
Theoretical Insight into Antioxidant Mechanisms of Trans-Isoferulic Acid in Aqueous Medium at Different pH


Abstract

1. Introduction
2. Results and Discussion
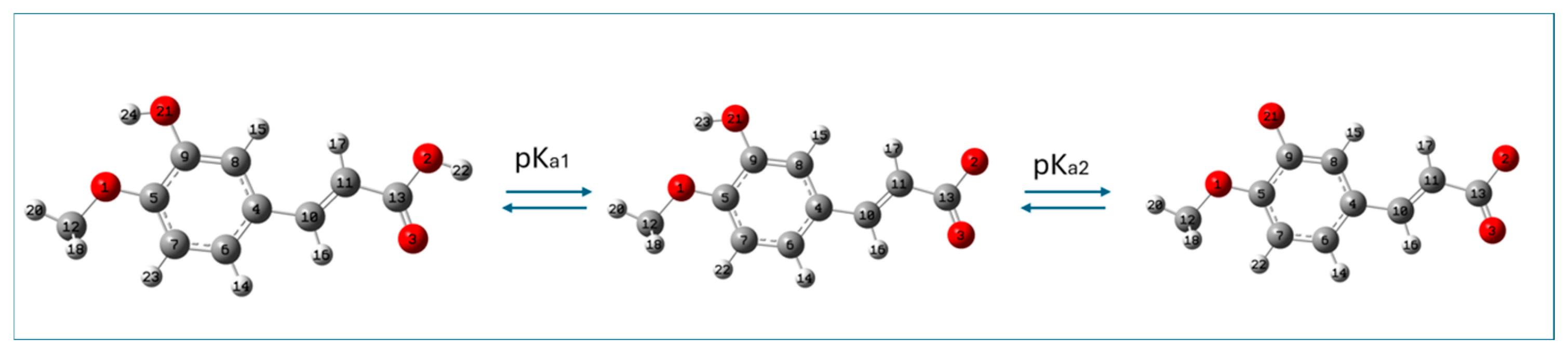
2.1. Theoretical Determination of Acid–Base Equilibria of Isoferulic Acid (pKa Values of Isoferulic Acid)
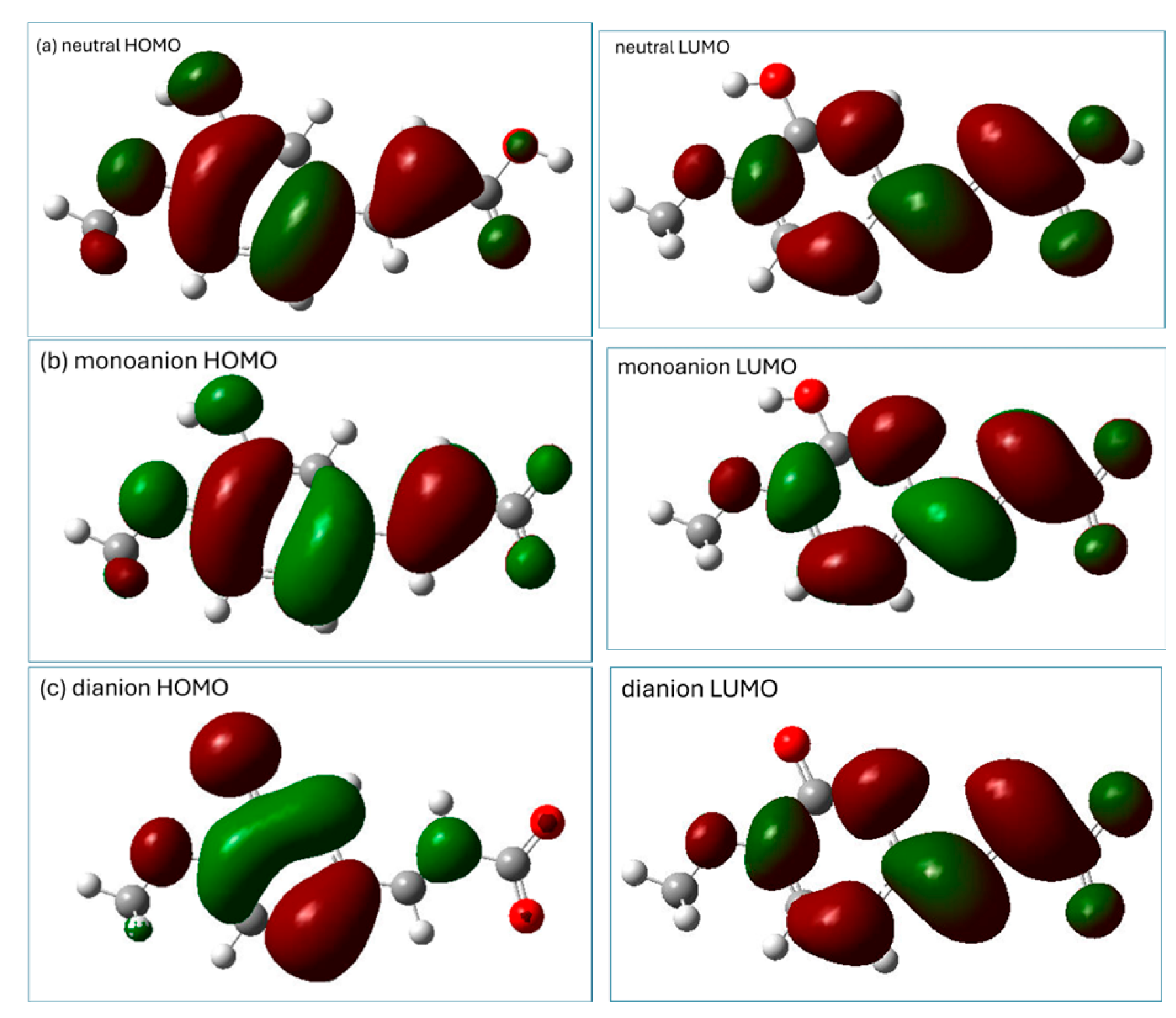
2.2. Prediction of Antioxidant Properties of Isoferulic Acid Based on the Indices Related to Frontier Molecular Orbitals Theory
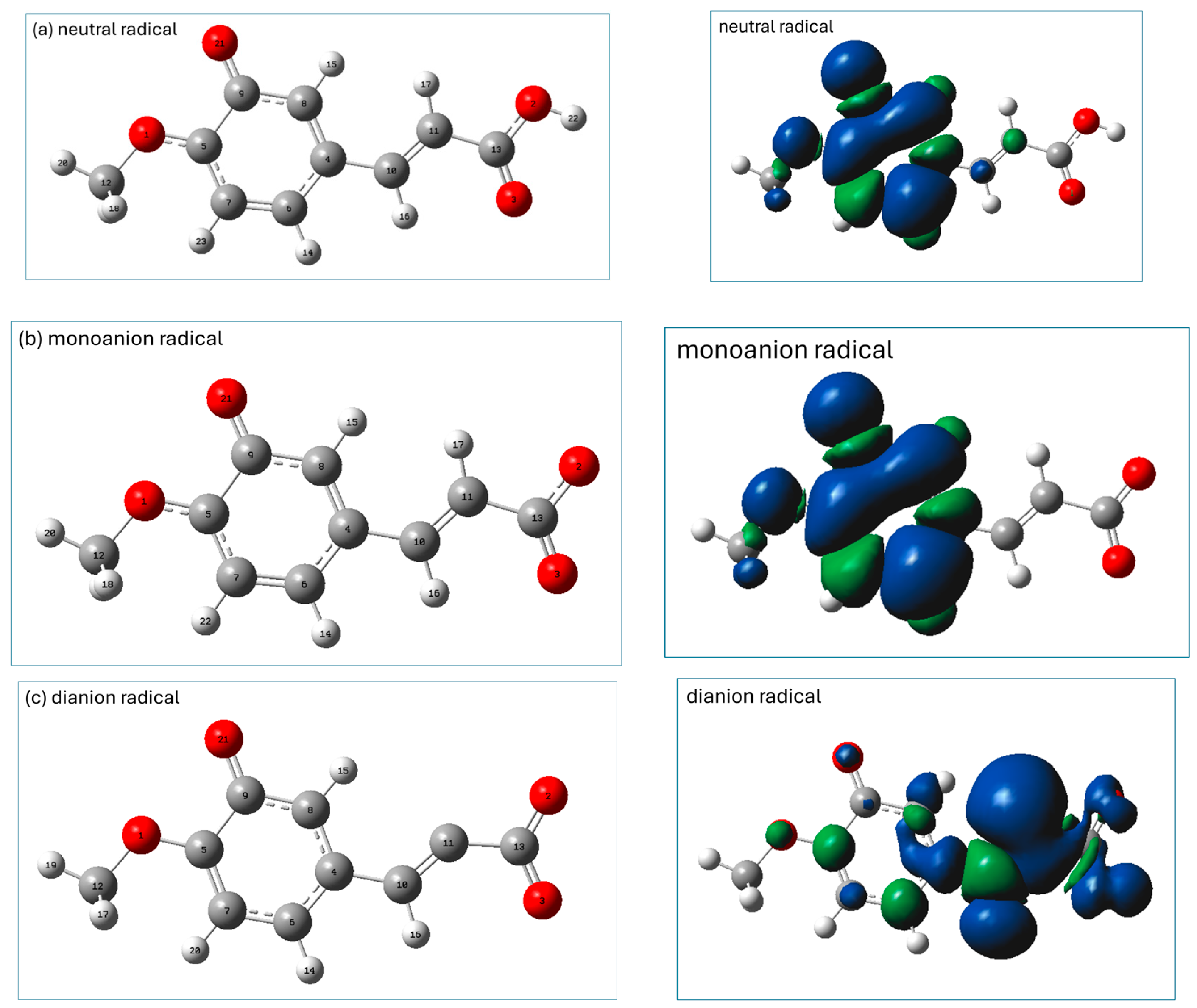
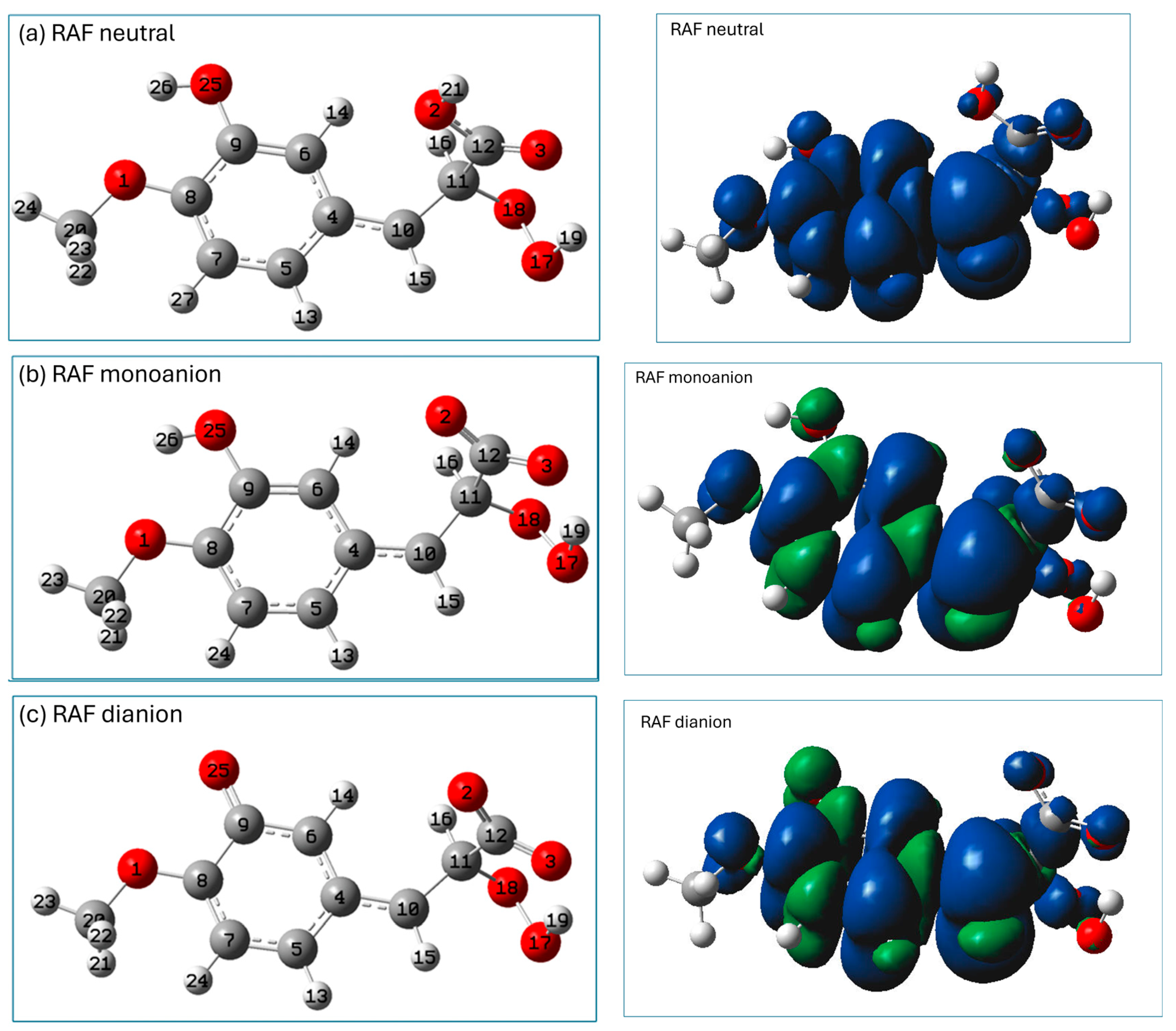
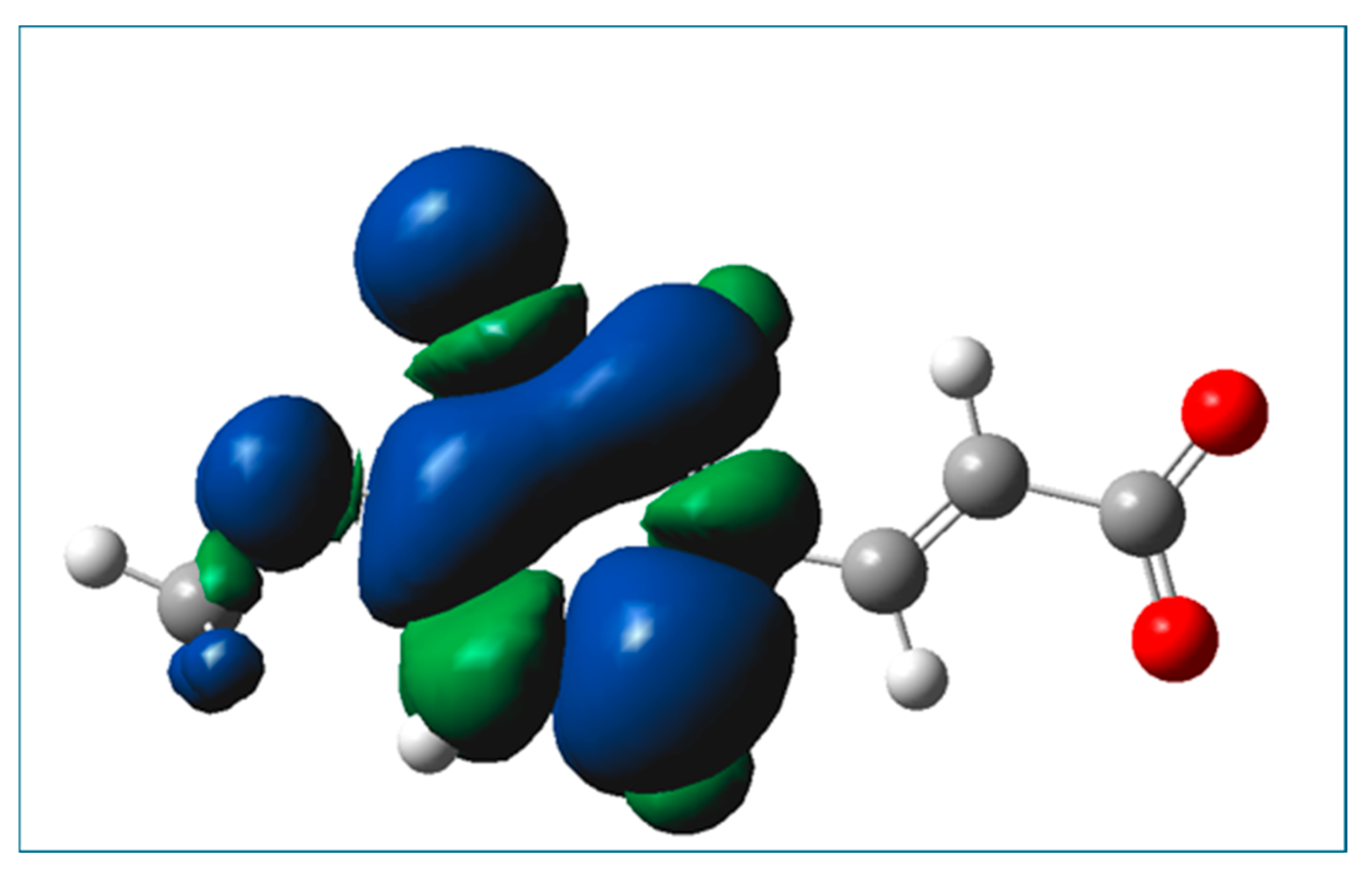
2.3. Prediction of Radical Attack Site
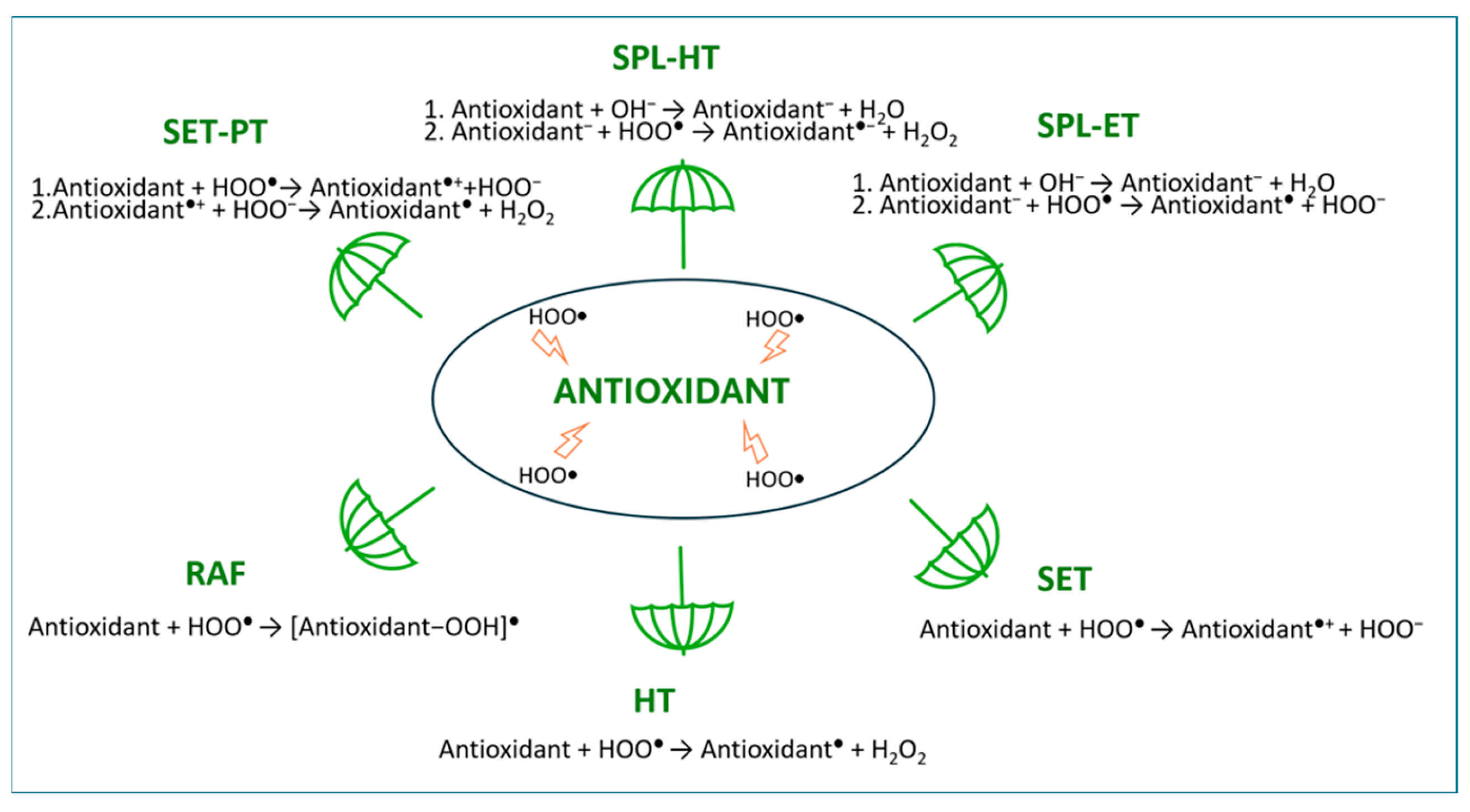
2.4. Evaluation of Free Radical Scavenging Ability via Intrinsic Thermochemical Parameters
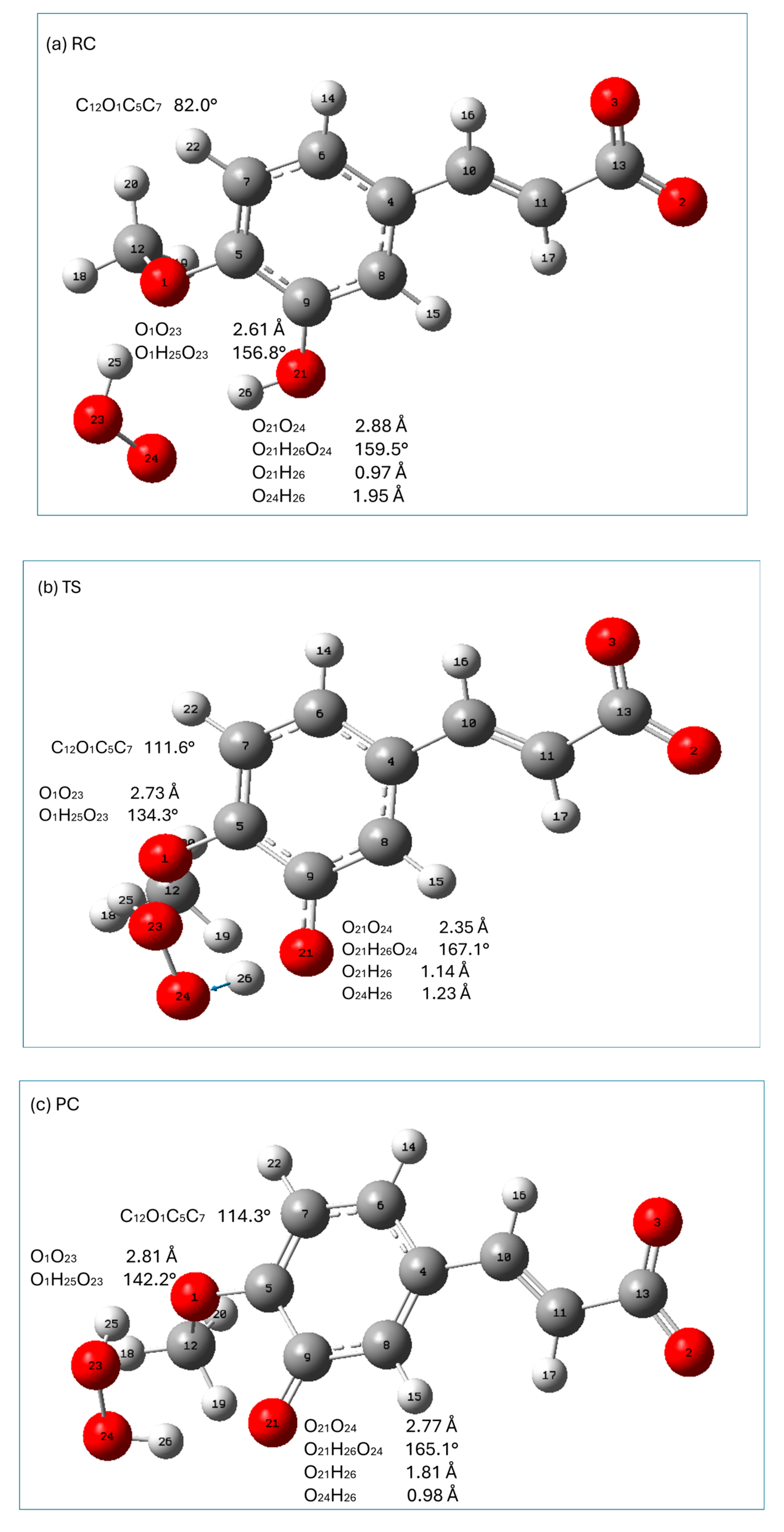
2.5. Possible Pathways of HOO● Free Radical Scavenging by Isoferulic Acid
2.6. Kinetics of Reactions Involved in the HT, SET, and RAF Mechanisms
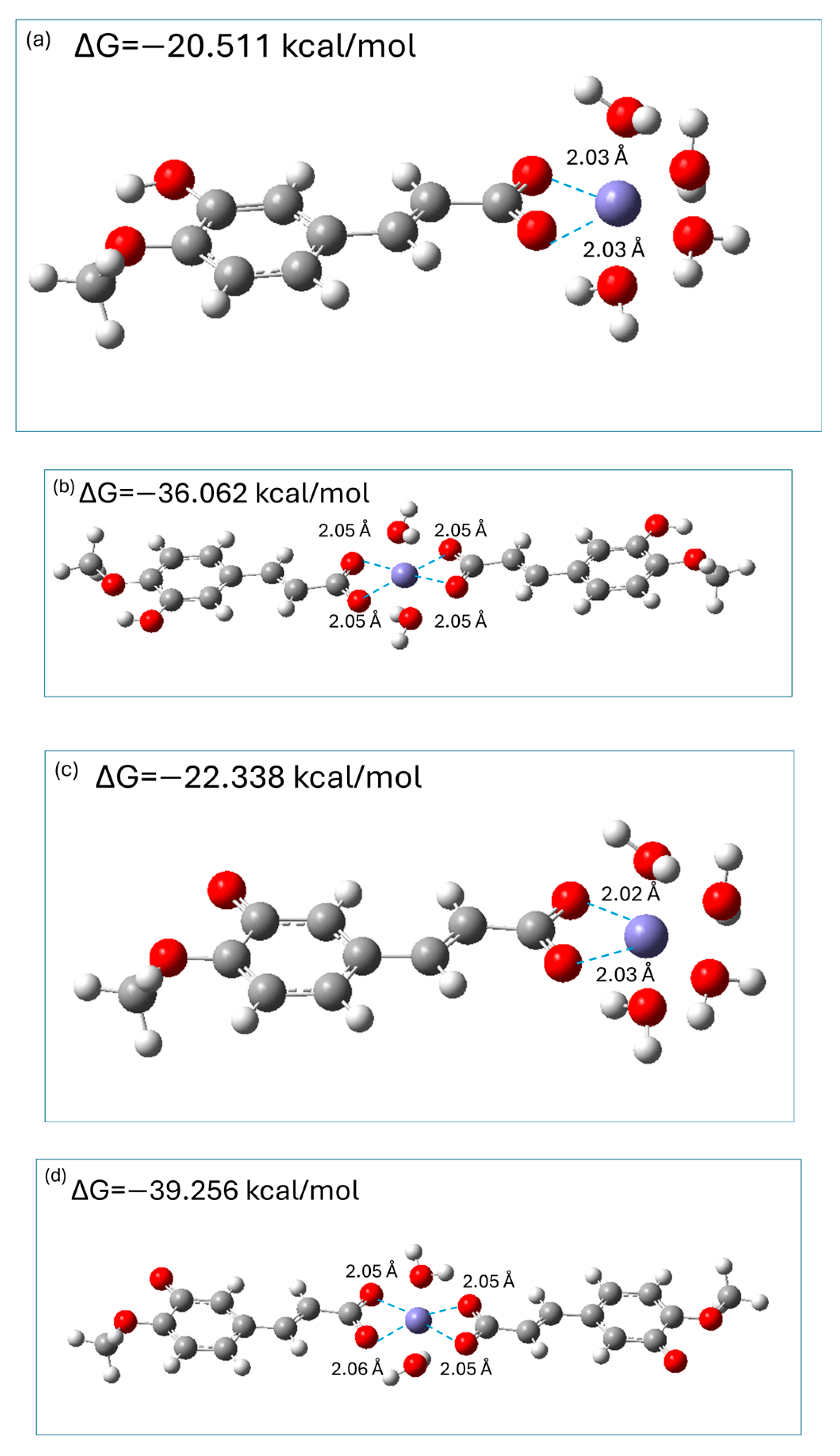
2.7. Fe2+ Ions Chelating Properties of Isoferulic Acid
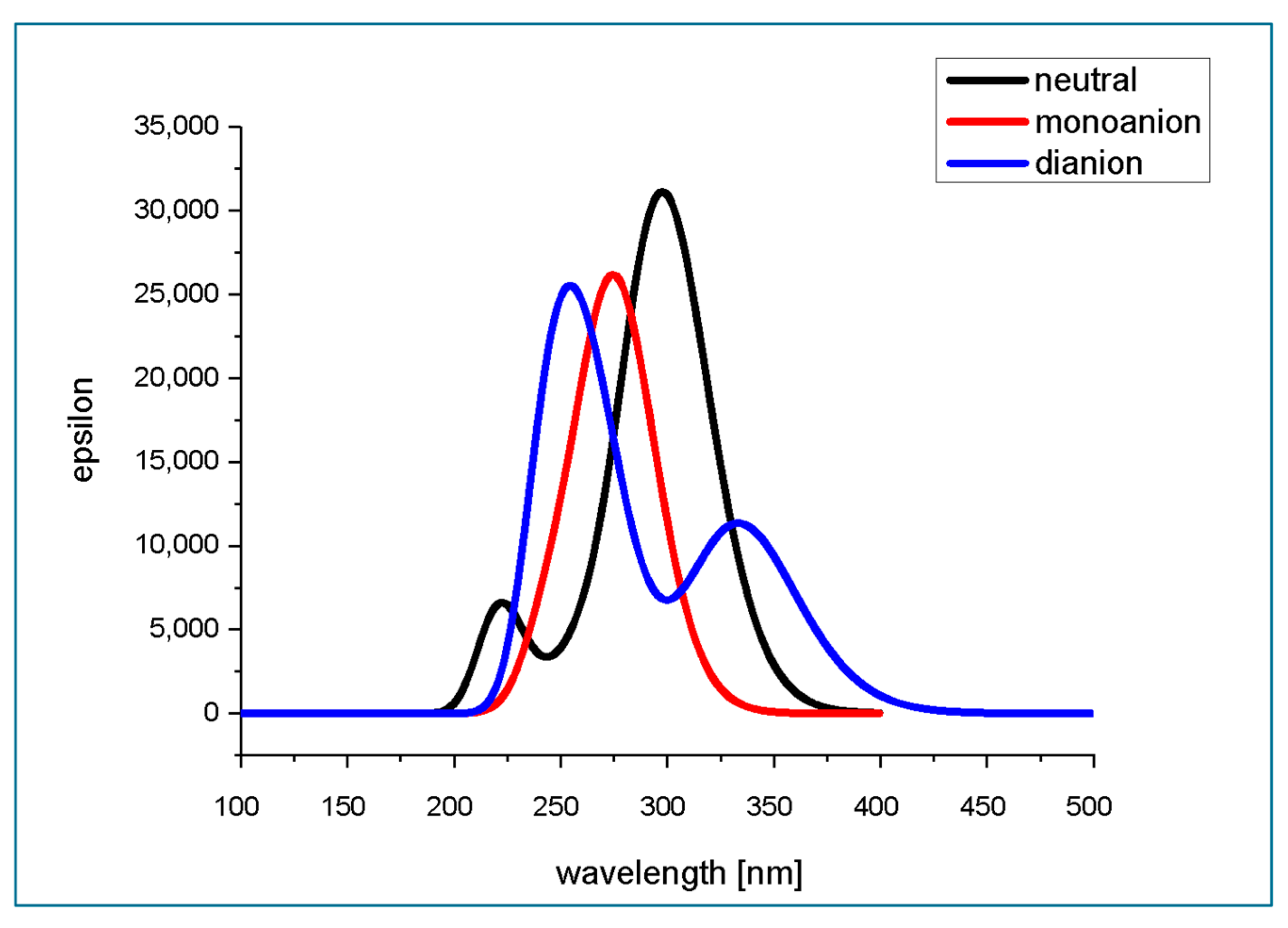
2.8. Theoretical Prediction of Absorption Spectra of Isoferulic Acid in Aqueous Medium at Different pH
3. Materials and Methods
3.1. Electronic and Geometrical Structure of Isoferulic Acid
3.2. Deprotonation Constants
3.2.1. Isodesmic Method
- (1)
- H2A + HA−(Ref) → HA− + H2A (Ref);
- (2)
- HA− + A2−(Ref) → A2− + HA− (Ref).
3.2.2. Parameter-Fitting Method
3.3. Indices Related to Frontier Molecular Orbitals Theory
3.4. Prediction of Radical Attack Site Based on Condensed Fukui Functions
3.5. Intrinsic Thermochemical Reactivity Indices
3.6. Thermodynamics of HOO● Free Radical Scavenging Reaction Pathways
3.7. Rate Constants for HT and RAF Reactions
3.8. Rate Constant for Single Electron Transfer (SET) Reaction
3.9. Correction for Diffusion-Controlled Rates
3.10. Chelation Ability of Isoferulic Acid
3.11. Theoretical Prediction of Absorption Spectra and Parameters of Electronic Transitions to the Excited Singlet States
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef]
- Mazzone, G. On the inhibition of hydroxyl radical formation by hydroxycinnamic acids: The case of caffeic acid as a promising chelating ligand of a ferrous ion. J. Phys. Chem. A 2019, 123, 9560–9566. [Google Scholar] [CrossRef] [PubMed]
- Tumilaar, S.G.; Hardianto, A.; Hirofumi, D.; Dikdik, K. A comprehensive review of free radicals, oxidative stress, and antioxidants: Overview, clinical applications, global perspectives, future directions, and mechanisms of antioxidant activity of flavonoid compounds. J. Chem. 2024, 1, 5594386. [Google Scholar] [CrossRef]
- Vo, Q.V.; Nam, P.C.; Bay, M.V.; Thong, N.M.; Hieu, L.T.; Mechler, A. A theoretical study of the radical scavenging activity of natural stilbenes. RSC Adv. 2019, 9, 42020–42028. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Raúl Alvarez-Idaboy, J. Computational strategies for predicting free radical scavengers’ protection against oxidative stress: Where are we and what might follow? Int. J. Quantum Chem. 2019, 119, e25665. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. A computational methodology for accurate predictions of rate constants in solution: Application to the assessment of primary antioxidant activity. J. Comput. Chem. 2013, 34, 2430–2445. [Google Scholar] [CrossRef]
- Spiegel, M. Current trends in computational quantum chemistry studies on antioxidant radical scavenging activity. J. Chem. Inf. Model. 2022, 62, 2639–2658. [Google Scholar] [CrossRef]
- Ou-Yang, H.; Stamatas, G.; Saliou, C.; Kollias, N. A chemiluminescence study of UVA-induced oxidative stress in human skin in vivo. J. Investig. Dermatol. 2004, 122, 1020–1029. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- León-Carmona, J.R.; Alvarez-Idaboy, J.R.; Galano, A. On the peroxyl scavenging activity of hydroxycinnamic acid derivatives: Mechanisms, kinetics, and importance of the acid–base equilibrium. Phys. Chem. Chem. Phys. 2012, 14, 12534–12543. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. Mechanism and kinetics studies on the antioxidant activity of sinapinic acid. Phys. Chem. Chem. Phys. 2011, 13, 11199–11205. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, M.; Cel, K.; Sroka, Z. The mechanistic insights into the role of pH and solvent on antiradical and prooxidant properties of polyphenols-nine compounds case study. Food Chem. 2023, 407, 134677. [Google Scholar] [CrossRef] [PubMed]
- Redžepović, I.; Marković, S.; Tošović, J. Antioxidative activity of caffeic acid: Mechanistic DFT study. Kragujev. J. Sci. 2017, 39, 109–122. [Google Scholar] [CrossRef]
- Chen, J.X.; Huang, S.H.; Wang, Y.; Shao, M.; Ye, W.C. Studies on the chemical constituents from Lobelia chinensis. Zhong Yao Cai. 2010, 33, 1721–1724. [Google Scholar]
- Ai, C.B.; Li, L.N. Salvianolic acids D and E: Two new depsides from Salvia miltiorrhiza. Planta Med. 1992, 58, 197–199. [Google Scholar] [CrossRef]
- Yapo, E.S.; Kouakou, H.; Kouakou, L.; Kouadio, J.Y.; Kouamé, P.; Mérillon, J.-M. Phenolic profiles of pineapple fruits (Ananas comosus L. Merrill) influence of the origin of suckers. Aust. J. Basic Appl. Sci. 2011, 5, 1372–1378. [Google Scholar]
- Kalinowska, M.; Gołębiewska, E.; Mazur, L.; Lewandowska, H.; Pruszyński, M.; Świderski, G.; Wyrwas, M.; Pawluczuk, N.; Lewandowski, W. Crystal Structure, spectroscopic characterization, antioxidant and cytotoxic activity of new Mg(II) and Mn(II)/Na(I) complexes of isoferulic acid. Materials 2021, 14, 3236. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Chen, D. Evaluation of antioxidant activity of isoferulic acid in vitro. Nat. Prod. Commun. 2011, 6, 1285–1288. [Google Scholar] [CrossRef]
- Karamać, M.; Kosińska, A. Comparison of radical-scavenging activities for selected phenolic acids. Pol. J. Food Nutr. Sci. 2005, 55, 165–170. [Google Scholar]
- Arfin, S.; Siddiqui, G.A.; Naeem, A.; Moin, S. Inhibition of advanced glycation end products by isoferulic acid and its free radical scavenging capacity: An in vitro and molecular docking study. Int. J. Biol. Macromol. 2018, 118, 1479–1487. [Google Scholar] [CrossRef]
- Karamać, M.; Koleva, L.; Kancheva, V.; Amarowicz, R. The structure—Antioxidant activity relationship of ferulates. Molecules 2017, 22, 527. [Google Scholar] [CrossRef]
- Sakai, S.; Kawamata, H.; Kogure, T.; Mantani, N.; Terasawa, K.; Umatake, M.; Ochiai, H. Inhibitory effect of ferulic acid and isoferulic acid on the production of macrophage inflammatory protein-2 in response to respiratory syncytial virus infection in RAW264.7 cells. Mediat. Inflamm. 1999, 8, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.M.; Chen, W.C.; Cheng, J.T. Mediation of β-Endorphin by Isoferulic Acid to Lower Plasma Glucose in Streptozotocin-Induced Diabetic Rats. J. Pharmacol. Exp. Ther. 2003, 307, 1196–1204. [Google Scholar] [CrossRef]
- Galano, A. Free radicals induced oxidative stress at a molecular level: The current status. Challenges and Perspectives of computational chemistry based protocols. J. Mex. Chem. Soc. 2015, 59, 231–262. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. Kinetics of radical-molecule reactions in aqueous solution: A benchmark study of the performance of Density Functional methods. J. Comput. Chem. 2014, 35, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Mardirossian, N.; Head-Gordon, M. How accurate are the Minnesota density functionals for noncovalent interactions, isomerization energies, thermochemistry, and barrier heights involving molecules composed of main-group elements? J. Chem. Theory Comput. 2016, 12, 4303–4325. [Google Scholar] [CrossRef]
- Available online: https://www.chemicalbook.com/ChemicalProductProperty_EN_CB9178579.htm (accessed on 28 February 2025).
- Erdemgil, F.Z.; Şanli, S.; Şanli, N.; Özkan, G.; Barbosa, J.; Guiteras, J.; Beltrán, J.L. Determination of pKa values of some hydroxylated benzoic acids in methanol–water binary mixtures by LC methodology and potentiometry. Talanta 2007, 72, 489–496. [Google Scholar] [CrossRef]
- Al Arni, S.; Drake, A.F.; Del Borghi, M.; Converti, A. Study of aromatic compounds derived from sugarcane bagasse. Part I: Effect of pH. Chem. Eng. Technol. 2010, 33, 895–901. [Google Scholar] [CrossRef]
- Bultinck, P.; Carbó-Dorca, R.; Langenaeker, W. Negative Fukui functions: New insights based on electronegativity equalization. J. Chem. Phys. 2003, 118, 4349–4356. [Google Scholar] [CrossRef]
- Melin, J.; Ayers, P.; Ortiz, J. Removing electrons can increase the electron density: A computational study of negative Fukui functions. J. Phys. Chem. A 2007, 111, 10017–10019. [Google Scholar] [CrossRef] [PubMed]
- Badhani, B.; Kakkar, R. Influence of intrinsic and extrinsic factors on the antiradical activity of gallic acid: A theoretical study. Struct. Chem. 2018, 29, 359–373. [Google Scholar] [CrossRef]
- Parise, A.; De Simone, B.C.; Marino, T.; Toscano, M.; Russo, N. Quantum mechanical predictions of the antioxidant capability of moracin C isomers. Front. Chem. 2021, 9, 666647. [Google Scholar] [CrossRef]
- Muraro, C.; Polato, M.; Bortoli, M.; Aiolli, F.; Orian, L. Radical scavenging activity of natural antioxidants and drugs: Development of a combined machine learning and quantum chemistry protocol. J. Chem.Phys. 2020, 153, 11411. [Google Scholar] [CrossRef]
- Piela, L. Chapter 14—Chemical Reactions. In Ideas of Quantum Chemistry, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 883–968. [Google Scholar]
- Mazzone, G.; Russo, N.; Toscano, M. Antioxidant properties comparative study of natural hydroxycinnamic acids and structurally modified derivatives: Computational insights. Comput. Theor. Chem. 2016, 1077, 39–47. [Google Scholar] [CrossRef]
- Babu, C.S.; Madhusoodanan, M.; Sridhar, G.; Tembe, B.L. Orientations of [Fe (H2O)6]2+ and [Fe(H2O)6]3+ complexes at a reactive separation in water. J. Am. Chem. Soc. 1997, 119, 5679–5681. [Google Scholar] [CrossRef]
- Amić, A.; Dimitrić Marković, J.M.; Marković, Z.; Milenković, D.; Milanović, Ž.; Antonijević, M.; Mastiľák Cagardová, D.; Rodríguez-Guerra Pedregal, J. Theoretical study of radical inactivation, LOX inhibition, and iron chelation: The role of ferulic acid in skin protection against uva induced oxidative stress. Antioxidants 2021, 10, 1303. [Google Scholar] [CrossRef]
- Truong, D.H.; Thi, N.; Nhung, A.; Quang Dao, D. Iron ions chelation-based antioxidant potential vs. pro-oxidant risk of ferulic acid: A DFT study in aqueous phase. Comput. Theor. Chem. 2020, 1185, 112905. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Galano, A.; Pérez-González, A.; Castañeda-Arriaga, R.; Muñoz-Rugeles, L.; Mendoza-Sarmiento, G.; Romero-Silva, A.; Ibarra-Escutia, A.; Rebollar-Zepeda, A.M.; León-Carmona, J.R.; Hernández-Olivares, M.A. Empirically fitted parameters for calculating pKa values with small deviations from experiments using a simple computational strategy. J. Chem. Inf. Model. 2016, 56, 1714–1724. [Google Scholar] [CrossRef]
- Janak, J.F. Proof that ∂E∂ni = ε in Density-Functional Theory. Phys. Rev. 1978, 18, 7165–7168. [Google Scholar] [CrossRef]
- Koopmans, T. Uber Die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- De Proft, F.; Martin, J.M.L.; Geerlings, P. Calculation of molecular electrostatic potentials and Fukui functions using density functional methods. Chem. Phys. Lett. 1996, 256, 400–408. [Google Scholar] [CrossRef]
- Kowalska-Baron, A. Theoretical Insight into Antioxidant Mechanism of Caffeic Acid Against Hydroperoxyl Radicals in Aqueous Medium at Different pH-Thermodynamic and Kinetic Aspects. Int. J. Mol. Sci. 2024, 25, 12753. [Google Scholar] [CrossRef]
- Marković, Z.; Tošović, J.; Milenković, D.; Marković, S. Revisiting the solvation enthalpies and free energies of the proton and electron in various solvents. Comput. Theor. Chem. 2016, 1077, 11–17. [Google Scholar] [CrossRef]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Hase, W.L.; Hynes, J.T. Current status of transition-state theory. J. Phys. Chem. 1983, 87, 2664. [Google Scholar] [CrossRef]
- Pollak, E.; Pechukas, P. Symmetry numbers, not statistical factors, should be used in absolute rate theory and in Bronsted relations. J. Am. Chem. Soc. 1978, 100, 2984. [Google Scholar] [CrossRef]
- Fernández-Ramos, A.; Ellingson, B.A.; Meana-Pañeda, R.; Marques, J.M.C.; Truhlar, D.G. Symmetry numbers and chemical reaction rates. Theor. Chem. Acc. 2007, 118, 813. [Google Scholar] [CrossRef]
- Eckart, C. The penetration of a potential barrier by electrons. Phys. Rev. 1930, 35, 1303. [Google Scholar] [CrossRef]
- Dzib, E.; Quintal, A.; Ortiz-Chi, F.; Merino, G. Eyringpy 2.0; Cinvestav: Merida, Mexico, 2021. [Google Scholar]
- Dzib, E.; Cabellos, J.L.; Ortiz-Chi, F.; Pan, S.; Galano, A.; Merino, G. Eyringpy: A program for computing rate constants in the gas phase and in solution. Int. J. Quantum Chem. 2019, 119, e25686. [Google Scholar] [CrossRef]
- Peng, C.; Schlegel, H.B. Combining synchronous transit and quasi-newton methods for finding transition states. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comp. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Collins, F.C.; Kimball, G.E. Diffusion-controlled reaction rates. J. Colloid Sci. 1949, 4, 425–437. [Google Scholar] [CrossRef]
- Von Smoluchowski, M. Versucheiner Mathematischen Theorie der Koagulations Kinetic Kolloider Lousungen. Z. Phys. Chem. 1917, 92, 129–168. [Google Scholar]
- Stokes, G.G. Mathematical and Physical Papers; University Press: Cambridge, UK, 1905. [Google Scholar]
- Einstein, A. On the motion of small particles suspended in liquids at rest required by the molecular-kinetic theory of heat. Ann. Phys. 1905, 17, 549–560. [Google Scholar] [CrossRef]
- Physical Properties of Common Liquids. Available online: http://trimen.pl/witek/ciecze (accessed on 11 July 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pKa | Isodesmic Method * | Parameter-Fitting Method | Literature Predicted Value [29] |
|---|---|---|---|
| pKa1 | 4.10 | 5.30 | 4.53 ± 0.11 |
| pKa2 | 6.30 | 10.80 | - |
| Isoferulic Acid | IP [kcal/mol] | EA [kcal/mol] | Χ [kcal/mol] | μ [kcal/mol] | Ƞ [kcal/mol] | S [kcal/mol | ω [kcal/mol] | ω− [kcal/mol] | ω+ [kcal/mol] |
|---|---|---|---|---|---|---|---|---|---|
| neutral | 172.00 | 28.96 | 100.48 | −100.48 | 71.52 | 35.76 | 70.58 | 129.76 | 17.65 |
| monoanion | 164.30 | 10.63 | 87.47 | −87.47 | 76.84 | 38.42 | 49.78 | 103.12 | 12.45 |
| dianion | 133.04 | 0.93 | 66.99 | −66.99 | 66.06 | 33.03 | 33.96 | 75.72 | 8.49 |
| Atom | Dianion |
|---|---|
| O1 | 0.034 |
| O2 | 0.043 |
| O3 | 0.039 |
| O21 | 0.170 |
| C4 | 0.007 |
| C5 | 0.109 |
| C6 | 0.125 |
| C7 | 0.000 |
| C8 | 0.132 |
| C9 | −0.007 |
| C10 | 0.076 |
| C11 | 0.143 |
| C12 | −0.006 |
| C13 | 0.010 |
| Isoferulic Acid | AIP [kcal/mol] | BDE [kcal/mol] | PA [kcal/mol] | PDE [kcal/mol] |
|---|---|---|---|---|
| neutral | 118.53 | 86.34 | 31.60 | 3.71 |
| monoanion | 110.98 | 84.70 | 42.74 | 9.63 |
| dianion | 77.87 | 106.78 | - | 64.83 |
| Radical | EA * [kcal/mol] |
|---|---|
| HOO• | −71.858 |
| NO2• | −96.043 |
| HO• | −93.008 |
| SO4•− | −114.48 |
| CH3O• | −78.429 |
| Cl3CO• | −142.315 |
| Isoferulic Acid | RAF ΔG [kcal/mol] | HT ΔG [kcal/mol] | SET ΔG [kcal/mol] | SET-PT ΔG [kcal/mol] | SPL-ET ΔG [kcal/mol] | SPL-HT ΔG [kcal/mol] |
|---|---|---|---|---|---|---|
| neutral | +2.858 (C11) | −2.332 (O21) | +44.126 | (1) 44.126 (2) −48.001 (O21) | (1) −32.093 * (2) +36.649 | (1) −32.093 * (2) −3.894 |
| monoanion | −6.245 (C11) | −3.894 (O21) | +36.649 | (1) 36.649 (2) −42.086 (O21) | (1) −20.646 (2) +3.793 | (1) −20.646 (2) +17.776 (C11) |
| dianion | −5.801 (C11) | +17.776 (C11) | +3.793 | (1) +3.793 (2) +12.440 | - | - |
| Bond Length [Å] | Neutral | Monoanionic | Dianionic |
|---|---|---|---|
| C4C10 | 1.41 | 1.41 | 1.42 |
| C10C11 | 1.49 | 1.49 | 1.49 |
| C11C12 | 1.53 | 1.56 | 1.56 |
| C12O2 | 1.33 | 1.24 | 1.24 |
| C12O3 | 1.20 | 1.26 | 1.26 |
| C11C18 | 1.42 | 1.43 | 1.44 |
| O18O17 | 1.42 | 1.43 | 1.43 |
| O2H16 | 2.59 | 2.57 | 2.58 |
| O3H19 | 2.10 | 1.68 | 1.68 |
| O1H26 | 2.09 | 2.09 | - |
| bond angle [°] | |||
| O1H26O25 | 113.7 | 114.1 | - |
| C4C10C11 | 124.9 | 125.4 | 125.9 |
| C10C11C12 | 112.1 | 112.2 | 111.9 |
| C11C12O2 | 111.5 | 116.0 | 116.4 |
| C11C12O3 | 124.2 | 116.3 | 116.6 |
| C11H16O2 | 65.9 | 68.0 | 67.5 |
| C11O18O17 | 109.6 | 109.3 | 109.5 |
| C10C11H16 | 111.5 | 111.3 | 111.3 |
| C10C11O18 | 113.2 | 111.4 | 112.1 |
| O17H19O3 | 123.5 | 145.4 | 145.9 |
| dihedral angle [°] | |||
| C4C10C11C12 | 91.8 | 87.3 | 100.2 |
| C6C4C10C11 | −1.06 | −2.40 | −0.59 |
| C10C11C12O2 | −64.5 | −74.9 | −75.9 |
| C4C10C11O18 | −144.2 | −146.8 | −134.1 |
| C10C11O18O17 | −57.6 | −63.4 | −62.8 |
| RC | TS | PC | Products | |||||
|---|---|---|---|---|---|---|---|---|
| H [kJ/mol] | G [kJ/mol] | H [kJ/mol] | G [kJ/mol] | H [kJ/mol] | G [kJ/mol] | H [kJ/mol] | G [kJ/mol] | |
| HT | −25.496 | 6.785 | 39.750 | 80.834 | −20.285 | 15.220 | −6.934 | −16.291 |
| RAF | −72.264 | −36.203 | 11.476 | 56.210 | −73.535 | −25.877 | −73.643 | −26.131 |
| pH 7 298.15 K | ΔGa# [kcal/mol] | κ | kD [M−1s−1] | kbim [M−1s−1] | kapp [M−1s−1] |
|---|---|---|---|---|---|
| HT | 19.3 | 3230.5 | 2.0 × 109 | 5.6 × 102 | 5.6 × 102 |
| RAF | 13.4 | 1.7 | 2.0 × 109 | 4.5 × 103 | 4.5 × 103 |
| Radical | ΔG [kJ/mol] | ΔGa# [kJ/mol] | λ [kJ/mol] | kbim [M−1s−1] | kD [M−1s−1] | kapp [M−1s−1] |
|---|---|---|---|---|---|---|
| HOO• | 15.871 | 20.591 | 45.028 | 1.53 × 109 | 7.84 × 106 | 7.80 × 106 |
| NO2• | −85.392 | 15.325 | 194.614 | 1.28 × 1010 | 7.89 × 106 | 7.88 × 106 |
| HO• | −71.833 | 0.706 | 87.552 | 4.67 × 1012 | 8.14 × 106 | 8.14 × 106 |
| SO4•− | −153.662 | 6.925 | 234.210 | 3.80 × 1011 | 7.57 × 106 | 7.57 × 106 |
| CH3O• | −10.707 | 4.847 | 37.767 | 8.79 × 1011 | 7.83 × 106 | 7.83 × 106 |
| Cl3CO• | −281.495 | 6.751 | 383.224 | 4.08 × 1011 | 7.51 × 106 | 7.51 × 106 |
| Neutral | Monoanion | Dianion | |||||||
|---|---|---|---|---|---|---|---|---|---|
| λ [nm] (eV) | f | μ [D] | λ [nm] (eV) | f | μ [D] | λ [nm] (eV) | f | μ [D] | |
| 1 | 298.29 (4.16) | 0.7577 | 7.4406 | 276.48 (4.48) | 0.6103 | 5.5546 | 333.81 (3.71) | 0.2783 | 3.0585 |
| 2 | 261.44 (4.74) | 0.0926 | 0.7967 | 257.95 (4.81) | 0.0000 | 0.0003 | 292.73 (4.24) | 0.0007 | 0.0064 |
| 3 | 241.20 (5.14) | 0.0000 | 0.0002 | 254.03 (4.88) | 0.0000 | 0.0000 | 267.54 (4.63) | 0.3373 | 2.9712 |
| 4 | 222.22 (5.58) | 0.1608 | 1.1760 | 248.08 (5.00) | 0.1708 | 1.3945 | 262.67 (4.72) | 0.0005 | 0.0047 |
| 5 | 220.44 (5.62) | 0.0130 | 0.0097 | 229.75 (5.40) | 0.0016 | 0.0119 | 253.41 (4.89) | 0.0000 | 0.0004 |
| 6 | 203.52 (6.09) | 0.0004 | 0.0026 | 212.39 (5.84) | 0.0005 | 0.0034 | 247.13 (5.02) | 0.4482 | 3.6466 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalska-Baron, A. Theoretical Insight into Antioxidant Mechanisms of Trans-Isoferulic Acid in Aqueous Medium at Different pH. Int. J. Mol. Sci. 2025, 26, 5615. https://doi.org/10.3390/ijms26125615
Kowalska-Baron A. Theoretical Insight into Antioxidant Mechanisms of Trans-Isoferulic Acid in Aqueous Medium at Different pH. International Journal of Molecular Sciences. 2025; 26(12):5615. https://doi.org/10.3390/ijms26125615
Chicago/Turabian StyleKowalska-Baron, Agnieszka. 2025. "Theoretical Insight into Antioxidant Mechanisms of Trans-Isoferulic Acid in Aqueous Medium at Different pH" International Journal of Molecular Sciences 26, no. 12: 5615. https://doi.org/10.3390/ijms26125615
APA StyleKowalska-Baron, A. (2025). Theoretical Insight into Antioxidant Mechanisms of Trans-Isoferulic Acid in Aqueous Medium at Different pH. International Journal of Molecular Sciences, 26(12), 5615. https://doi.org/10.3390/ijms26125615