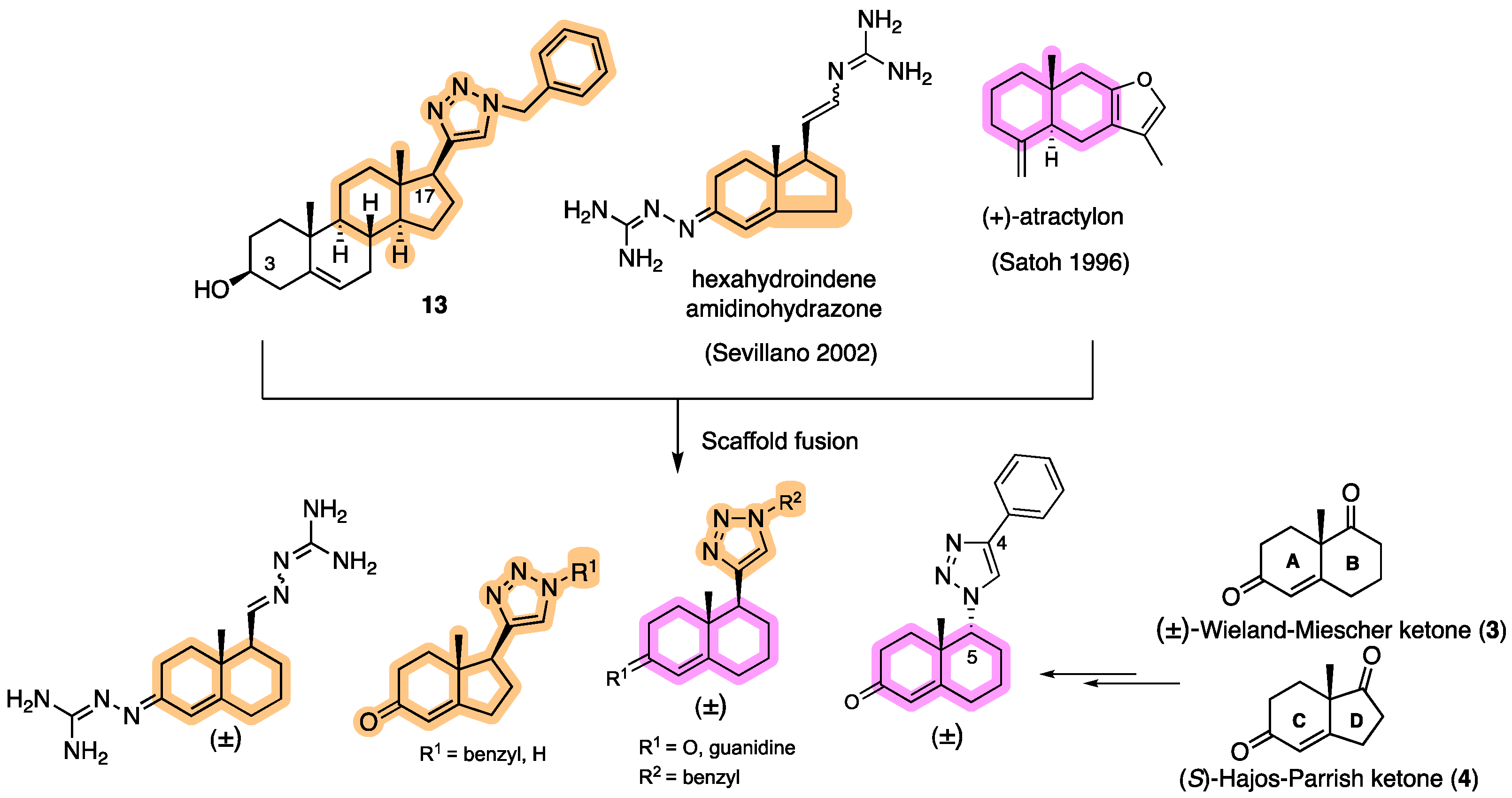
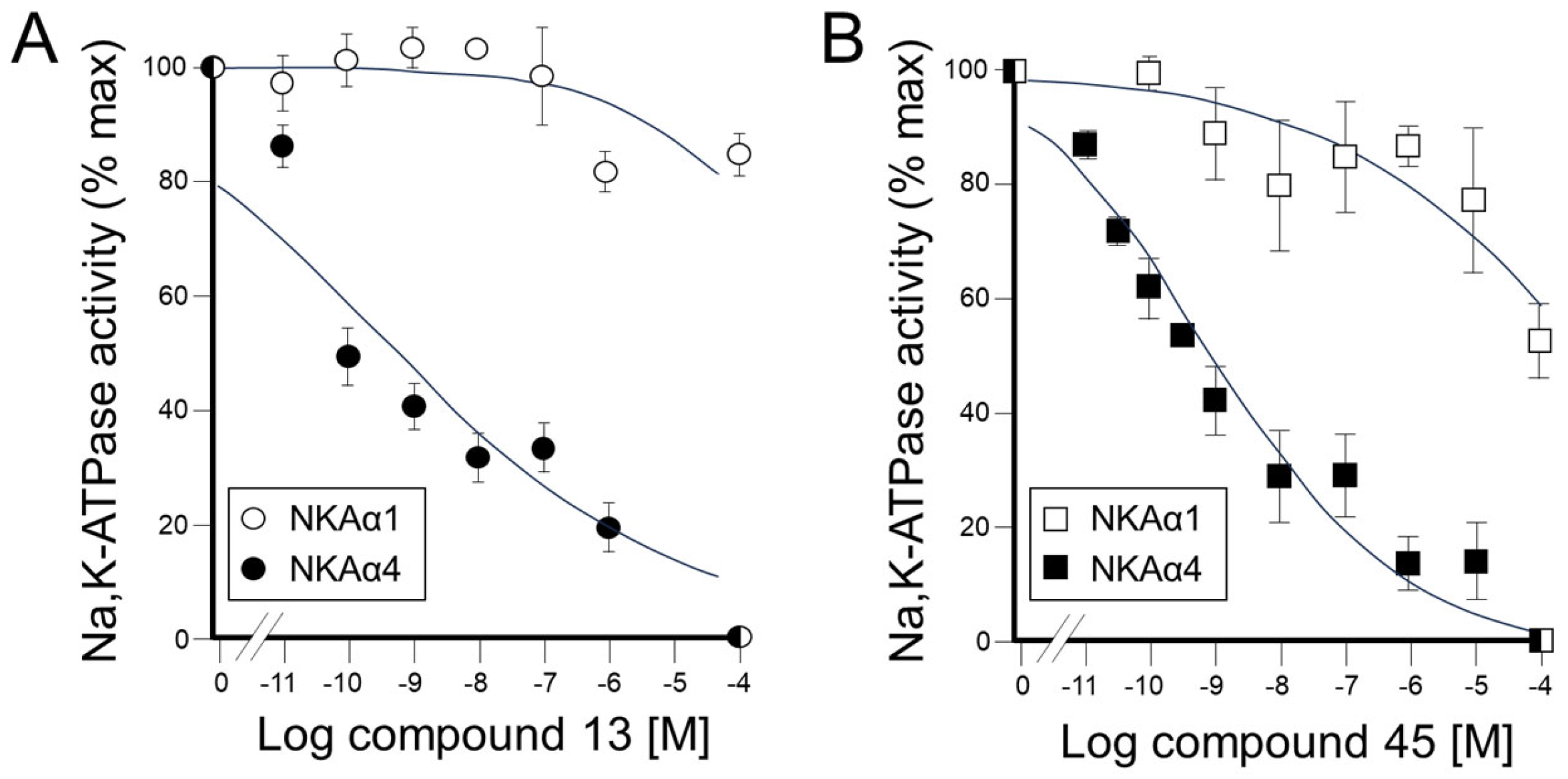
(3S,8S,9S,10R,13S,14S,17S)-17-(1-Benzyl-1H-1,2,3-triazol-4-yl)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-ol (13): Compound 13 was prepared following general procedure C using silylated benzyl triazole 9 (80 mg, 0.12 mmol) in 1 N HCl in MeOH (5 mL). The solid was washed with DCM to give pure 13 (41 mg, 80%) as a white solid: mp 286–287 °C; –9.41 (c 1.55, CHCl3: MeOH (3:1); 1H NMR (400 MHz, CDCl3+CD3OD) δ 7.36–7.26 (m, 4H), 7.25–7.15 (m, 2H), 5.48 (s, 2H), 5.31 (dt, J = 4.1, 1.9 Hz, 1H), 3.85 (s, 1H), 3.42 (tt, J = 10.7, 4.8 Hz, 1H), 2.73 (t, J = 9.8 Hz, 1H), 2.19 (dtdd, J = 15.8, 13.2, 5.3, 2.3 Hz, 2H), 2.10–1.87 (m, 3H), 1.84–1.66 (m, 4H), 1.60–0.97 (m, 10H), 0.96 (s, 3H), 0.44 (s, 3H); 13C NMR (100 MHz, CDCl3+CD3OD) δ 153.0, 144.8, 138.7, 132.9, 132.5, 131.6, 125.3, 125.1, 75.0, 59.8, 57.8, 54.1, 51.7, 47.3, 45.6, 41.38, 41.15, 40.4, 36.2, 35.6, 34.9, 30.5, 28.3, 24.5, 23.1, 16.6; HRMS (ESI) calcd for C28H38N3O (M + H)+ 432.3015, found 432.3018.
2-(4-((3S,8S,9S,10R,13S,14S,17S)-3-Hydroxy-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-1H-1,2,3-triazol-1-yl)acetic acid (14): Compound 14 was prepared following general procedure C using silylated triazole 10 (60 mg, 0.092 mmol) in HCl in MeOH (1 N, 4 mL). The residue was purified by column chromatography (acetone/hexanes, 4:6) to obtain 14 (27 mg, 70%) as a white solid: mp 221–223 °C; –15.68 (c 0.38 CHCl3: MeOH (3:1)); 1H NMR (400 MHz, CDCl3+CD3OD) δ 7.40 (s, 1H), 5.28 (d, J = 5.2 Hz, 1H), 5.09 (s, 2H), 3.73 (s, 3H), 3.29 (s, 1H), 2.74 (t, J = 9.8 Hz, 1H), 2.17 (p, J = 12.8 Hz, 2H), 2.08–1.90 (m, 3H), 1.75 (dd, J = 18.0, 11.6 Hz, 4H), 1.58–1.10 (m, 9H complex), 1.07–0.85 (m, 5H), 0.83–0.71 (m, 1H), 0.44 (s, 3H); HRMS (ESI) calcd for C24H36N3O3 (M + H)+ 414.2757, found 414.2767.
(3
S,8
S,9
S,10
R,13
S,14
S,17
S)-10,13-Dimethyl-17-(1
H-1,2,3-triazol-4-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1
H-cyclopenta[
a]phenanthren-3-ol (
15): Compound
15 was prepared following general procedure
C using silylated triazole
12 (0.07 g, 0.12 mmol in 1 N HCl in MeOH (6 mL). The residue was purified by column chromatography (acetone/hexanes, 4:6) to obtain
15 (0.027 g, 67%) as a white solid: mp. 236–238 °C. Spectroscopic data for compound
15 were consistent with previously reported data [
42].
(3
S,8
S,9
S,10
R,13
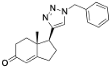
S,14
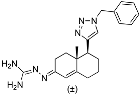
S,17
R)-17-Ethynyl-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1
H-cyclopenta[
a]phenanthren-3-ol (
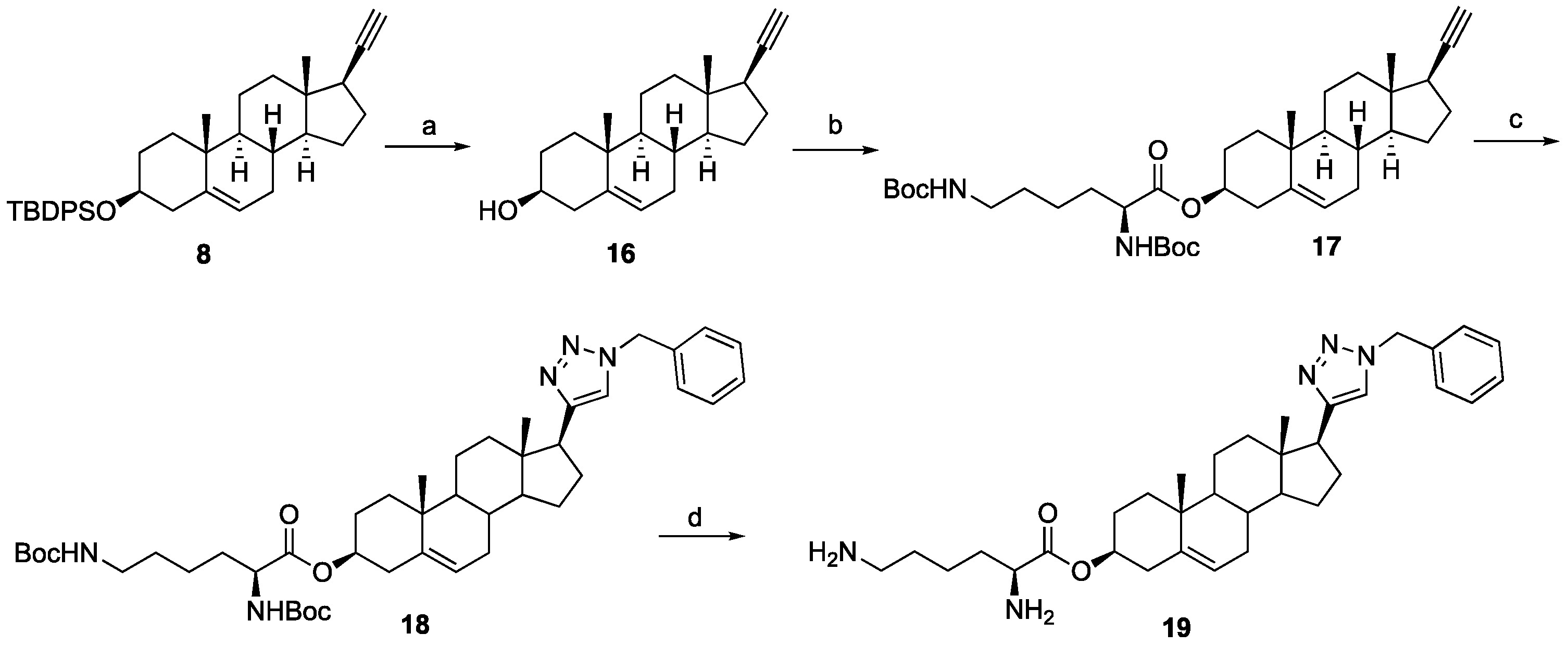
16): Silylated alkyne
8 (0.48 g, 0.89 mmol) was dissolved in 1 N HCl in MeOH (10 mL) and the solution was stirred for 5 h at room temperature. The reaction mixture was concentrated in vacuo. The residue was purified by column chromatography (silica gel, acetone/hexanes, 2:7) to give compound
16 (0.168 g, 63%) as a white solid.
1H NMR (400 MHz, CDCl
3) δ 5.36–5.29 (m, 1H), 3.59–3.43 (m, 1H), 2.38–2.10 (m, 3H), 2.09–1.94 (m, 3H), 1.93–1.77 (m, 3H), 1.76–1.37 (m, 8H), 1.34–1.14 (m, 1H), 1.16–0.87 (m, 7H), 0.79 (s, 3H);
13C NMR (100 MHz, CDCl
3) δ 140.8, 121.4, 85.9, 71.7, 69.8, 54.9, 50.1, 43.6, 42.2, 41.9, 37.3, 37.1, 36.6, 32.4, 31.8, 31.6, 29.0, 24.6, 20.8, 19.4, 13.3. Spectroscopic data for compound
12 were consistent with previously reported data [
43].
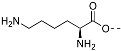
(3S,8S,9S,10R,13S,14S,17R)-17-Ethynyl-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl N2,N6-bis(tert-butoxycarbonyl)-L-lysinate (17): To a solution of compound 16 (0.19 g, 0.63 mmol), N,N-diBoc-L-lysine (0.24 g, 0.70 mmol), and 4-dimethylaminopyridine (7 mg, 0.006 mmol) in anhydrous THF (12 mL) dicyclohexylcarbodiimide (0.10 g, 0.70 mmol) was added at room temperature. The solution was stirred overnight and then filtered through Celite. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by column chromatography (acetone/hexanes, 3:7) to give compound 17 (0.30 g, 76%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 5.37 (d, J = 4.5 Hz, 1H), 5.07 (d, J = 7.0 Hz, 1H), 4.73–4.51 (m, 2H), 4.22 (d, J = 4.8 Hz, 1H), 3.11 (d, J = 6.2 Hz, 2H), 2.31 (d, J = 7.6 Hz, 2H), 2.25–2.13 (m, 1H), 2.12–1.95 (m, 2H), 1.89–1.34 (m, 34H), 1.27–1.21 (m, 1H), 1.16–0.91 (m, 7H), 0.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.1, 156.0, 155.4, 139.4, 122.6, 85.8, 79.7, 79.1, 74.8, 69.8, 54.8, 53.3, 50.0, 43.6, 41.8, 40.1, 37.9, 37.0, 36.9, 36.6, 32.5, 32.3, 31.8, 29.6, 29.0, 28.4, 28.3, 27.6, 24.6, 22.4, 20.8, 19.3, 13.3.
(3S,10R,13S,17S)-17-(1-Benzyl-1H-1,2,3-triazol-4-yl)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl N2,N6-bis(tert-butoxycarbonyl)-L-lysinate (18): Compound 18 was prepared following general procedure A using alkyne 17 (0.31 g, 0.50 mmol), benzyl azide (46 mg, 0.18 mmol), sodium ascorbate (39 mg, 0.20 mmol), and CuSO4.5H2O (24 mg, 0.10 mmol) to yield 18 (0.29 g, 78%) as a solid: 1H NMR (400 MHz, CDCl3) δ 7.49–7.39 (m, 3H), 7.34 (d, J = 5.8 Hz, 1H), 7.32 (dd, J = 7.5, 1.8 Hz, 2H), 5.65–5.55 (m, 2H), 5.47 (d, J = 3.9 Hz, 1H), 5.18 (d, J = 7.5 Hz, 1H), 4.85–4.59 (m, 2H), 4.31 (d, J = 4.6 Hz, 1H), 3.19 (d, J = 6.1 Hz, 2H), 2.87 (t, J = 9.8 Hz, 1H), 2.40 (d, J = 7.7 Hz, 2H), 2.23–2.02 (m, 3H), 1.99–1.56 (m, 14H), 1.55–1.50 (m, 18H), 1.47–1.23 (m, 6H), 1.16–1.00 (m, 4H), 0.57 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.1, 156.0, 155.4, 149.2, 139.4, 135.1, 129.0, 128.5, 127.7, 122.7, 120.8, 79.7, 79.1, 74.9, 55.9, 53.9, 53.3, 50.1, 47.8, 43.5, 40.1, 37.9, 37.6, 36.9, 36.6, 32.4, 32.2, 31.8, 29.5, 28.4, 28.3, 27.6, 26.6, 24.5, 22.4, 20.6, 19.3, 12.9.
(3S,10R,13S,17S)-17-(1-Benzyl-1H-1,2,3-triazol-4-yl)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl L-Lysinate Dihydrochloride (19): Compound 18 (0.20 g, 0.26 mmol) was dissolved in 2 N HCl in ether (10 mL), and the reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the resulting residue was purified by column chromatography (silica gel, MeOH/DCM, 2:8) to obtain 19 (0.10 g, 71%) as a yellowish solid: mp 258–260 °C; − 0.50 (c 0.59, MeOH). 1H NMR (400 MHz, CD3OD) δ 8.02 (s, 1H), 7.36 (s, 5H), 5.56 (d, J = 52.2 Hz, 2H), 5.45 (s, 1H), 4.75–4.70 (m, 1H), 4.02 (s, 1H), 2.91 (m, 3H), 2.31 (m, 2H), 2.30–0.84 (m, 26H), 0.53 (s, 3H); LCMS (ESI) m/z 560.52 (M + H)+.
(3
S,8
R,9
S,10
R,13
S,14
S)-3-((
tert-Butyldiphenylsilyl)oxy)-10,13-Dimethyl-1,2,3,4,7,8,9,10,11,12,13,14,15,16-tetradecahydro-17
H-cyclopenta[
a]phenanthren-17-one (
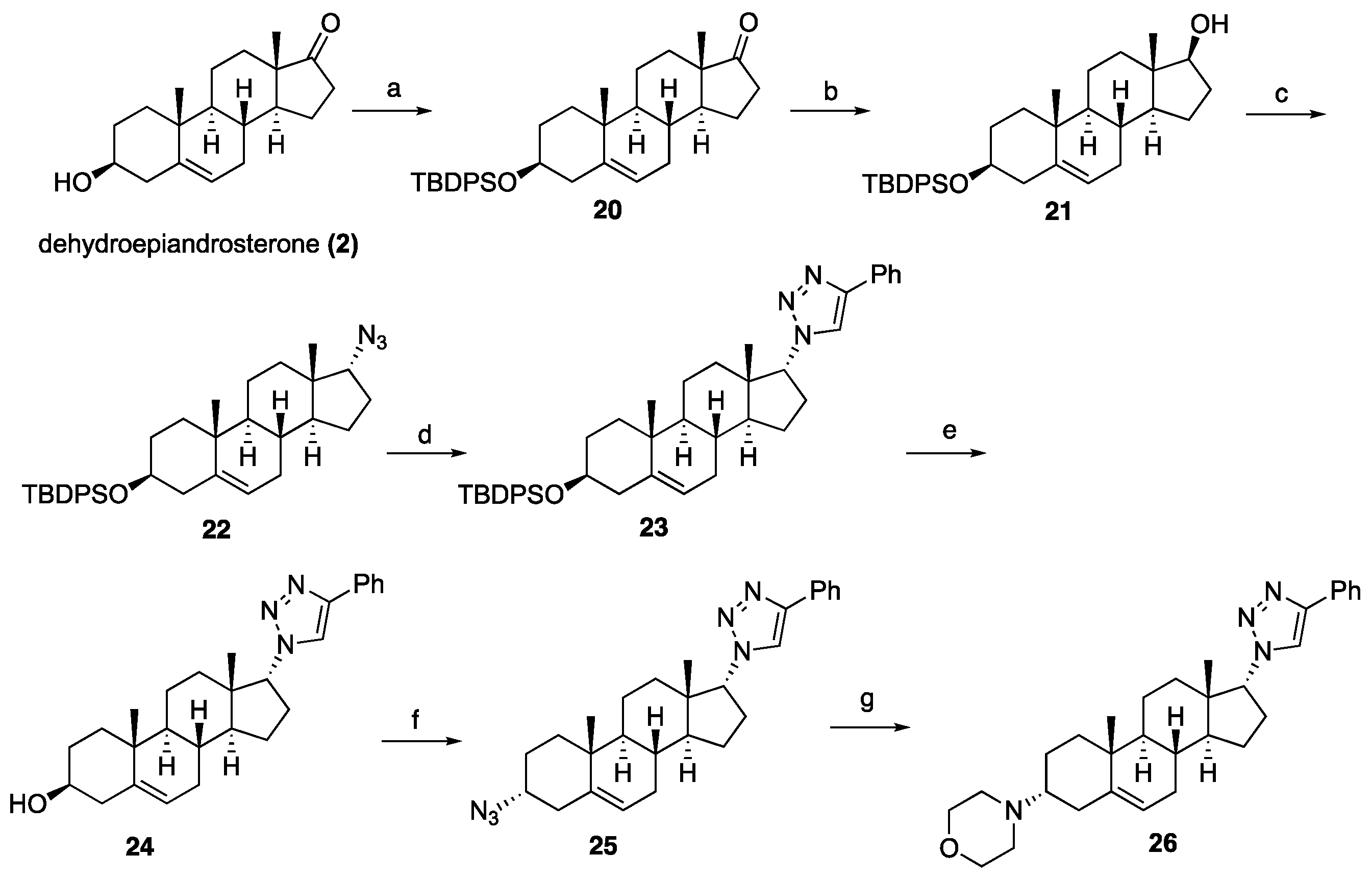
20): To a solution of dehydro-epi-androsterone (3.00 g, 10.41 mmol) in CH
2Cl
2 (75 mL) at 0 °C, imidazole (1.41 g, 20.83 mmol) and TBDPSCl (2.92 mL, 11.45 mmol) were added. After being stirred at ambient temperature for 5 h, the reaction mixture was quenched with saturated aqueous NaHCO
3 (50 mL) and extracted with CH
2Cl
2 (2 × 75 mL). The combined organic layers were dried over Na
2SO
4 and concentrated in vacuo to give a residue, which was purified by column chromatography (silica gel, EtOAc/hexanes, 1:9) to give compound
20 (5.37 g, 98%) as a white solid. Spectroscopic data for compound
20 were consistent with previously reported data [
44].
(3
S,8
R,9
S,10
R,13
S,14
S,17
S)-3-((
tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1
H-cyclopenta[
a]phenanthren-17-ol (
21): To a solution of ketone
20 (1.70 g, 3.23 mmol) in MeOH:THF (30 mL, 1:1), NaBH
4 (0.36 g, 9.69 mmol) was added at 0 °C. After stirring for 1 h, the reaction mixture was quenched by the addition of saturated aqueous NH
4Cl (50 mL). The mixture was extracted with EtOAc (3 × 50 mL), washed with brine (50 mL), and dried over Na
2SO
4. The volatiles were evaporated and purified by flash chromatography (silica gel, EtOAc/hexane 2:8) to afford the alcohol
21 (1.50 g, 88%) as a white solid. Spectroscopic data for compound
21 were consistent with previously reported data [
44].
General procedure D for the Mitsunobu azidation: To a solution of triphenylphosphine (1.3 equiv) in THF (30 mL) diethyl azodicarboxylate (1 equiv, 40% solution in toluene) was added at 0 °C, and the resulting orange solution was stirred for 10 min. The alcohol (1.0 equiv) in THF (10 mL) was added to the above solution. After stirring for 10 min, diphenylphosphoryl azide (1.7 equiv) was added. The reaction mixture was allowed to warm to room temperature and stirred for 10 h. The solvent was evaporated, and the resulting residue was purified by column chromatography (silica gel, EtOAc/hexanes, 1:9) to yield the azides.
(((3S,8R,9S,10R,13S,14S,17R)-17-Azido-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)oxy)(tert-butyl)diphenylsilane (22): Compound 22 was prepared following general procedure D using triphenylphosphine (1.09 g, 4.18 mmol), diethyl azodicarboxylate (2.20 mL, 4.82 mmol, 40% solution in toluene), alcohol 21 (1.70 g, 3.21 mmol), and diphenyl phosphoryl azide (1.18 g, 4.82 mmol) to obtain 22 (1.26 g, 71%) as a viscous oil. 1H NMR (400 MHz, CDCl3) δ 7.77–7.59 (m, 4H), 7.48–7.29 (m, 6H), 5.18–5.05 (m, 1H), 3.53 (ddd, J = 16.4, 10.5, 5.4 Hz, 2H), 2.44–2.23 (m, 1H), 2.24–2.04 (m, 2H), 2.03–1.84 (m, 1H), 1.81–1.29 (m, 11H (overlapped with H2O), 1.28–1.12 (m, 2H), 1.06 (s, 9H), 0.98 (s, 3H), 0.92–0.81 (m, 2H), 0.73 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.2, 135.7 (2C), 134.7, 129.4 (2C), 127.4 (2C), 120.8, 73.1, 71.4, 49.8, 49.6, 45.6, 42.4, 37.2, 36.5, 32.4, 32.1, 32.0, 31.8, 28.6, 27.0, 24.7, 20.5, 19.4, 19.1, 17.4.
1-((3S,8R,9S,10R,13S,14S,17R)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-4-phenyl-1H-1,2,3-triazole (23): Compound 23 was prepared following general procedure A using azide 22 (0.900 g, 1.62 mmol) and phenylacetylene (0.330 g, 3.25 mmol), sodium ascorbate (128 mg, 0.64 mmol), and CuSO4.5H2O (80 mg, 0.32 mmol) to yield 23 (0.71 g, 67%) as a solid: 1H NMR (400 MHz, CDCl3) δ 7.82 (dd, J = 8.2, 1.2 Hz, 2H), 7.69–7.60 (m, 5H), 7.47–7.28 (m, 9H), 5.21–5.04 (m, 1H), 4.62 (dt, J = 14.0, 7.0 Hz, 1H), 3.61–3.35 (m, 1H), 2.66–2.41 (m, 1H), 2.26 (tt, J = 22.9, 11.6 Hz, 2H), 2.19–1.91 (m, 3H), 1.74–1.31 (m, 10H(overlapped with H2O)), 1.12–0.99 (m, 9H), 0.97 (s, 3H), 0.95 (s, 3H), 0.83–0.62 (m, 2H), 0.28 (dd, J = 18.3, 10.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 146.9, 141.2, 135.7 (2C), 134.7 (2C), 129.4 (2C), 128.7, 127.9, 127.4 (2C), 125.6, 120.6, 119.5, 73.1, 70.3, 50.2, 49.3, 46.1, 42.4, 37.0, 36.4, 32.4, 32.1, 31.9, 31.7, 28.6, 26.9, 25.3, 20.3, 19.3, 19.1, 18.3; HRMS (ESI) calcd for C43H54N3OSi (M + H)+ 656.4036, found 656.4038.
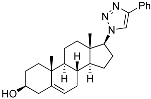
(3S,8R,9S,10R,13S,14S,17R)-10,13-Dimethyl-17-(4-phenyl-1H-1,2,3-triazol-1-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-ol (24): Compound 24 was prepared following general procedure C using 23 (0.50 g, 0.76 mmol) dissolved in 1 N HCl in MeOH (15 mL). The residue was purified by column chromatography (silica gel, acetone/hexanes, 4:6) to give 24 (0.23 g, 72%) as a white solid: mp 234–236 °C; – 25.36 (c 0.40, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 7.2 Hz, 2H), 7.67 (s, 1H), 7.42 (t, J = 7.6 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 5.43–5.28 (m, 1H), 4.64 (dd, J = 8.5, 1.6 Hz, 1H), 3.60–3.37 (m, 1H), 2.55 (ddd, J = 18.7, 10.8, 2.5 Hz, 1H), 2.43–1.97 (m, 5H), 1.87–1.33 (m, 11H (overlapped with H2O)), 1.08–0.77 (m, 8H), 0.46–0.22 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 146.9, 140.7, 130.7, 128.8, 128.0, 125.6, 121.2, 119.7, 71.6, 70.3, 50.2, 49.4, 46.1, 42.1, 37.1, 36.4, 32.4, 32.2, 31.9, 31.5, 28.6, 25.3, 20.4, 19.3, 18.4; HRMS (ESI) calcd for C27H36N3O (M + H)+ 418.2858, found 418.2842.
1-((3R,8R,9S,10R,13S,14S,17R)-3-Azido-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-4-phenyl-1H-1,2,3-triazole (25): Compound 25 was prepared following general procedure D using triphenylphosphine (0.16 g, 0.62 mmol), diethyl azodicarboxylate (0.12 g, 0.71 mmol, 40% solution in toluene), alcohol 24 (0.20 g, 0.47 mmol), and diphenyl phosphoryl azide (0.22 mL, 0.81 mmol) to obtain 25 (0.12 g, 58%) as a yellow foam. 1H NMR (400 MHz, CDCl3) δ 7.89–7.79 (m, 2H), 7.67 (d, J = 5.7 Hz, 1H), 7.42 (t, J = 7.6 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 5.39 (dd, J = 13.5, 11.0 Hz, 1H), 4.69 (dd, J = 8.6, 1.8 Hz, 1H), 3.88–3.80 (m, 1H), 2.72–2.39 (m, 2H), 2.41–1.97 (m, 4H), 1.90–1.21 (m, 12H), 1.08–0.90 (m, 6H), 0.45–0.21 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 147.0, 138.0, 128.7, 128.0, 125.6 (2C), 122.6, 119.3, 70.2, 57.9, 50.3, 49.2, 46.0, 37.0, 35.9, 33.4, 32.4, 32.0, 31.9, 28.7, 26.0, 25.2, 20.0, 18.9, 18.3; HRMS (ESI) calcd for C27H35N6 (M + H)+ 443.2923, found 443.2911.
4-((3R,8R,9S,10R,13S,14S,17R)-10,13-Dimethyl-17-(4-phenyl-1H-1,2,3-triazol-1-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)morpholine (26): To a solution of azide 25 (0.13 g, 0.30 mmol) in THF, triphenyl phosphine (0.10 g, 0.39 mmol) was added at ambient temperature. After stirring for 1 h, the reaction mixture was diluted with water and stirred at room temperature for 10 h. The solvent was evaporated under reduced pressure to give the corresponding amine, which was used for the next step without purification.
To a solution of the crude amine (0.13 g, 0.32 mmol) in toluene (10 mL), 2-bromomethyl ether (0.11 g, 0.48 mmol) and K2CO3 (0.050 g, 0.64 mmol) were added. The reaction mixture was refluxed for 24 h. After cooling to room temperature, the reaction mixture was filtered and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, CHCl3/MeOH, 1:9) to afford 26 (0.11 g, 77%) as a white solid: mp 178–180 °C; –15.56 (c 0.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.94–7.78 (m, 2H), 7.68 (s, 1H), 7.43 (dd, J = 10.4, 4.7 Hz, 2H), 7.32 (dd, J = 16.2, 8.7 Hz, 1H), 5.18 (s, 1H), 4.71–4.59 (m, 1H), 3.65 (s, 4H), 2.66–1.98 (m, 10H), 1.86–1.19 (m, 12H (overlapped with H2O), 0.99 (d, J = 9.7 Hz, 7H), 0.35 (dd, J = 12.3, 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 146.9, 140.6, 130.8, 128.8, 128.0, 125.6, 119.9, 119.6, 70.3, 67.1, 59.9, 50.3 (2C), 49.0, 46.1, 36.9, 34.5, 33.3, 32.5, 32.1, 31.8, 28.7, 25.2, 24.2, 20.0, 19.9, 18.4; HRMS (ESI) calcd for C31H43N4O (M + H)+ 487.3437, found 487.3437.
(3S,8R,9S,10R,13S,14S,17R)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl 4-nitrobenzoate (27): To a solution of triphenylphosphine (3.87 g, 14.77 mmol) in THF (100 mL), diethyl azodicarboxylate (2.87 g, 16.25 mmol) was added at 0 °C. The resulting orange solution was stirred for 10 min. Alcohol 21 (3.90 g, 7.38 mmol) in THF (20 mL) was added to the above solution. After stirring for 10 min, a solution of 4-nitrobenzoic acid (2.96 g, 17.72 mmol) in THF (15 mL) was added. The reaction mixture was allowed to warm to room temperature and stirred for 12 h. The solvent was evaporated under reduced pressure, and the residue was purified by column chromatography (silica gel, EtOAc/hexanes, 15:85) to give impure 30 (3.90 g) as a yellowish foam, which was used directly in the next step.
(3S,8R,9S,10R,13S,14S,17R)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-ol (28): 4-Nitrobenzoate 27 (3.10 g, 4.64 mmol) was dissolved in THF (30 mL) and MeOH (10 mL). Powdered K2CO3 (1.28 g, 9.29 mmol) was added, and the reaction mixture was stirred at room temperature overnight. EtOAc (100 mL) was added to dilute the reaction and filtered through a pad of Celite. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by column chromatography (EtOAc/hexanes 1:2) to obtain corresponding inverted alcohol 28 (2.07 g, 53% for two steps) as a viscous oil: 1H NMR (400 MHz, CDCl3) δ 7.79–7.61 (m, 4H), 7.51–7.29 (m, 6H), 5.26–5.07 (m, 1H), 3.73 (d, J = 5.9 Hz, 1H), 3.67–3.46 (m, 1H), 2.51–2.25 (m, 1H), 2.23–2.09 (m, 2H), 2.03–1.90 (m, 1H), 1.81–1.28 (m, 13H), 1.22–1.07 (m, 10H), 1.01 (s, 3H), 0.94–0.83 (m, 2H), 0.66 (s, 3H).
1-((3S,8R,9S,10R,13S,14S,17S)-3-((tert-Butyldiphenylsilyl)oxy)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-4-phenyl-1H-1,2,3-triazole (29): To a solution of triphenylphosphine (1.22 g, 4.67 mmol) in THF (50 mL), diethyl azodicarboxylate (0.93 g, 5.39 mmol) was added at 0 °C, and the resulting orange solution was stirred for 10 min. Alcohol (1.90 g, 3.59 mmol) in THF (10 mL) was added to the above solution. After being stirred for 10 min, a solution of diphenylphosphoryl azide (1.68 g, 6.11 mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred for 12 h. The solvent was evaporated under reduced pressure, and the residue was purified by column chromatography (silica gel, EtOAc/hexanes) to give azide (0.85 g, 43%) as a yellow oil. LCMS (ESI) m/z 576.24 (M + Na)+.
To a mixture of the azide (0.40 g, 0.72 mmol) and phenylacetylene (0.14 g, 1.44 mmol) in DMF (6 mL), sodium ascorbate (57 mg, 0.29 mmol) in H2O (3 mL) was added and stirred for two min at ambient temperature. Then, a CuSO4·5H2O (35 mg, 0.14 mmol) in H2O (3 mL) was added to the above mixture. The mixture was stirred at room temperature for 12 h and extracted with EtOAc (3 × 20 mL). The evaporation of combined organic extracts under reduced pressure afforded a green solid, which was purified by flash column chromatography (silica gel, acetone/hexanes, 2:8) to give silyl protected triazole 29 (0.27 g, 58%) as a viscous oil: 1H NMR (400 MHz, CDCl3) δ 7.86–7.80 (m, 2H), 7.73 (s, 1H), 7.68 (ddd, J = 7.9, 3.4, 1.5 Hz, 4H), 7.47–7.31 (m, 9H), 5.14 (d, J = 5.1 Hz, 1H), 4.41 (t, J = 9.5 Hz, 1H), 3.63–3.45 (m, 1H), 2.69–2.46 (m, 1H), 2.41–2.22 (m, 2H), 2.15 (dd, J = 13.3, 2.8 Hz, 1H), 2.09–1.76 (m, 3H), 1.77–1.43 (m, 8H overlapped with H2O), 1.40–1.12 (m, 4H), 1.06 (s, 9H), 1.00 (d, J = 11.1 Hz, 3H), 0.97–0.80 (m, 2H), 0.59 (s, 3H).
(3S,8R,9S,10R,13S,14S,17S)-10,13-Dimethyl-17-(4-phenyl-1H-1,2,3-triazol-1-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-ol (30): Compound 30 was prepared following general procedure C using silylated triazole (0.17 g, 0.25 mmol) in 1 N HCl in MeOH (10 mL) and the solution was stirred for 5 h at room temperature. The reaction mixture was concentrated under reduced pressure. The residue was purified by column chromatography (silica gel, acetone/hexanes, 3:7) to give compound 30 (70 mg, 65%) as a white solid: mp 278–280 °C. 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 7.77 (d, J = 6.8 Hz, 2H), 7.38 (t, J = 7.1 Hz, 2H), 7.30 (t, J = 7.9 Hz, 1H), 5.28 (d, J = 23.0 Hz, 1H), 4.42 (t, J = 8.9 Hz, 1H), 3.40 (m, 1H), 2.53 (m, 1H), 2.22 (dt, J = 23.8, 15.3 Hz, 3H), 2.01 (d, J = 15.0 Hz, 1H), 1.79 (d, J = 13.0 Hz, 4H), 1.69–1.14 (m, 8H), 1.13–0.87 (m, 5H), 0.56 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 150.5, 144.9, 133.4, 132.8, 132.4, 129.6, 124.7, 124.0, 75.0, 74.9, 57.0, 53.9, 48.2, 45.6, 41.1, 40.6, 40.4, 35.9, 35.3, 34.9, 29.6, 27.3, 24.4, 23.1, 15.8. LCMS (ESI) m/z 418.41 (M + H)+.
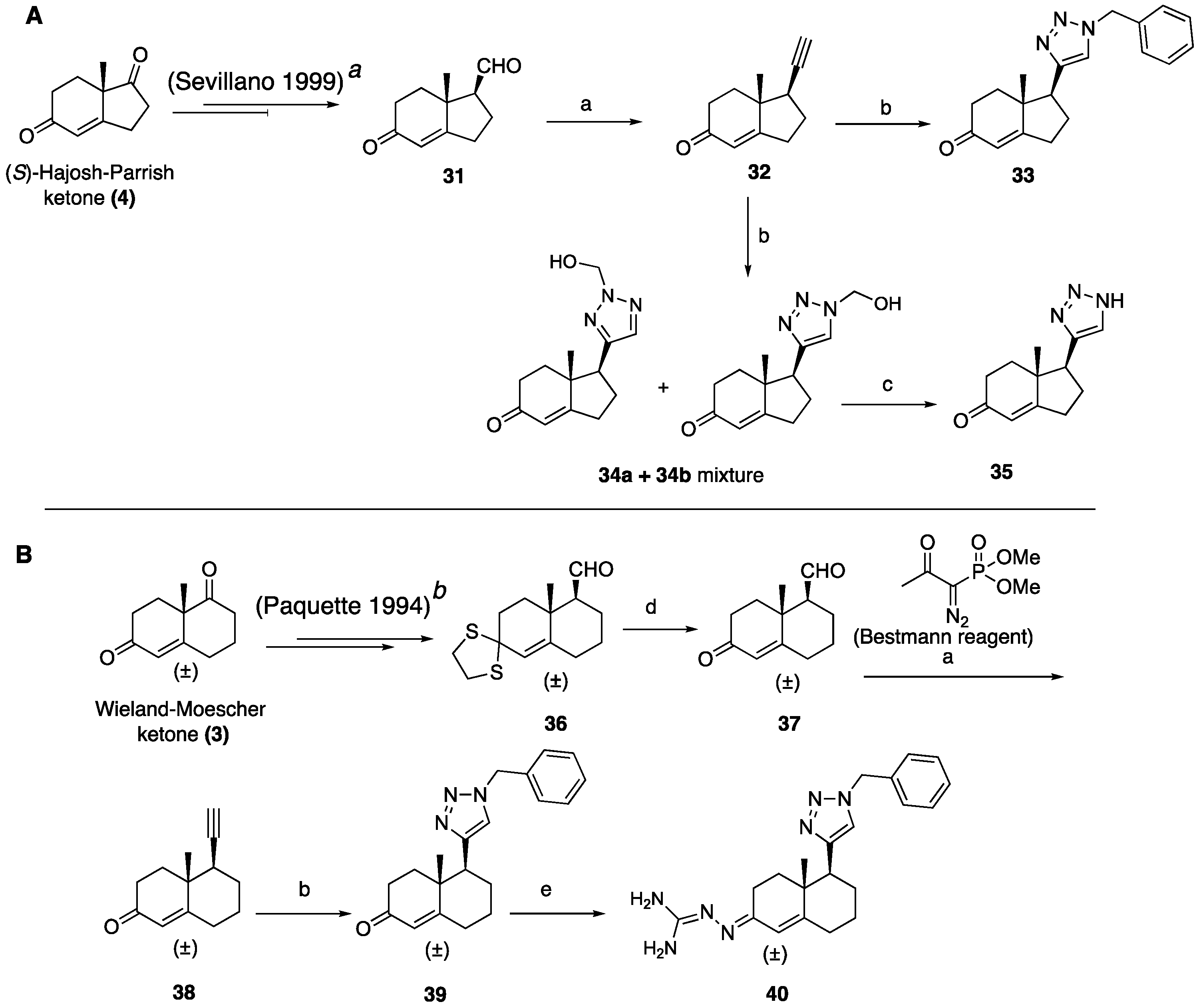
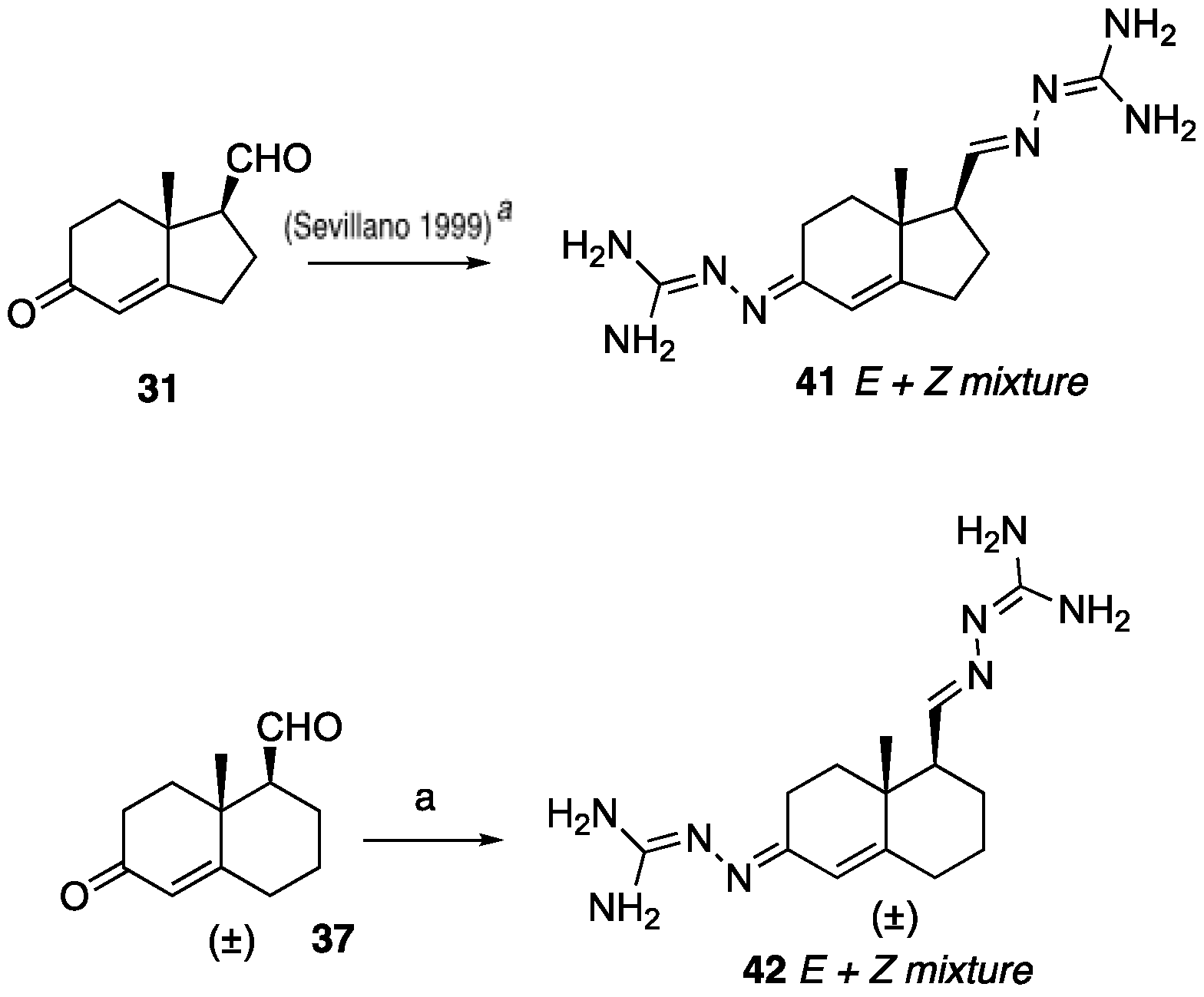
(1R,7aR)-1-Ethynyl-7a-methyl-1,2,3,6,7,7a-hexahydro-5H-inden-5-one (32): To a solution of Bestmann reagent (0.13 g, 0.71 mmol) in MeOH (10 mL), K2CO3 (0.19 g, 1.43 mmol) was added and the mixture was stirred for 10 minutes at 0 °C. Then, a solution of aldehyde 31 (85 mg, 0.47 mmol) in MeOH (5 mL) was added. After stirring at room temperature for 8 h, the reaction mixture was treated with saturated aqueous NaHCO3, and then the methanol was removed under reduced pressure. The residue was dissolved in EtOAc (30 mL) and washed with water, dried over Na2SO4, and concentrated under reduced pressure to give 32 (54 mg, 65%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 5.77 (s, 1H), 2.72 (dddd, J = 9.0, 7.2, 5.0, 2.7 Hz, 1H), 2.59–2.33 (m, 4H), 2.23–2.11 (m, 3H), 2.04–1.89 (m, 1H), 1.78 (td, J = 13.7, 5.2 Hz, 1H), 1.23 (d, J = 12.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 198.8, 175.3, 122.5, 82.6, 71.7, 45.3, 42.3, 34.5, 33.3, 28.7, 28.2, 17.6; HRMS (ESI) calcd for C12H15O (M + H)+ 175.1123, found 175.1128.
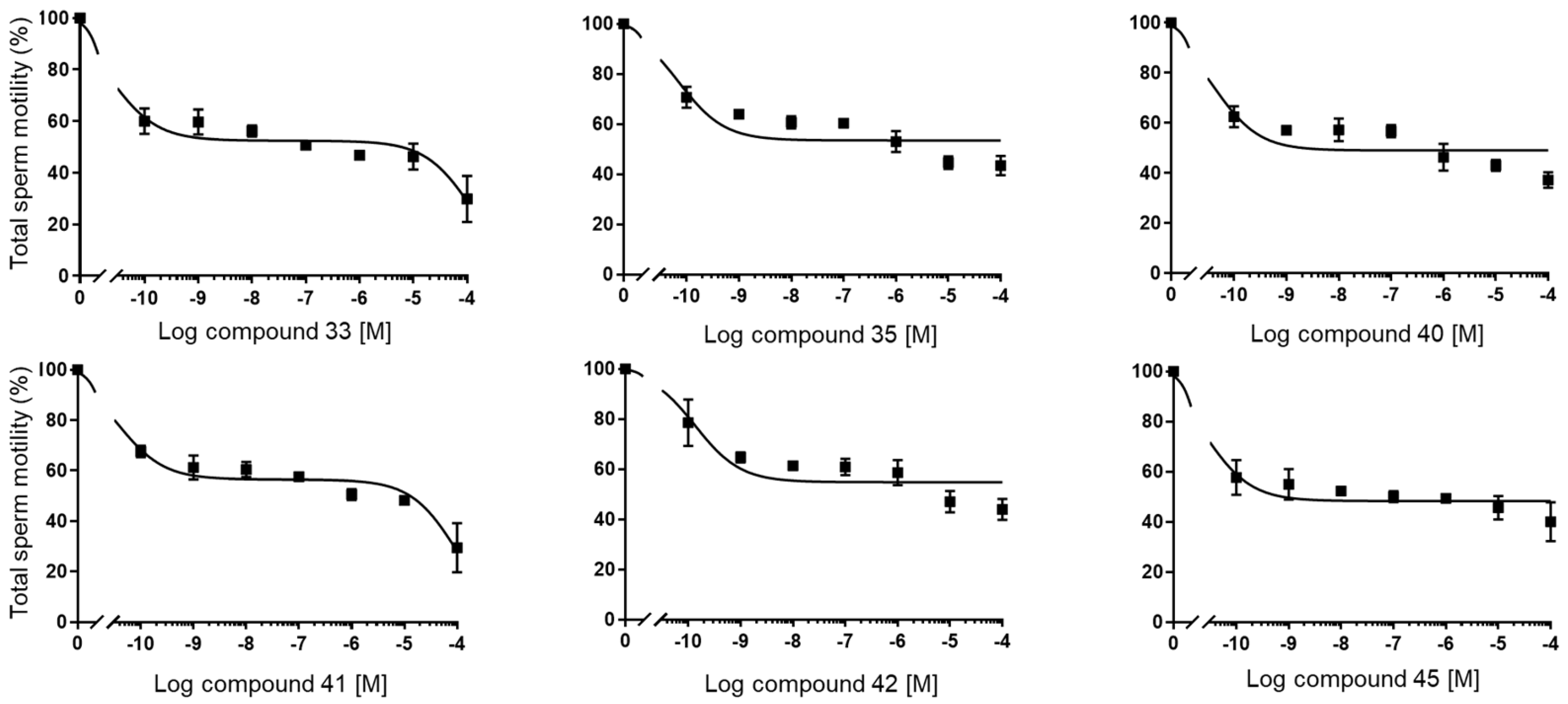
(1S,7aR)-1-(1-Benzyl-1H-1,2,3-triazol-4-yl)-7a-methyl-1,2,3,6,7,7a-hexahydro-5H-inden-5-one (33): Compound 33 was prepared following general procedure A using benzyl azide (57 mg, 0.43 mmol) and alkyne 32 (50 mg, 0.28 mmol), sodium ascorbate (22 mg, 0.11 mmol), and CuSO4.5H2O (13 mg, 0.056 mmol) to yield 33 as a white solid (59 mg, 67%). mp 122–125 °C; + 71.2 (c 0.40, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.41–7.24 (m, 3H), 7.25–7.13 (m, 3H), 5.76 (d, J = 10.1 Hz, 1H), 5.53–5.37 (m, 2H), 2.96 (dd, J = 12.3, 7.4 Hz, 1H), 2.82–2.65 (m, 1H), 2.59–2.00 (m, 6H), 1.96–1.86 (m, 1H), 0.82 (s, 3H); HRMS (ESI) calcd for C19H22N3O (M + H)+ 308.1763, found 308.1766.
(1S,7aR)-1-(2-(Hydroxymethyl)-2H-1,2,3-triazol-4-yl)-7a-methyl-1,2,3,6,7,7a-hexahydro-5H-inden-5-one (34a+34b): To a mixture of HCHO (0.41 mL, 5.17 mmol, 10 equiv, 37% aq.), glacial AcOH (0.04 mL, 0.77 mmol), and 1,4- dioxane (0.41 mL), stirred for 15 min, NaN3 (0.05 g, 0.77 mmol) was added, followed by alkyne 32 (0.09 g, 0.51 mmol). After an additional 10 min of stirring, sodium ascorbate (38.8 mg, 0.196 mol, 20 mol %) was added, followed by CuSO4.5H2O (39.6 mg, 0.20 mmol). The mixture was stirred for 18 h at room temperature and extracted with CHCl3 (3 × 10 mL). Combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to obtain the residue, which was a regioisomeric mixture of 34a and 34b (7:3) (0.07 g, 54%). The crude product was sufficiently pure to be used without further purification: 1H NMR (400 MHz, CD3OD) δ 7.44 (s, 0.7H), 7.19 (s, 0.3H), 5.88–5.56 (m, 2H), 4.97–4.55 (m, 2H), 2.94 (dd, J = 12.3, 7.4 Hz, 1H), 2.83–2.63 (m, 1H), 2.65–1.66 (m, 7H), 1.17 (s, 1H), 0.83 (s, 2H).
(1S,7aR)-7a-Methyl-1-(1H-1,2,3-triazol-4-yl)-1,2,3,6,7,7a-hexahydro-5H-inden-one (35): The mixture of 34a and 34b (65 mg, 0.26 mmol) and active MnO2 (220 mg, 2.63 mmol) in CHCl3 (10 mL) was stirred under reflux for 20 h. The reaction was filtered through Celite and washed with CHCl3: MeOH (1:1, 20 mL). The solvents were removed under reduced pressure. The residue was purified by column chromatography (silica gel, acetone/hexanes, 3:7) to obtain 35 (34 mg, 60%) as a white solid: mp 126–129 °C; + 2.25 (c 0.31, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 5.89 (t, J = 1.9 Hz, 1H), 3.09 (dd, J = 12.3, 7.4 Hz, 1H), 2.86 (ddt, J = 19.8, 10.7, 2.3 Hz, 1H), 2.65 (dtd, J = 19.8, 9.0, 1.8 Hz, 1H), 2.57–2.31 (m, 3H), 2.25 (dddd, J = 13.1, 9.4, 7.4, 2.3 Hz, 1H), 2.13–1.97 (m, 2H), 0.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 199.8, 177.7, 144.4, 131.9, 122.5, 47.5, 45.7, 35.2, 33.4, 29.0, 26.0, 17.4; HRMS (ESI) calcd for C12H16N3O (M + H)+ 218.1293, found 218.1279.
8a-Methyl-6-oxo-1,2,3,4,6,7,8,8a-octahydronaphthalene-1-carbaldehyde (37): To a solution of dithiolane 36 (0.40 g, 1.49 mmol) in CH3CN: H2O (10:1, 20 mL) at room temperature, bis(trifuoroacetoxy)iodobenzene (0.96 g, 2.23 mmol) was added in one portion. After stirring for 10 min, a mixture of water, saturated aqueous NaHCO3, and saturated aqueous Na2S2O3 (1:1:1, 18 mL) was added. The aqueous phase was extracted with EtOAc (3 × 20 mL). The combined organics were dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by column chromatography (silica gel, EtOAc/hexanes, 3:7) to obtain 37 (0.26 g, 92%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 9.83 (dd, J = 6.4, 3.0 Hz, 1H), 6.01–5.55 (m, 1H), 2.60–2.20 (m, 6H), 2.19–1.96 (m, 2H), 1.96–1.73 (m, 2H), 1.54–1.36 (m, 1H), 1.32 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 203.1, 198.6, 167.6, 125.0, 60.0, 38.5, 36.1, 33.6, 32.6, 25.2, 22.2, 18.3.
5-Ethynyl-4a-methyl-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-one (38): Compound 38 was synthesized using the same procedure employed for 32 using Bestmann reagent (0.60 g, 3.12 mmol), K2CO3 (0.860 g, 6.25 mmol), and 37 (0.400 g, 2.08 mmol) to obtain a crude residue, which was purified by column chromatography (silica gel, EtOAc/hexanes, 2:8) to obtain 38 (0.266 g, 68%) as a solid: 1H NMR (400 MHz, CDCl3) δ 5.79–5.73 (m, 1H), 2.57–2.18 (m, 6H), 1.96–1.74 (m, 4H), 1.43 (ddd, J = 17.2, 8.6, 2.9 Hz, 1H), 1.31 (s, 3H), 1.25 (t, J = 7.1 Hz, 1H).
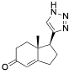
5-(1-Benzyl-1H-1,2,3-triazol-4-yl)-4a-methyl-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-one (39): Compound 39 was prepared following general procedure A using benzyl azide (0.21 g, 1.59 mmol) and alkyne 38 (0.20 g, 1.06 mmol), sodium ascorbate (84 mg, 0.42 mmol), and CuSO4.5H2O (52 mg, 0.21 mmol) to yield 39 as a white solid, (0.17 g, 51%). 1H NMR (400 MHz, CDCl3) δ 7.49–7.37 (m, 3H), 7.35–7.26 (m, 4H), 5.85 (d, J = 1.6 Hz, 1H), 5.64–5.48 (m, 2H), 2.89 (dd, J = 13.1, 3.4 Hz, 1H), 2.60–2.46 (m, 1H), 2.45–2.31 (m, 3H), 2.16 (qd, J = 13.4, 3.7 Hz, 1H), 2.09–1.96 (m, 2H), 1.85 (d, J = 13.6 Hz, 1H), 1.76 (dt, J = 13.6, 4.3 Hz, 1H), 1.56 (ddd, J = 16.2, 10.2, 5.5 Hz, 1H), 1.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 199.5, 169.3, 148.2, 134.8, 129.1, 128.7, 127.8, 124.8, 121.5, 54.0, 46.3, 39.8, 35.9, 33.7, 32.7, 27.6, 26.3, 17.8; HRMS (ESI) calcd for C20H24N3O (M + H)+ 322.1919, found 322.1910.
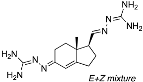
N’-(5-(1-Benzyl-1H-1,2,3-triazol-4-yl)-4a-methyl-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-ylidene)amidinohydrazone (40): To a solution of 39 (116 mg, 0.36 mmol) in EtOH (10 mL), aminoguanidine hydrochloride (39 mg, 0.36 mmol, 1.0 equiv) and HCl (2 N, 1.0 equiv) in EtOH (0.18 mL) were added. The mixture was heated to reflux for 45 min, then cooled to room temperature, concentrated under reduced pressure, and crystallized to obtain the corresponding amidinohydrazone 40 (97 mg, 71%) as a yellow foam: 1H NMR (400 MHz, CD3OD) δ 7.99 (d, J = 3.7 Hz, 1H), 7.58–7.08 (m, 5H), 6.35 (d, J = 1.8 Hz, 0.3H), 6.13–5.91 (m, 0.7H), 5.62 (d, J = 2.3 Hz, 2H), 2.81 (dd, J = 13.1, 3.3 Hz, 1H), 2.72–2.07 (m, 4H), 1.77 (tt, J = 13.4, 7.4 Hz, 2H), 1.64–1.36 (m, 2H), 1.09 (s, 1H), 1.03 (s, 2H); HRMS (ESI) calcd for C21H28N7 (M + H)+ 378.2406, found 378.2396.
(
E+Z)-2-(((1
S,7a
R,
E)-5-(2-Carbamimidoylhydrazineylidene)-7a-methyl-2,3,5,6,7,7a-hexahydro-1
H-inden-1-yl)methylene)hydrazine-1-carboximidamide (
41): To a solution of the ketone-aldehyde
31 (57 mg, 0.32 mmol) in EtOH (6 mL), 2.00 equiv of the aminoguanidine hydrochloride (70 mg, 0.64 mmol) and 1 equiv of 2 N HCl (2 N, 1.0 equiv) in EtOH were added. The mixture was heated to reflux for 45 min, then cooled to room temperature, concentrated under reduced pressure, and the residue was washed with hexanes and EtOAc (8:2, 30 mL) to obtain the corresponding amidinohydrazone
41 (70 mg, 78%) as a yellow foam. Spectroscopic data for compound
41 were consistent with reported data [
27,
28].
(E+Z)-2-((6-(2-Carbamimidoylhydrazineylidene)-8a-methyl-1,2,3,4,6,7,8,8a-octahydronaphthalen-1-yl)methylene)hydrazine-1-carboximidamide (42): Compound 42 was synthesized using the same procedure employed for 41 using ketone-aldehyde 37 (40 mg, 0.20 mmol), aminoguanidine hydrochloride (48 mg, 0.43 mmol), and 2 N HCl (2 N, 1.0 equiv). After the completion of the reaction, the residue was washed with hexanes and EtOAc (8:2, 30 mL) to obtain the corresponding amidinohydrazone 42 (51 mg, 80%) as a white foam: 1H NMR (400 MHz, CD3OD) δ 7.59 (dq, J = 15.2, 8.6, 8.1 Hz, 2H), 7.29 (s, 1H), 6.43 (s, 0.2H), 5.95 (s, 0.8H), 3.22–2.92 (m, 1H), 2.76 (d, J = 17.1 Hz, 1H), 2.57–2.16 (m, 4H), 1.81 (dt, J = 74.0, 13.4 Hz, 5H), 1.49–1.03 (m, 4H). HRMS (ESI) calcd for C14H25N8 (M + H)+ 305.2202, found 305.2204.
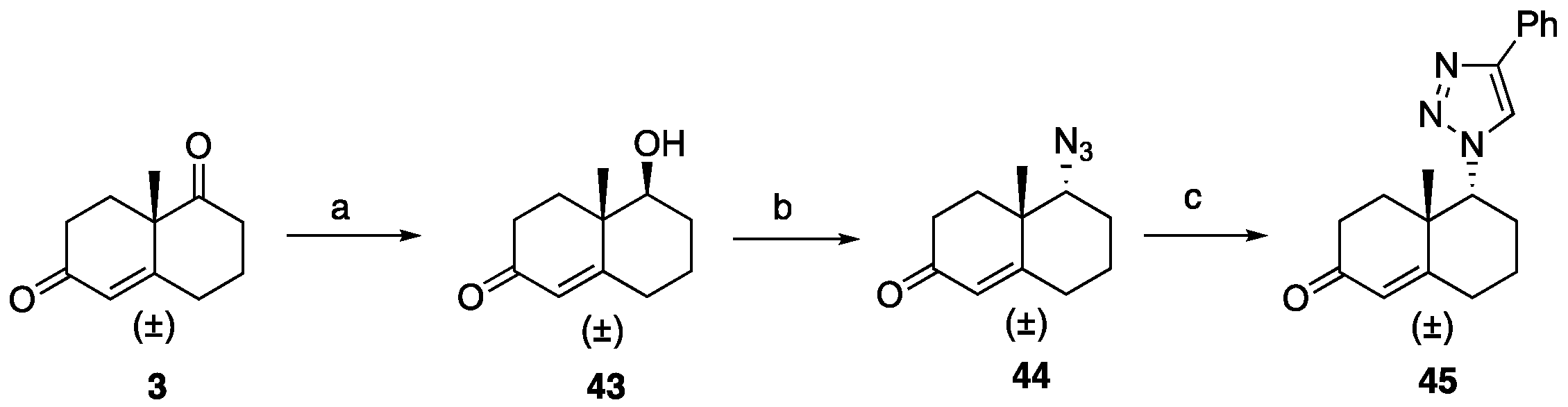
5-Hydroxy-4a-methyl-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-one (43): To a solution of ketone 3 (0.50 g, 2.80 mmol) in EtOH (18 mL), NaBH4 (40 mg, 1.10 mmol) was added at 0 °C, and the reaction was stirred for 5 min. AcOH (0.5 mL) was then introduced to the reaction mixture and stirred for an additional 5 min. Volatiles were removed under reduced pressure. To the resulting residue, EtOAc (30 mL) was added, followed by washing with brine and drying over Na2SO4. Purification through flash column chromatography (EtOAc: hexanes 1:3) yielded hydroxy ketone 43 (0.41 g, 82%) as a colorless liquid: 1H NMR (400 MHz, CDCl3) δ 5.74 (d, J = 1.8 Hz, 1H), 3.39 (dd, J = 11.6, 4.3 Hz, 1H), 2.66–2.51 (m, 1H), 2.48–2.23 (m, 3H), 2.25–2.08 (m, 2H), 1.90–1.74 (m, 3H), 1.73–1.61 (m, 1H), 1.46–1.28 (m, 1H), 1.16 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 200.0, 169.1, 125.3, 78.1, 41.7, 34.2, 33.7, 32.1, 30.2, 23.2, 15.3.
5-Azido-4a-methyl-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-one (44): Compound 44 was prepared following general procedure D using triphenylphosphine (0.16 g, 0.64 mmol), diethyl azodicarboxylate (0.11 mL, 0.64 mmol, 40% solution in toluene), alcohol 43 (0.10 g, 0.58 mmol), and diphenyl phosphoryl azide (0.13 mL, 0.64 mmol) to obtain 44 (0.057 g, 50%) as a yellow liquid. 1H NMR (400 MHz, CDCl3) δ 5.81 (d, J = 1.9 Hz, 1H), 3.56 (t, J = 2.8 Hz, 1H), 2.55–2.35 (m, 4H), 2.33–2.24 (m, 1H), 2.17–1.93 (m, 2H), 1.90–1.69 (m, 2H), 1.59–1.50 (m, 1H), 1.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 198.8, 165.7, 126.9, 68.6, 40.1, 34.0, 32.1, 31.4, 25.5, 22.4, 20.4.
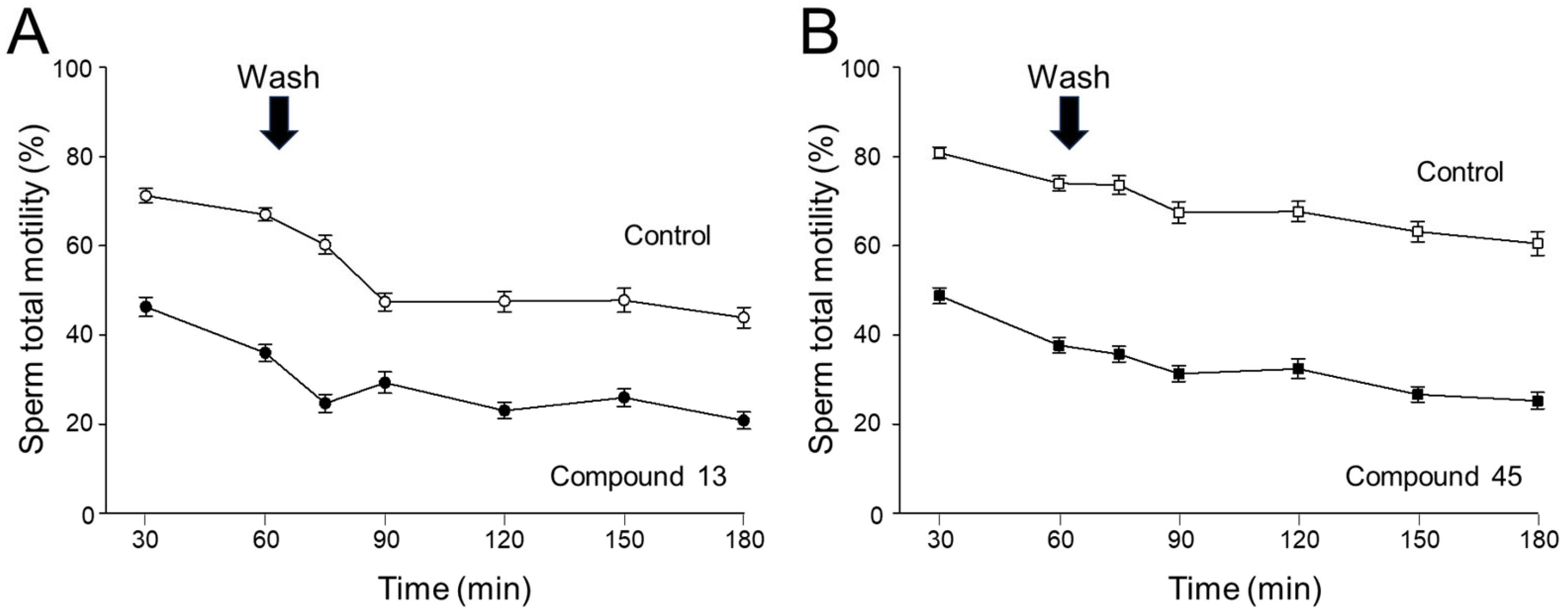
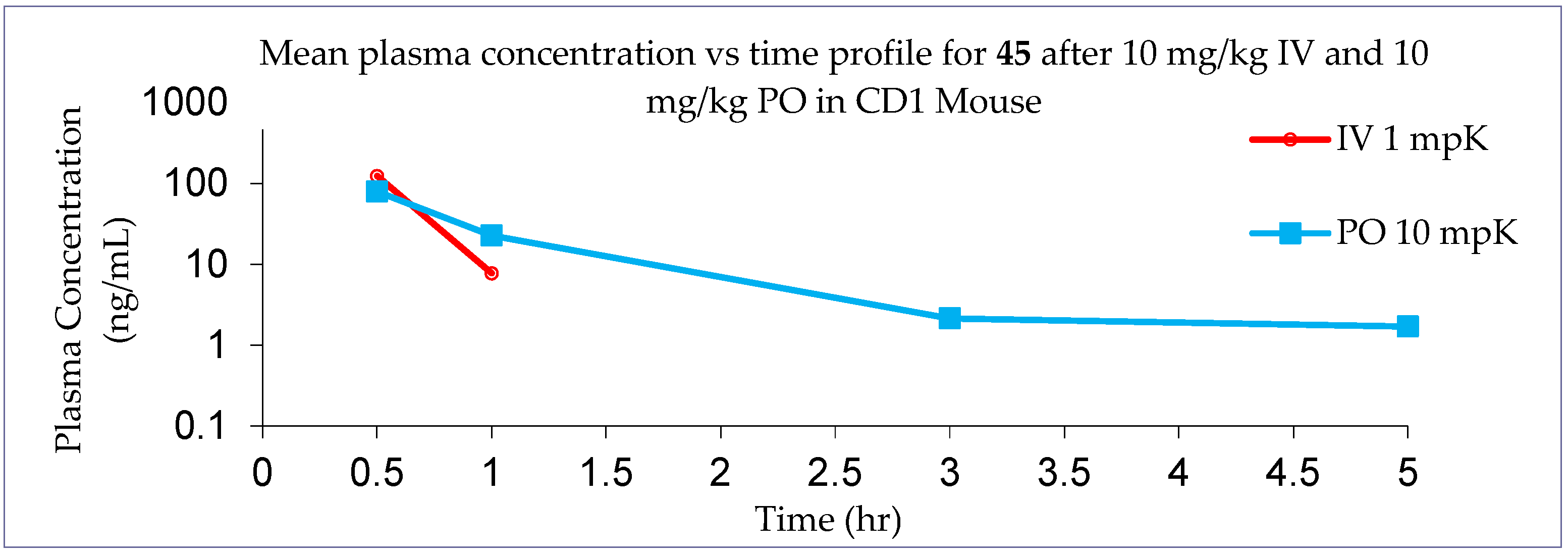
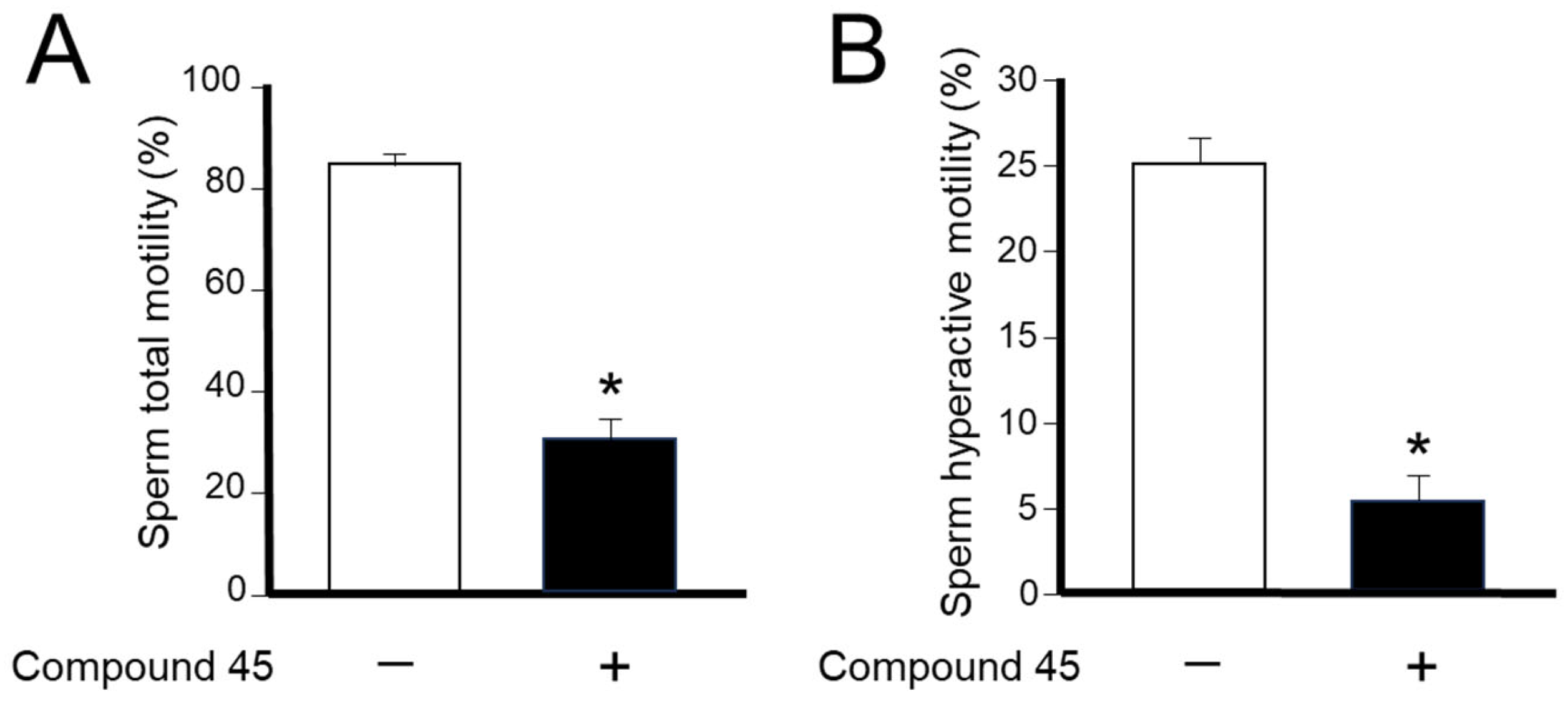
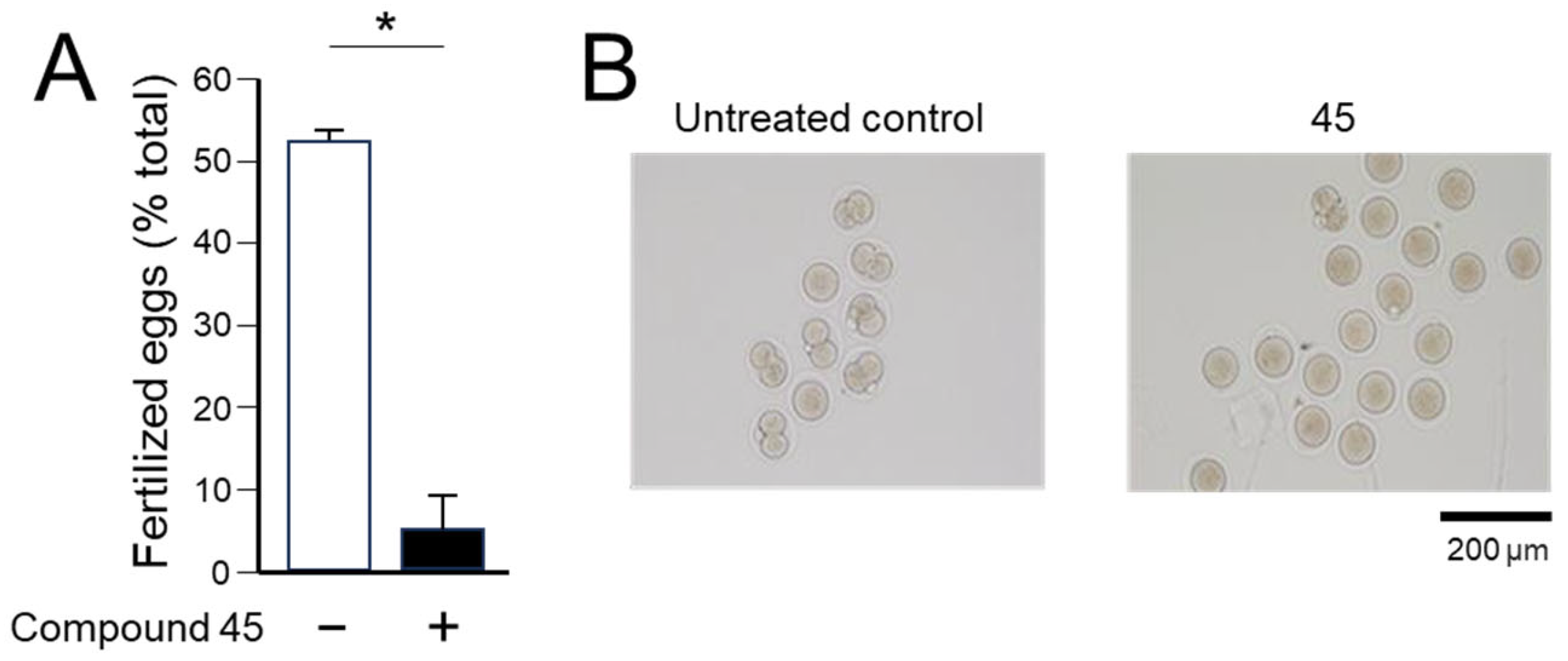
4a-Methyl-5-(4-phenyl-1H-1,2,3-triazol-1-yl)-4,4a,5,6,7,8-hexahydronaphthalen-2(3H)-one (45): To a mixture of azide 44 (49 mg, 0.22 mmol) and phenylacetylene (36 mg, 0.33 mmol) in DMF (2 mL), sodium ascorbate (17 mg, 0.088 mmol) in H2O (1 mL) was added and stirred for 2 min at ambient temperature. Then, a CuSO4.5H2O (11 mg, 0.044 mmol) in H2O (1 mL) was added to the above mixture. The reaction was stirred at room temperature for 12 h, quenched with water (3 mL), and extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a greenish residue, which was purified by flash chromatography (silica gel, EtOAc/hexanes, 4:6) to give 45 (52 mg, 71%) as a white solid: mp 178–180 °C; 1H NMR (400 MHz, CDCl3) δ 7.79–7.72 (m, 2H), 7.68 (s, 1H), 7.41–7.33 (m, 2H), 7.32–7.25 (m, 1H), 5.93 (d, J = 1.5 Hz, 1H), 4.79 (t, J = 3.6 Hz, 1H), 2.71–2.27 (m, 4H), 2.22–2.12 (m, 1H), 2.10–1.81 (m, 4H), 1.51 (s, 3H), 1.49–1.41 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 197.6, 164.9, 147.6, 130.3, 128.9, 128.4, 127.2, 125.8, 119.6, 66.5, 40.0, 33.6, 31.0, 30.8, 26.9, 24.0, 20.5; HRMS (ESI) calcd for C19H22N3O (M + H)+ 308.1763, found 308.1776.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}