1. Introduction
The genus
Echinochloa, commonly known as barnyard grass, is a particularly widespread and problematic weed species found in paddy fields across the globe [
1]. It poses a significant threat to paddy production, often leading to substantial yield losses. The taxonomic classification of the
Echinochloa genus is notably intricate, primarily due to its pronounced morphological variability. Traditionally,
E. crus-galli has been regarded as the predominant
Echinochloa species in Ningxia (China). However, our surveys conducted in Ningxia’s paddy fields have revealed the concurrent presence of
E. colona and
E. oryzoide populations alongside
E. crus-galli, underscoring the complexity of barnyard grass taxonomy in the region. Phenotypic and genetic diversities are pivotal factors influencing weed adaptation to agroecosystems [
2]. Notably, herbicide resistance has been reported to correlate with the morphological characteristics of the weed [
3]. For instance, Tsuji et al. [
4] tested 15 different herbicides on eight
Echinochloa phylopogon and found that the morphological differences in plant height, blade length, and spikelet length among the populations were consistent with the level of sensitivity to different herbicides. Therefore, a comprehensive understanding of barnyard grass diversity within a specific field represents an essential prerequisite for effective weed management.
Genetic diversity encompasses the total genetic information carried by all organisms on Earth, encompassing genetic and chromosomal variations. It underlies biological diversity and reflects the genetic variability and diversity among species [
5]. Morphological diversity, on the other hand, refers to the diversity manifested in organisms through their morphology, structure, physiological functions, and other characteristics [
6]. It serves as the visible expression of organisms adapted to their environment. Both morphological methods and DNA techniques can aid in identifying genetic variations. However, each approach has its strengths and limitations, and no single technique can fully replace the other. Utilizing morphological traits to assess genetic variation remains a straightforward and uncomplicated approach, providing a fundamental basis for barnyard grass taxonomy. Nevertheless, changes in agricultural activities, herbicide application, and shifting climatic conditions have induced significant alterations in barnyard grass morphology [
7], leading to varying perspectives among researchers regarding its classification. Consequently, establishing a standardized criterion for the taxonomic identification of
Echinochloa remains a challenge.
The identification of molecular markers represents a research method that enables the direct analysis of nucleotide sequence variations [
8]. Compared to morphological identification, this approach remains unaffected by seasonal changes, environmental factors, or the developmental stage of the plant. On the other hand, DNA barcoding serves as a rapid method for identifying and characterizing plant species using one or a few standardized DNA fragments [
9]. The application of plant DNA barcoding allows for an accurate and swift identification of plants on a large scale, irrespective of individual plant morphology and growth stage. However, for DNA barcoding to be effective, there must be a sufficient degree of sequence variation between species to enable clear differentiation, while within a species, the variation must be limited to define distinct thresholds for intraspecific and interspecific genetic differences. Zhang et al. [
10] used the chloroplast DNA barcode
psbA to categorize over 200 barnyard grass specimens into four groups. Similarly, Amaguchi et al. [
11] utilized the chloroplast barcode
trnT-L-F to classify nine barnyard grass species into five groups. Lahaye et al. [
12] successfully amplified 1667 plant materials using the chloroplast barcode
matK. Taberlet et al. [
13] recommended the chloroplast barcode
trnL intron as a suitable plant barcode. Another commonly used fragment for plant DNA barcoding is the intra-ribosomal transcribed spacer region
ITS. In the present study, we selected chloroplast DNA barcodes (
psbA,
trnL-F, and
matK) and the nuclear DNA barcode (
ITS) as candidate sequences to evaluate their discriminative capacity for
Echinochloa spp. in Ningxia to establish standardized DNA barcode sequences applicable to
Echinochloa spp., providing a valuable reference for molecular identification.
Traditional random DNA molecular markers (RDMs) have demonstrated their effectiveness and reliability through extensive applications. One such targeted gene molecular marker is the Start Codon-Targeted Polymorphism (SCoT) marker, which was developed by Collard and Mackill [
14]. SCoT molecular markers exploit the highly conserved and consistent sequence flanking the ATG start codon, similar to RAPD and ISSR markers. SCoT primers are designed to bind to the ATG translation initiation site on both the positive and negative DNA strands, facilitating the amplification of DNA fragments between these binding sites. SCoT markers represent valuable complements to RAPD and ISSR markers [
15]. SCoT markers can be used to trace specific traits and identify trait-related target genes, providing a clearer depiction of the relationships and genetic structure among diverse resources. The technology involving SCoT gene targeting markers has found widespread application in plant genetics and genomics research, spanning various domains, including, Iraqi barley [
16], medicinal plants [
17], vegetables [
18], trees [
19,
20,
21], and crops [
22]. However, few studies have been conducted to identify the biodiversity of barnyard plants based on SCoT molecular markers.
In Ningxia, there are usually four to five distinct species of the Echinochloa genus in paddy fields. However, there is a lack of information on barnyard grass populations’ genetic and morphological diversity, and these weed populations remain poorly characterized. To address this knowledge gap, we employed a comprehensive approach integrating morphological analysis, SCoT molecular markers, and DNA barcoding technology to thoroughly assess the genetic and morphological diversity among 46 distinct barnyard grass populations in Ningxia’s paddy fields.
3. Discussion
Genetic diversity plays an important role in overall biodiversity. Species with greater genetic diversity are better equipped to adapt to changing environmental conditions [
24]. As observed in prior studies, our research confirmed the extensive variability among barnyard grass species based on numerous morphological and biological characteristics. These findings are consistent with the outcomes of a genetic diversity analysis of barnyard grass by Lu [
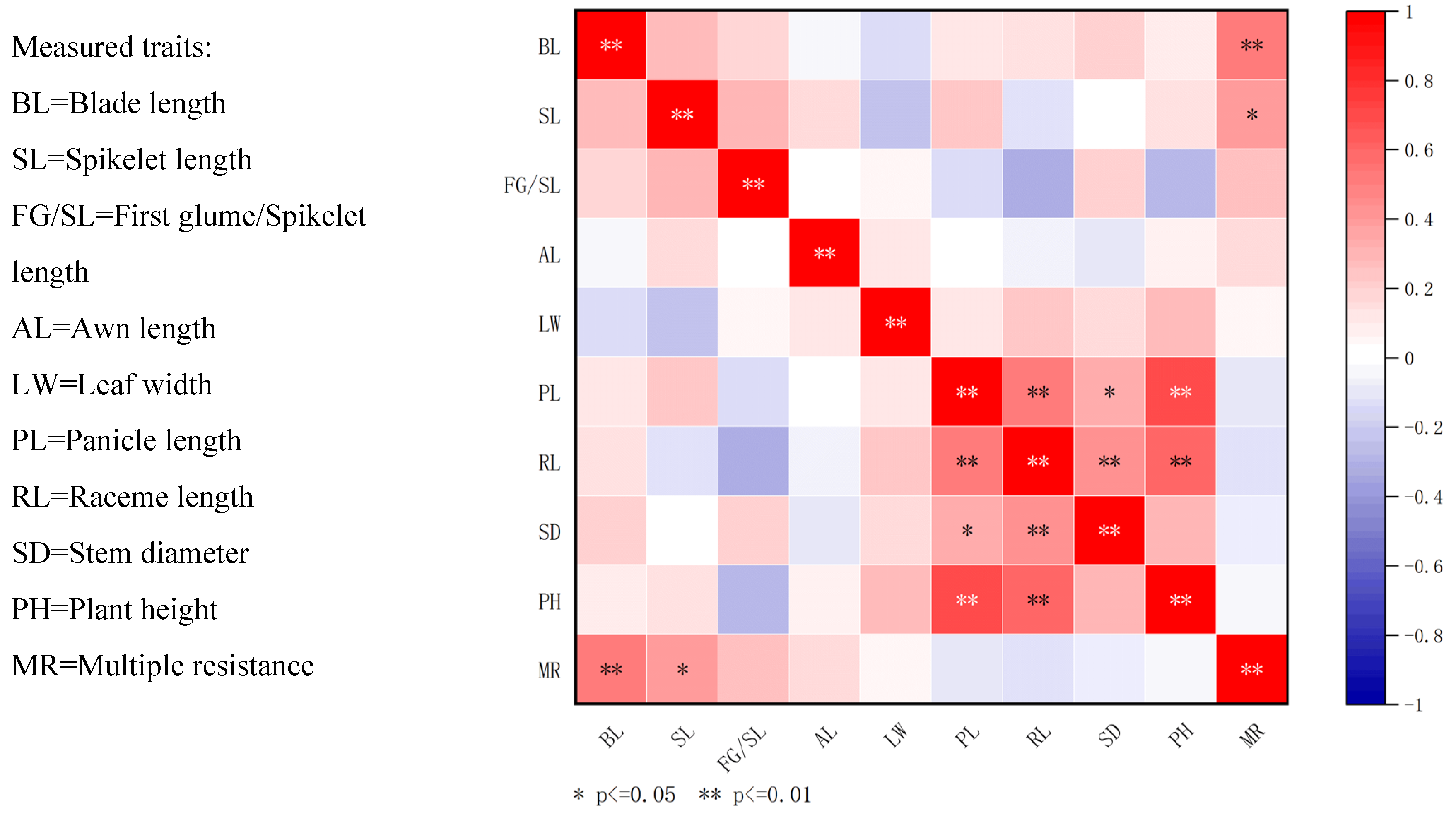
25]. Furthermore, we identified a positive correlation between certain phenotypic traits and the number of herbicide resistance sites among the tested biotypes. Herbicides play a role in driving morphological variations in weeds and favoring the dominance of specific populations.
DNA barcoding relies on standardized short genomic regions with sufficient sequence variation to distinguish between species [
26,
27]. In this study,
ITS demonstrated superior distinctiveness compared to other sequences, making it more suitable for barnyard plant identification. In our experiment, the combination of
ITS and
psbA barcodes provided enhanced discrimination of
Echinochloa spp. The recognition ability of DNA barcodes is related to the number of combinations [
28]. SCoT marker technology, effective in generating trait-linked genetic markers, shows significant value in genetics. To establish a stable, reproducible system, we used orthogonal design to optimize these key variables, successfully constructing a robust SCoT reaction system. Therefore, in this study, we used six SCoT primers to analyze 46 barnyard grass populations, resulting in a high percentage of polymorphic loci (86.1%), thereby highlighting the effectiveness of SCoT markers in assessing barnyard grass biodiversity.
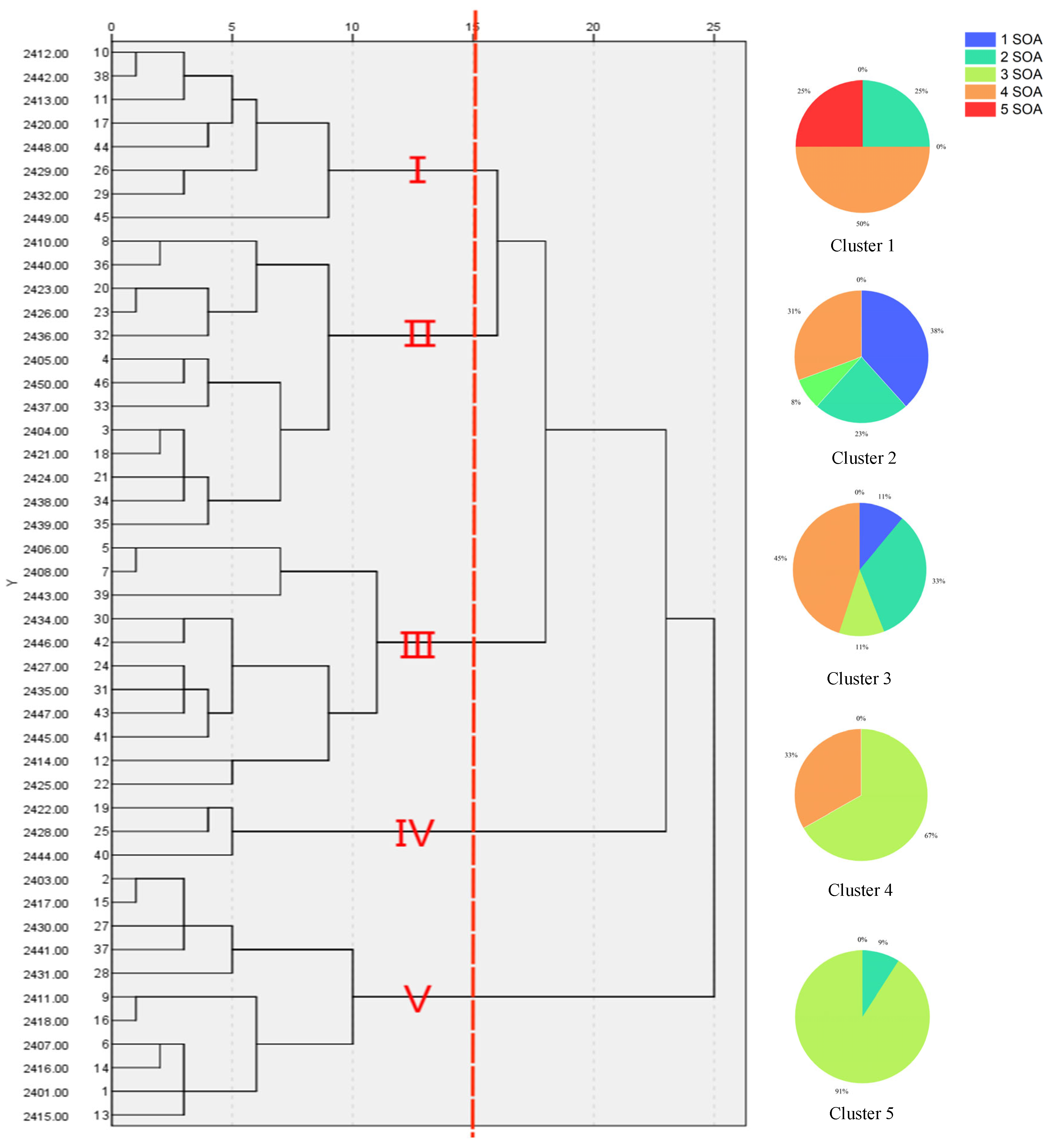
When comparing genetic diversity assessed by molecular markers with the results obtained from morphological analysis, we often observe discrepancies between the two. The fundamental cause of the discrepancies observed in cluster analysis lies in their inherently distinct genetic information bases [
29]. This is because genetic similarity primarily hinges on relatively stable genomic information, as supported by the research conducted by Hamrick and Liu [
3,
30]. Molecular marker technology detects variations at the DNA sequence level of organisms, encompassing base substitutions, insertions, or deletions within the genetic material [
31]. These variations are marked by high stability and heritability, remaining unaffected by the organism’s developmental stage, environmental conditions, or phenotypic complexity. For example, molecular marker technology can precisely elucidate genetic disparities and kinship among geographically distinct populations of the same species, regardless of their climatic adaptations. Molecular marker technology can scan the entire genome and detect numerous genetic variation loci, offering a more comprehensive view of an organism’s genetic diversity [
32]. Many traits in organisms are jointly controlled by multiple genes with complex interrelationships [
33]. Molecular marker technology, through methods such as association analysis, can identify multiple gene loci associated with specific traits and analyze their interaction patterns, providing a robust tool for understanding the genetic mechanisms of organisms.
In contrast, the manifestation of morphological traits arises from the interplay between gene expression and environmental conditions, with even minor environmental changes potentially inducing substantial alterations in these traits. For instance, in this study, the leaf length (M1) and plant height (M9) of barnyard grass were identified as two pivotal morphological traits that are significantly influenced by environmental factors, thereby demonstrating notable adaptive variations. Specifically, within the Yinchuan region, which is characterized by favorable irrigation practices and abundant soil moisture (e.g., sampling sites ID 2401, 2404), barnyard grass exhibited elongated leaves and increased plant stature. The data revealed that the mean leaf length in these areas surpassed 80 cm. Conversely, in arid environments, such as the Shapotou District in Zhongwei City (e.g., sampling sites ID 2421, 2422), barnyard grass adapted to the harsh conditions by reducing leaf length and plant height. Quantitative data indicated that the average leaf length in these regions fell below 60 cm. These morphological alterations constitute an adaptive strategy employed by plants to mitigate drought stress, where reduced leaf area minimizes water loss and decreased plant height mitigates the effects of wind and soil erosion, ultimately enhancing their prospects for survival under drought conditions.
In contrast, morphological trait measurement typically only reflects the effects of a few key genes or gene combinations. While the formation of morphological traits is influenced by multiple genes, in practice, we often select only a few easily observable and measurable morphological traits for analysis. These traits may only represent the expression effects of a subset of genes, limiting the ability to fully reveal an organism’s genetic complexity. Additionally, morphological traits can be affected by epigenetic modifications [
34], such as epigenetic marks that alter gene expression patterns without changing the DNA sequence, further complicating the use of morphological traits in reflecting an organism’s genetic essence.
Phenotypic differentiation in
Echinochloa spp. is shaped by the interplay of genotype, environment, and their interactions, exhibiting a hierarchical adaptive mechanism. Phenotypic plasticity serves as the primary mediator of environmental responses [
35]: under high-water conditions in Yinchuan (e.g., Helan County), barnyard grass elongates leaves (M1 = 104.40 cm) and increases plant height to enhance photosynthetic efficiency. Conversely, in drought-stressed Zhongwei (e.g., Shapotou District), populations shorten leaves and thicken stems (M8) to reduce transpiration, with significant height differences observed between samples 2411 (64.62 cm) and 2415 (104.40 cm), confirming environment-driven phenotypic divergence. Soil nutrient gradients further amplify morphological variation; for instance, nitrogen enrichment in Shizuishan (Pingluo County) promotes tillering (M3) and spikelet length (M4), while nutrient-poor soils in Wuzhong (Litong District) inhibit morphological development.
Herbicide selection pressure and microenvironmental stressors accelerate adaptive morphological evolution: prolonged exposure to 34% propanil in populations (e.g., Cluster 1) enhances metabolic detoxification via increased leaf length (M1 = 39.0 cm) and biomass accumulation, supporting the hypothesis of a positive correlation between leaf length and herbicide resistance. In contrast, windblown sand in Shapotou District induces stem thickening (M8) without genetic variation detected by ITS markers, indicating that morphological changes are primarily stress-induced.
Geographic isolation and restricted gene flow accelerate population differentiation, yet phenotypic plasticity may obscure genetic differences [
36]. For example,
ITS-based genetic divergence exists between Yinchuan and Lingwu populations separated by the Helan Mountains, despite morphological convergence due to similar irrigation practices. In contrast, Zhongwei’s arid populations have shortened awns (M5) to adapt to drought while retaining genetic diversity in SCoT markers, reflecting the decoupling of morphological and genetic adaptation.
Genotype-by-environment interactions (G×E) result in context-dependent phenotypic expression within the same genetic background. In Wuzhong, diurnal temperature fluctuations induce variation in tillering (M4) and spikelet morphology, yet SCoT markers remain genetically similar to Yinchuan populations, suggesting that phenotypic changes are environmentally driven. Intensive agricultural management in Yinchuan reduces plant height (e.g., Cluster 5, 43.35 cm) without detectable genetic differentiation in ITS analysis, indicating that anthropogenic disturbances accelerate adaptive evolution through phenotypic selection, with genetic divergence lagging behind.
The divergences between morphological and molecular classification reflect the adaptive strategies of Echinochloa species in complex environments. The combined effects of geographic isolation, herbicide selection pressure, and phenotypic plasticity have led to a decoupling between phenotypic traits and genetic backgrounds. In the future, it is essential to integrate environmental data with multi-omics technologies to comprehensively elucidate the evolutionary dynamics of Echinochloa, thereby providing a theoretical basis for precise prevention and control measures.
4. Materials and Methods
4.1. Plant Materials
Barnyard grass seeds were collected from rice cultivation areas across four regions in Ningxia province, Yinchuan, Zhongwei, Wuzhong, and Shizuishan, during September 2021. Seeds displaying a uniform morphology and maturity harvested from the same field were categorized into a single population group. Within each sampling site, the barnyard grass species remained consistent, representing dominant populations that had adapted to and withstood long-term environmental pressures. Seeds from the same sampling site were pooled and stored separately, with a minimum 10 km separation distance or distinct geographical barriers such as rivers and mountains between collection sites to minimize potential genetic mixing. A total of 46 barnyard grass populations were collected across the study region (
Figure 10,
Table A6), with detailed collection information provided in
Appendix A. Mature barnyard grass seeds were placed in labeled nylon mesh bags, dried in the sun, and subsequently stored at room temperature for use.
Seeds from all 46 barnyard grass populations were soaked in water for 24 h, followed by surface drying on filter paper. For each population, ten uniformly mature seeds were sown in 14 cm diameter plastic pots filled with potting mix comprising 1:1 (v/v) peat and sand. Seeds were covered with approximately 0.5 cm layer of soil to optimize germination conditions while accommodating complete life cycle development. The pots were cultivated in the greenhouse of the Institute of plant Protection, Chinese Academy of Agricultural Sciences, China. Sampling was conducted until the plants reached the 2–3 leaf stage of growth. We collected leaf tissue samples from 10 samples per population for DNA extraction and stored at −20 °C for subsequent analysis. After sampling, the remaining plants were carefully transplanted into individual 14 cm pots containing growth substrate. Plants were maintained in a green house until full maturity, at which point the morphological indexes of the plants were assessed.
Sampling locations (n = 46) are marked with black dots corresponding to different populations.
4.2. Multi-Resistance Assay
Barnyard grass samples cultured in 4.1 were used to determine susceptibility to six different herbicides at the three-leaf stage using whole-plant experimental assays. The six herbicides and the maximum recommended field dosage were 10% metamifop 120 g a.i/hm2, 25% penoxsulam 300 g a.i/hm2, 10% pyribenzoxim 45 g a.i/hm2, 34% propani 4498.2 g a.i/hm2, 25% quinclorac 300 g a.i./hm2, and 40% cyhalofop-butyl 120 g a.i/hm2, respectively. Each herbicide was treated using a 3WP-2000 type self-propelled spray tower (manufactured by Nanjing Institute of Agricultural Mechanization, Nanjing, China) with a flat–fat nozzle under the following operational conditions: a travel distance of 1320 mm and a constant speed of 412 mm/s. The cultivation conditions are set as L/D = 13 h, (30 ± 5) °C // 11 h, (25 ± 5) °C. Fourteen days after herbicide application, the aboveground fresh weight of each barnyard grass sample was weighed. The fresh weight suppression rate of different barnyard grass populations was then calculated to untreated controls. The biotypes with a fresh weight inhibition rate lower than 78% were defined as resistant biotypes. Three replicate pots containing 10 healthy and vigorous plants per pot were used, and the entire experiment was conducted twice.
4.3. Morphological Traits
The morphological characteristics of 46 barnyard grass samples were observed and described in accordance with taxonomic standards from the
Flora of China and the morphological framework established by Zou Manyu et al. [
37]. Nine quantitative traits were measured at the reproductive growth stage, denoted as M1 to M9: (M1) leaf length, (M2) leaf width, (M3) panicle length, (M4) raceme length, (M5) awn length, (M6) spikelet length, (M7) first glume length relative to spikelet length, (M8) main stem diameter, and (M9) plant height (
Table 4). For each population, 10 healthy-growing plants are selected for measurement, with the mean value calculated for analysis.
4.4. DNA Extraction
To prepare the samples, 100 mg of tender apical leaves tissue was collected and transferred to individual 1.5 mL centrifuge tubes. Additionally, we added two sterile steel beads, each with a diameter of 3 mm, into each tube. Samples were immediately frozen in liquid nitrogen for 2 min and transformed into a fine powder using a TissueLyser II sample crusher (Beijing Tiangen Biochemical Technology Co., Ltd., Beijing, China) at a frequency of 30 Hz for 45 s. Genomic DNA extraction was performed following the protocol outlined in the Tiangen New Plant Genomic DNA Extraction Kit (DP320-03, Tiangen Biochemical Technology Co., Ltd., Beijing, China). To ensure the integrity of the DNA, we conducted a 1% agarose gel electrophoresis analysis of the samples and assessed the concentration and purity of the extracted DNA using an ultraviolet spectrophotometer (Gene Co., Ltd., Beijing, China). The DNA was then diluted to a working concentration of 50 ng/μL and safely stored in a −20 °C refrigerator for future use.
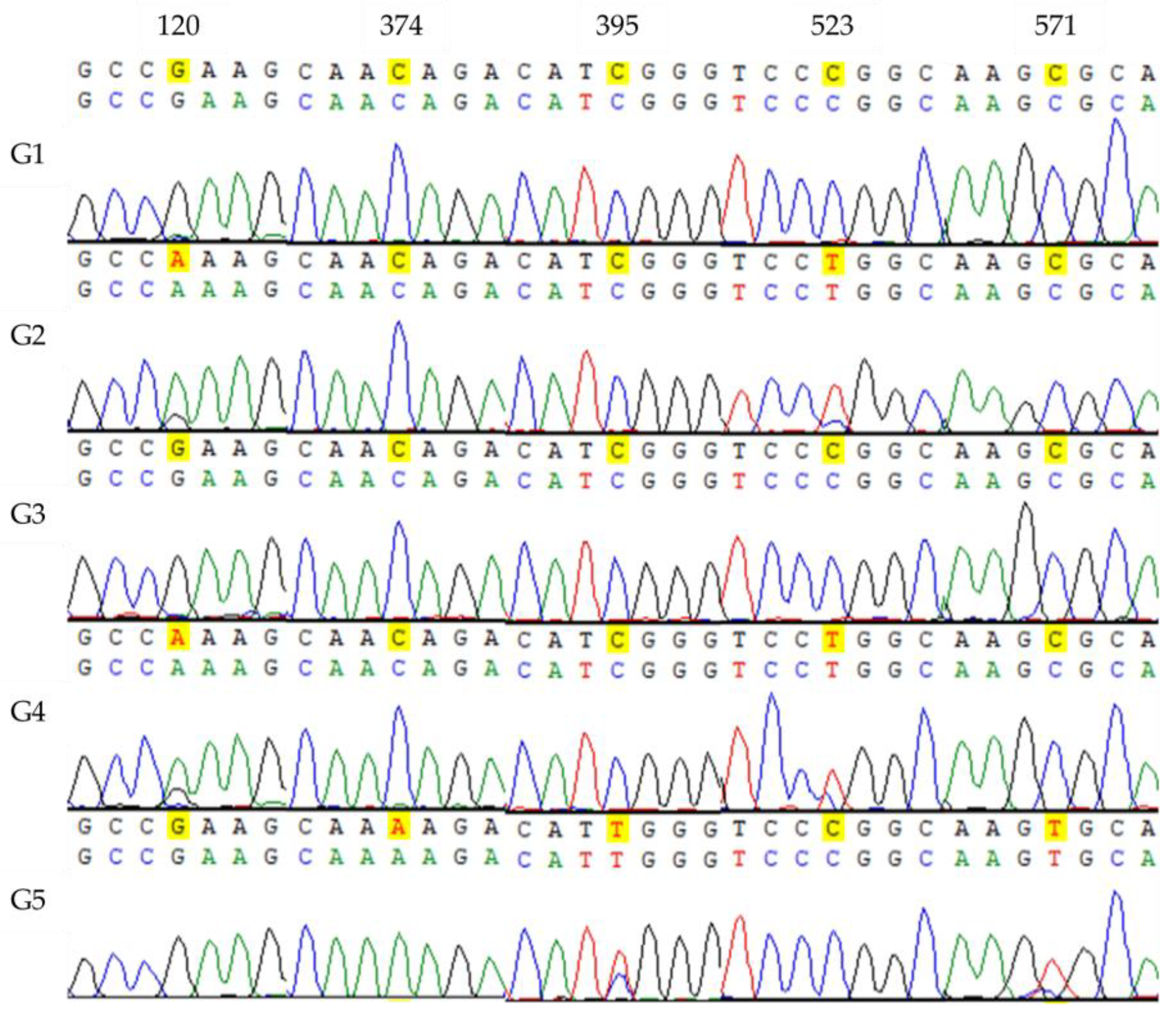
4.5. PCR Amplification of DNA Barcode Primers
In this study, the DNA barcoding genes of 10 samples from each of the 46 barnyard grass (
Echinochloa spp.) populations were successfully amplified. In a 25 μL PCR reaction system, 1 μL (50 ng/μL) genomic DNA, 1 μL (10 μM) each of forward and reverse primers (for primer details, see the
Table 5 provided in Gao and Yuan [
38,
39]), 12.5 μL 2× TIANGEN Taq Plus PCR mix, and 9.5 μL ddH
2O were used. After 10-s centrifugation for separation, the PCR process was continued with an initial denaturation at 94°C for 3 min, followed by 34 cycles of denaturation at 94°C for 30 s, annealing at the optimal temperature for 30 s, extension at 72°C for 40–60 s, and a final extension step at 72°C for 5 min. Subsequently, the PCR products were assessed using 1% agarose gel electrophoresis, and the outcomes were analyzed with a gel image analysis system. Clear and singular bands in the PCR products were selected for subsequent bidirectional sequencing, which was performed by Beijing Biomad Gene Technology Co., Ltd., Beijing, China
4.6. SCoT Molecular Marker Primer Screening and PCR Amplification
4.6.1. Primer Selection
The primers used for SCoT molecular markers were screened by referring to 36 SCoT primers published by Collard and Mackill [
14], from which SCoT primers with strong polymorphisms and clear bands were selected.
4.6.2. Single-Factor Optimization of PCR Reaction System
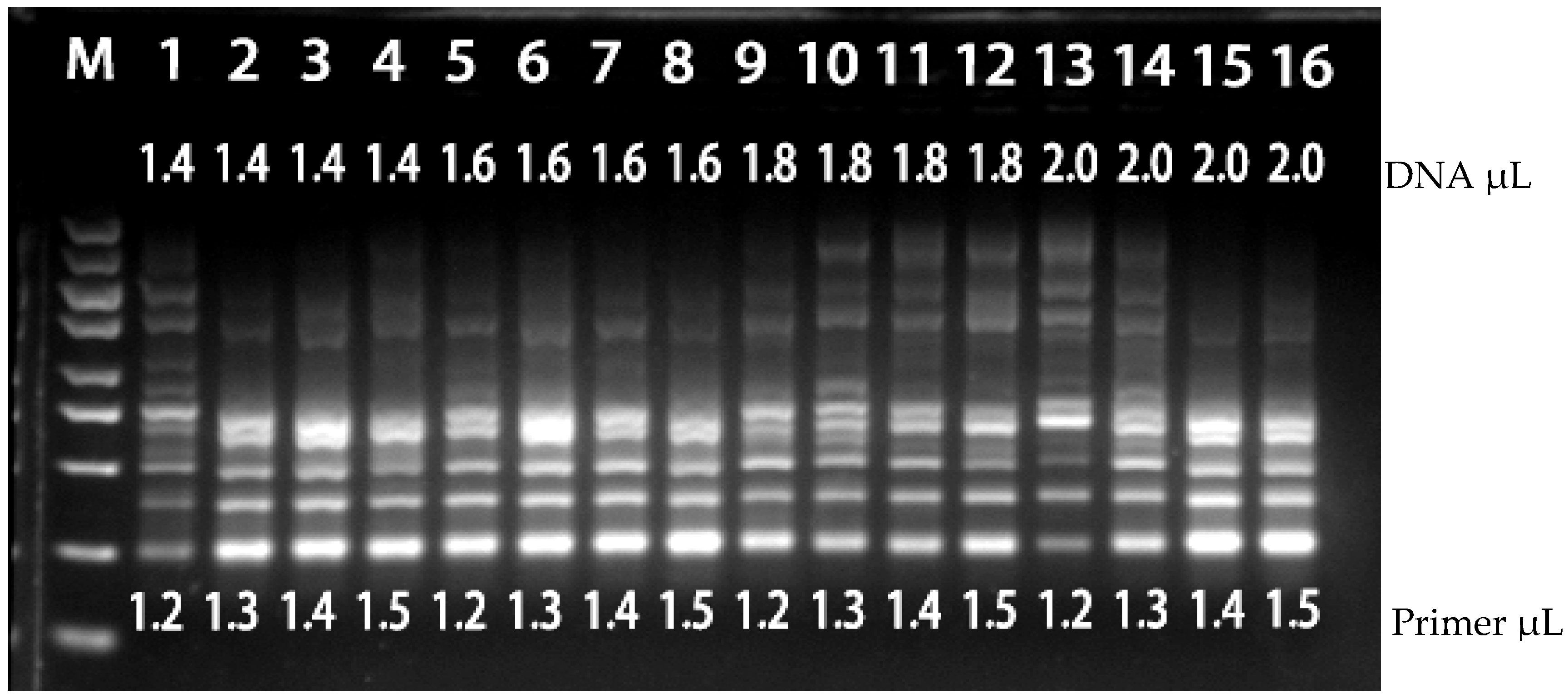
The 2× TIANGEN Taq Plus PCR mix was kept constant as a fixed component of the PCR system. However, we conducted univariate optimization for other variables within the system, including the quantity of barnyard grass DNA template, the addition of SCoT primers, and annealing temperatures. To ensure the reliability of our results and minimize the impact of operational errors, the experiment was replicated twice.
The six selected primers, SCoT6, SCoT11, SCoT12, SCoT20, SCoT29, and SCoT31, were configured to a concentration of 10 μM for PCR optimization (
Table 6). The 12 single-factor variables for barnyard grass DNA template addition (50 ng/μL) were 0.2 μL–2.4 μL; the 12 single-factor variables for SCoT primer addition (10 μM) were 0.5 μL–1.6 μL. For each primer pair, eight temperature gradients were set up to investigate the optimal annealing temperature of each primer at Tm ± 4 °C. PCR products were detected using 1.5% agarose gel, and the results were observed using a gel imaging analysis system.
4.6.3. PCR Orthogonal Optimization
We implemented a two-factor, four-level orthogonal experimental design
Table 7, specifically denoted as L
16 (4
2), encompassing a total of 16 unique experimental groups, each repeated twice. Our analysis indicates that the evaluation of experimental results extends beyond a mere enumeration of bands. Factors such as band clarity and background characteristics play a crucial role in our assessment. Our evaluation relied on direct observation to discern the strengths and weaknesses of the treatment condition settings.
4.7. Data Analysis
4.7.1. Morphological Trait Diversity and Cluster Analysis
Data processing and analysis were conducted using Excel 2019 to evaluate the degree of variation and diversity across each trait. Morphological diversity was quantified using the Shannon–Wiener Diversity Index (H′), calculated as follows:
Here, Pi represents the frequency of occurrence of the i-th variant type. Initially, the mean values of quantitative traits of 46 populations were divided into 10 levels, 1 level < X − 2 s, and 10 levels ≥ X + 2 s, with an interval of 0.5 s between each level, where X represents the mean and s signifies the standard deviation [
40]. The distribution frequency of the ten levels in each trait was calculated separately.
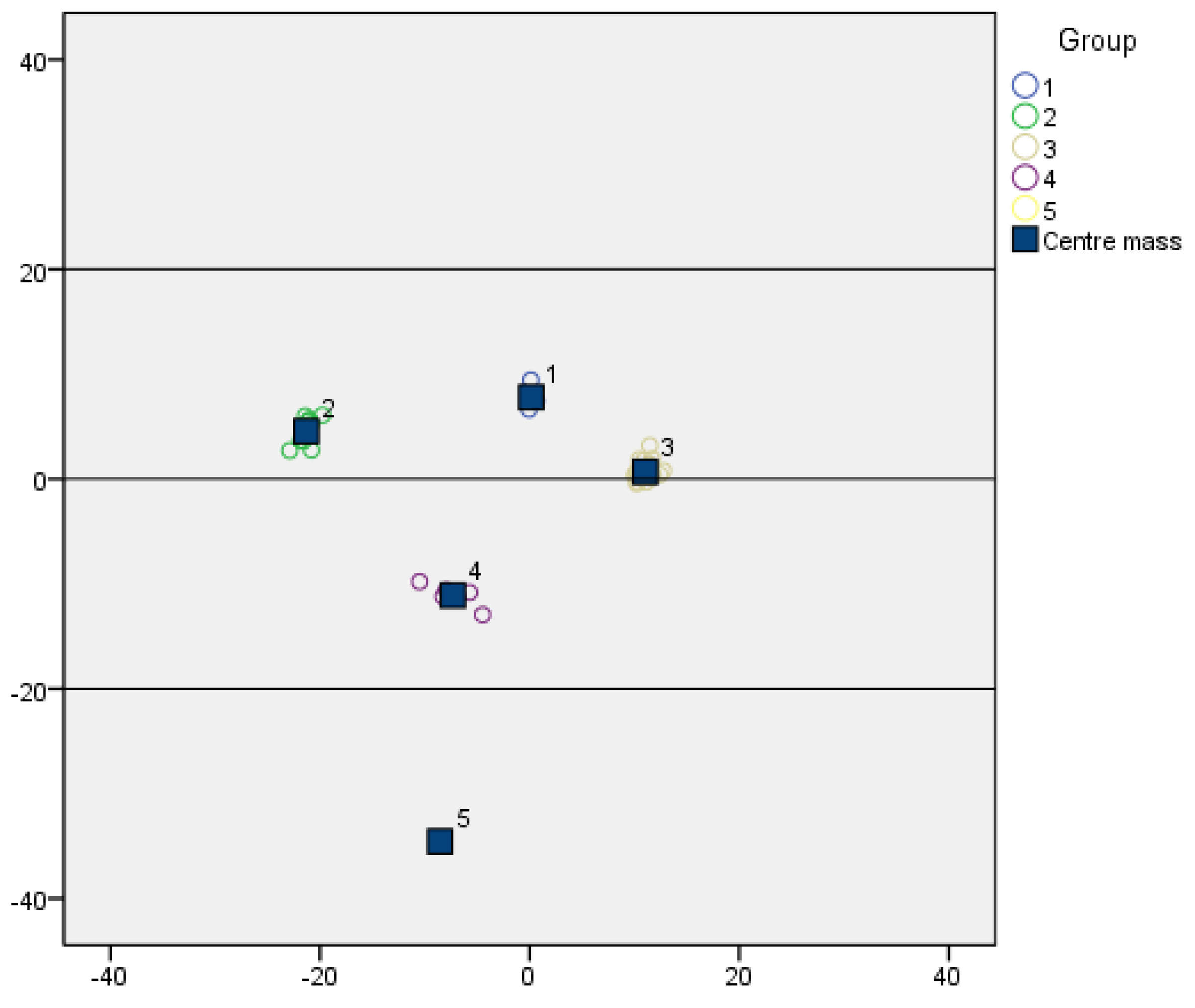
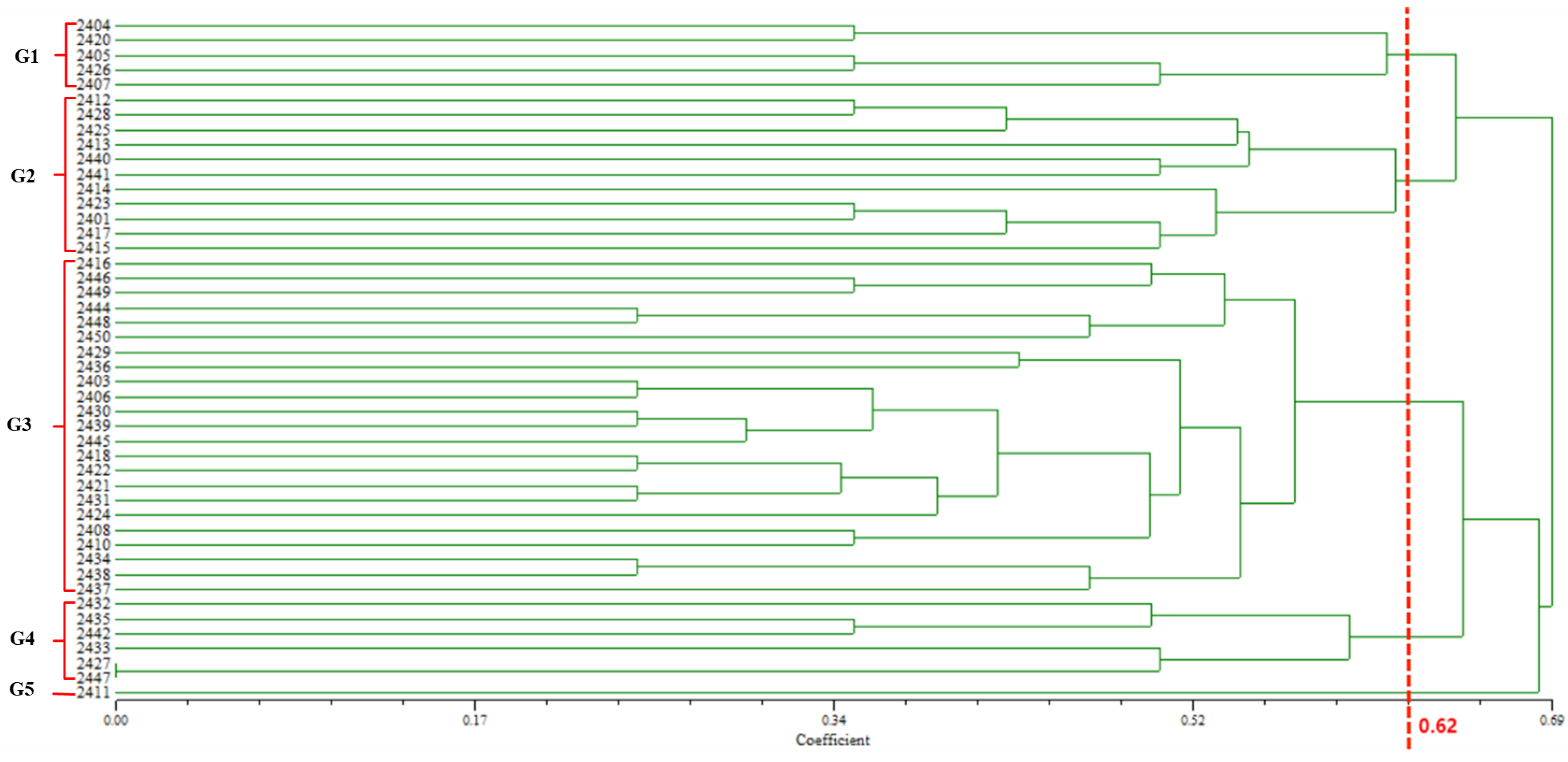
Nine quantitative indexes of the 46 barnyard grass populations were standardized separately, and the results of the standardized quantitative indicators were used for cluster analysis. Ward’s method was employed for clustering, with Euclidean distance used as the measure of dissimilarity between germplasms. Based on the results of cluster analysis, a one-way analysis of variance (ANOVA) was conducted using SPSS 24 to statistically test the significance of differences among different geographical groups. The Student–Newman–Keuls (S-N-K) method was selected for the post hoc analysis.
4.7.2. DNA Barcoding Cluster Analysis
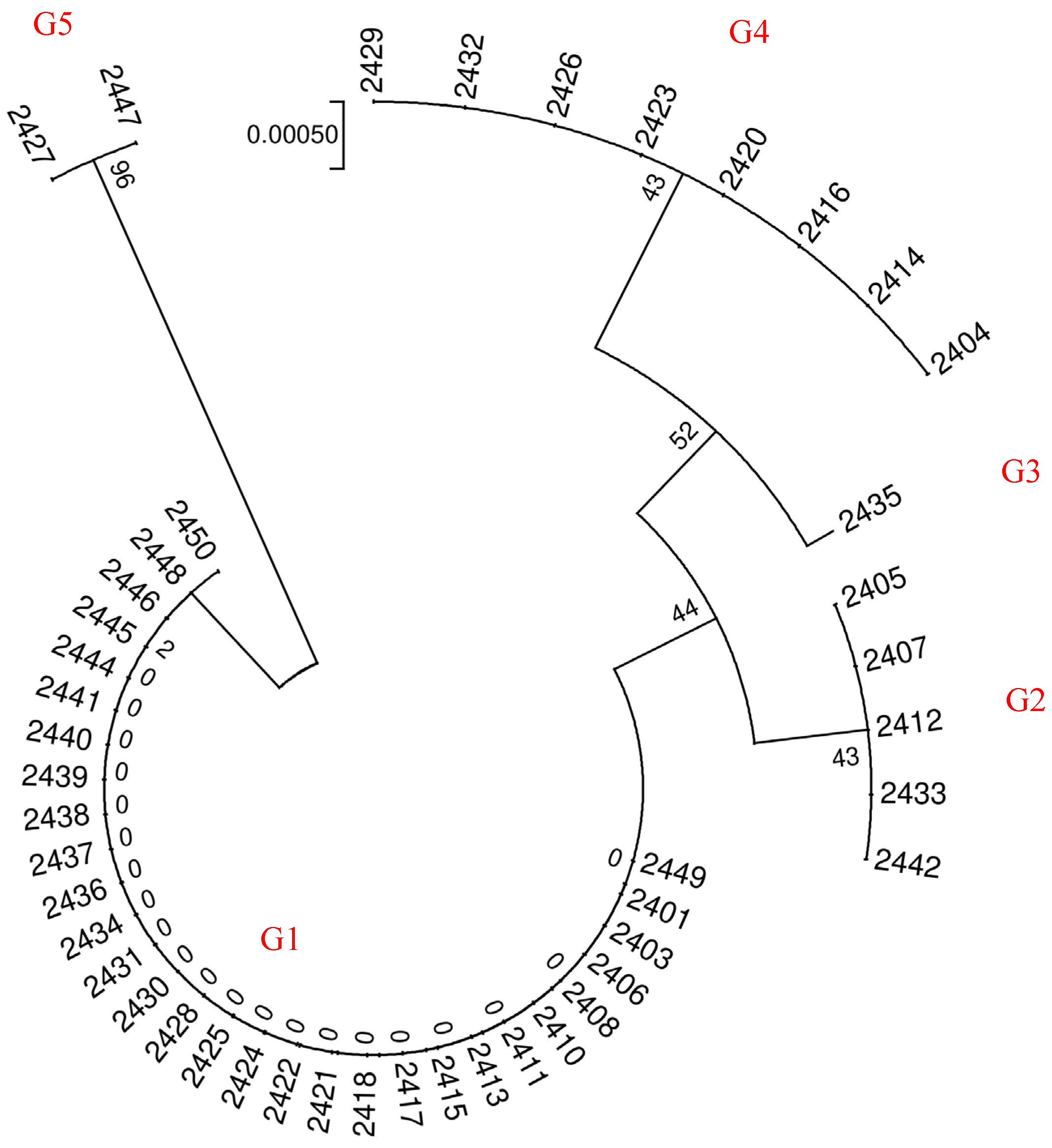
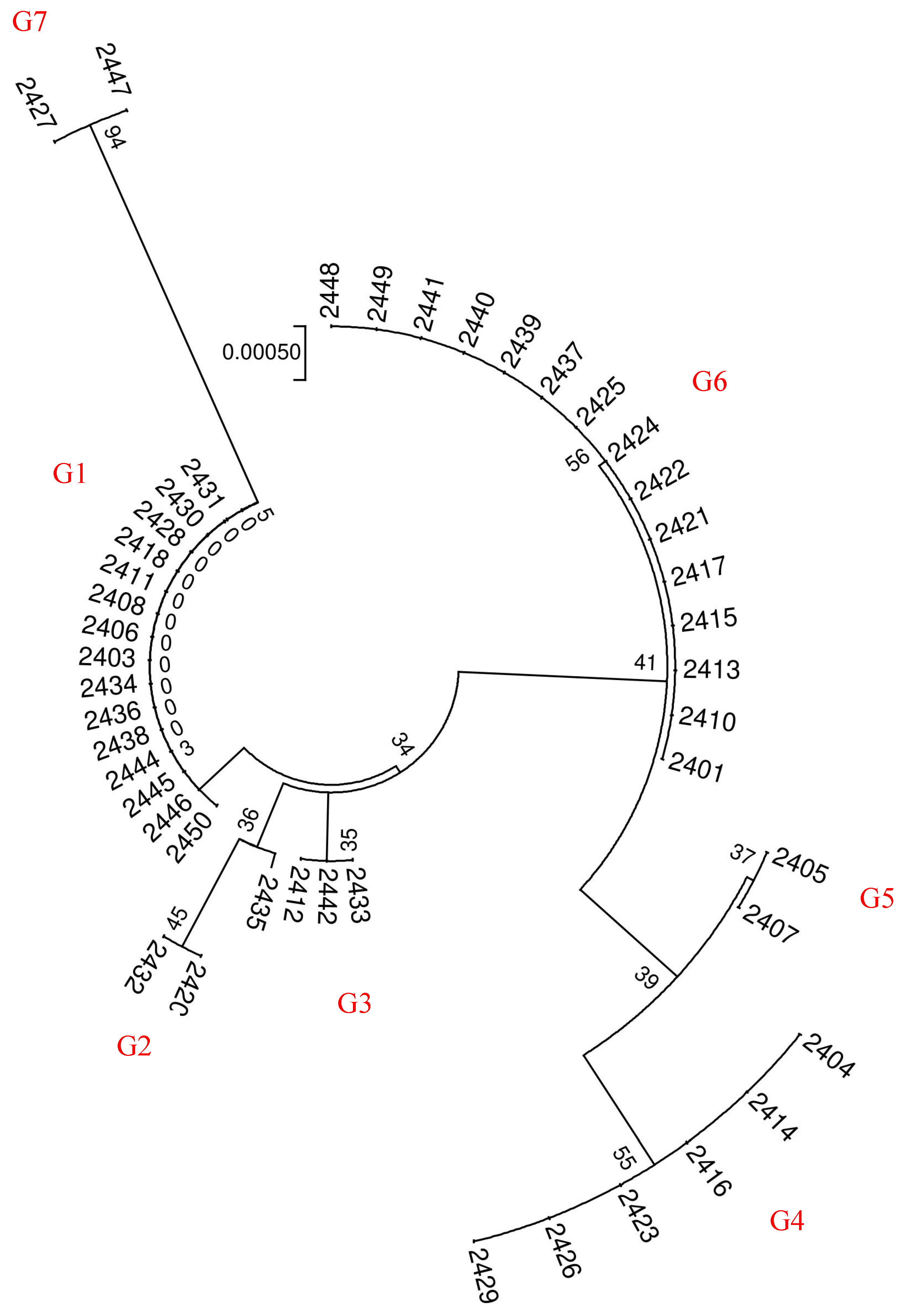
Sequencing peaks from each sample were subjected to quality analysis using Vector NTI 11.5 software. Low-mass sequence regions were excluded, and the sequences were aligned, checked, and proofread base by base. Subsequently, multiple-sequence alignment was conducted using MEGA 6.0 software. To obtain information on DNA barcode sequence variation, the Kimura 2-Parameter (K2P) genetic distance was computed between samples. Using the K2P distance model, we constructed a phylogenetic tree through the Neighbor-Joining (NJ) method to analyze the genetic relationships among the samples. Furthermore, the Bootstrap confidence value for each branch was tested with 1000 replications to enhance the reliability of our findings.
4.7.3. SCoT Molecular Marker Cluster Analysis
After the SCoT-PCR amplification, we conducted gel electrophoresis and captured images of the resulting band patterns. We then counted the distinct and clearly visible bands and scored them for each SCoT primer. These polymorphic bands served as reference points for individual random SCoT primers. Bands sharing the same position during migration were considered similar. We focused on selecting bands with well-defined and prominent backgrounds for subsequent statistical analysis, which were carefully analyzed with the Gel-Pro Analyzer 3.0 band analysis software. A control marker was introduced to simplify the analysis, which allowed us to assign values to specific migration positions based on the presence (assigned as “1”) or absence (assigned as “0”) of a band. This process yielded the initial “0/1” matrix, which formed the basis for subsequent analyses and comparisons.
The “0/1” matrix was reformatted to meet the input specifications of NTSYSpc 2.10e. To ensure data compatibility and facilitate subsequent analyses, the dataset was systematically structured and compiled into an Excel spreadsheet. The processed spreadsheet was imported into NTSYSpc 2.10e, where the Simple Matching (SM) similarity coefficient was employed to quantify pairwise genetic resemblances. The SM coefficient compares binary (“0/1”) profiles across samples, generating a genetic similarity matrix that encapsulates all pairwise genetic relationships within the dataset.
The Polymorphism Information Content (
PIC) is used to quantify the polymorphic value of SCoT marker loci. The calculation formula is as follows:
n: total number of alleles. pi: frequency of the i-th allele, satisfying the condition.
Prior to clustering, phenotypic data were standardized to mitigate confounding effects due to measurement scale disparities. Euclidean distances were computed based on the standardized phenotypic values, providing a quantitative metric of inter-sample dissimilarity. This distance matrix served as the basis for hierarchical agglomerative clustering (HAC) analysis.
The genetic similarity matrix and the Euclidean distance matrix were integrated to perform comprehensive clustering and systematic analysis, yielding a hierarchical clustering dendrogram that visually represents the genetic and phenotypic relationships among samples.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}