
Paradoxical SERCA2a Dysregulation Contributes to Atrial Fibrillation in a Model of Diet-Induced Obesity
 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
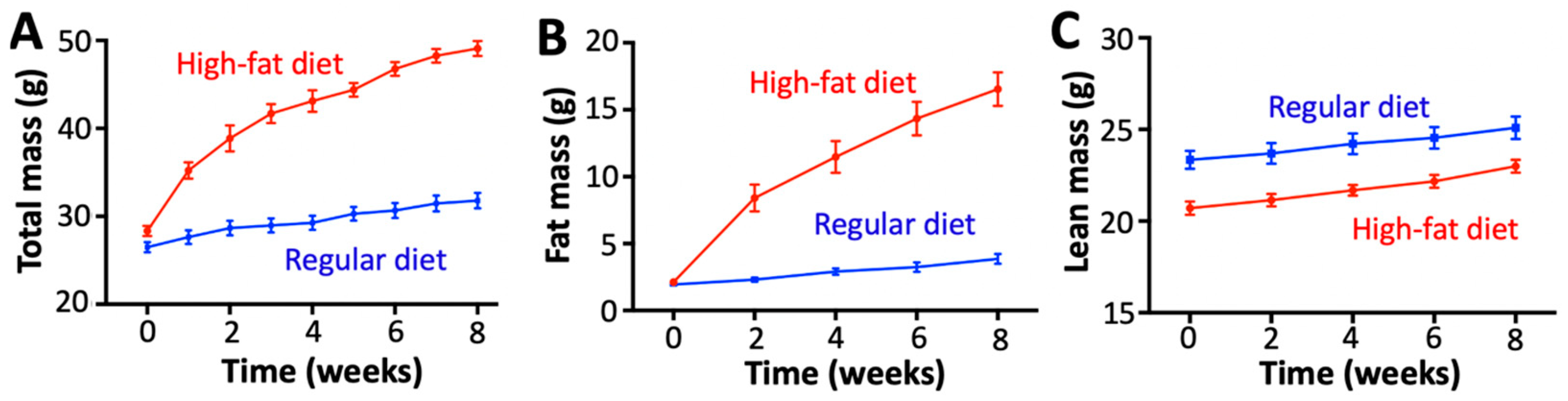
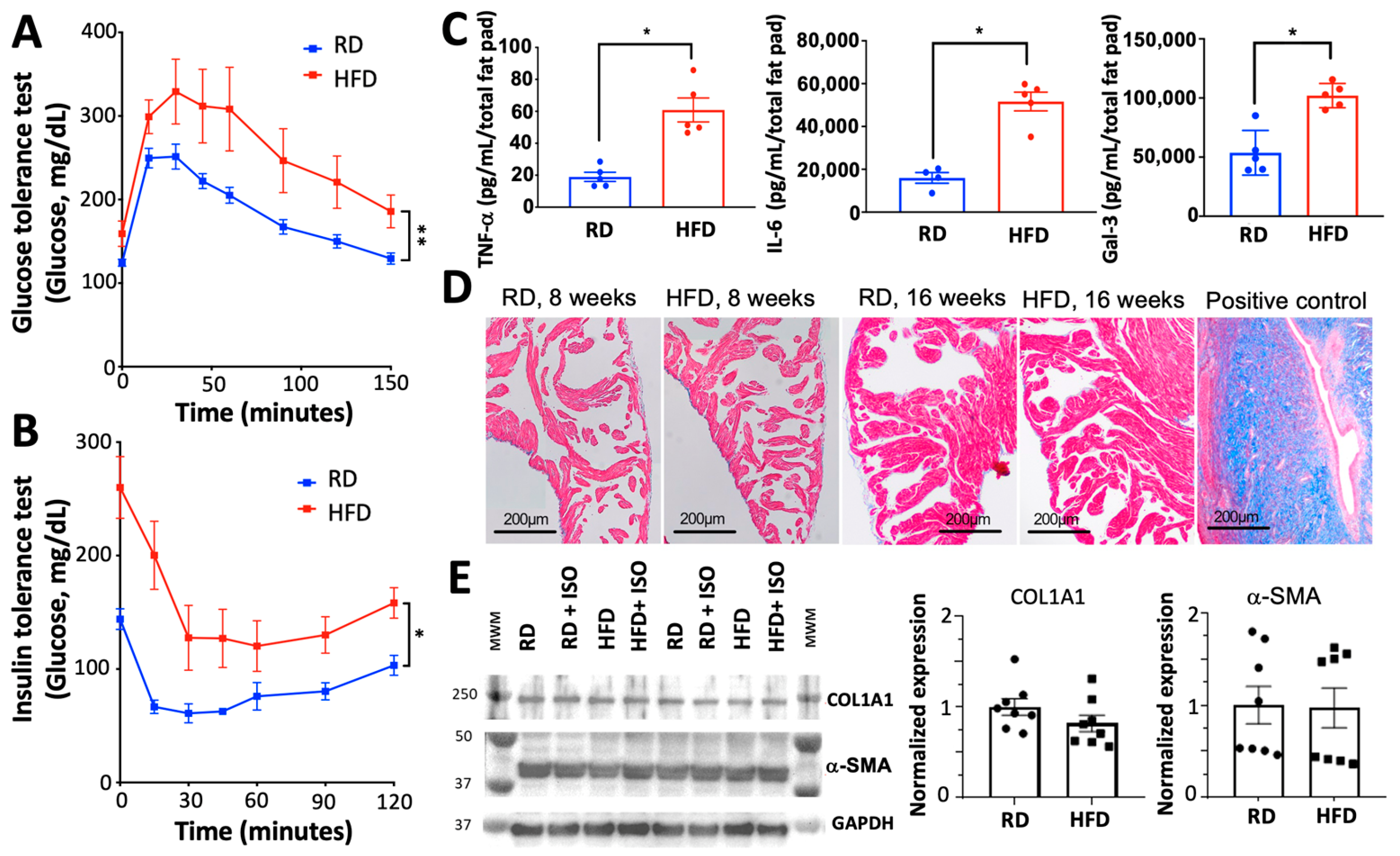
2.1. Diet-Induced Obesity Induces Metabolic and Inflammatory Imbalances Without Fibrosis or Alterations in Cardiovascular Hemodynamics
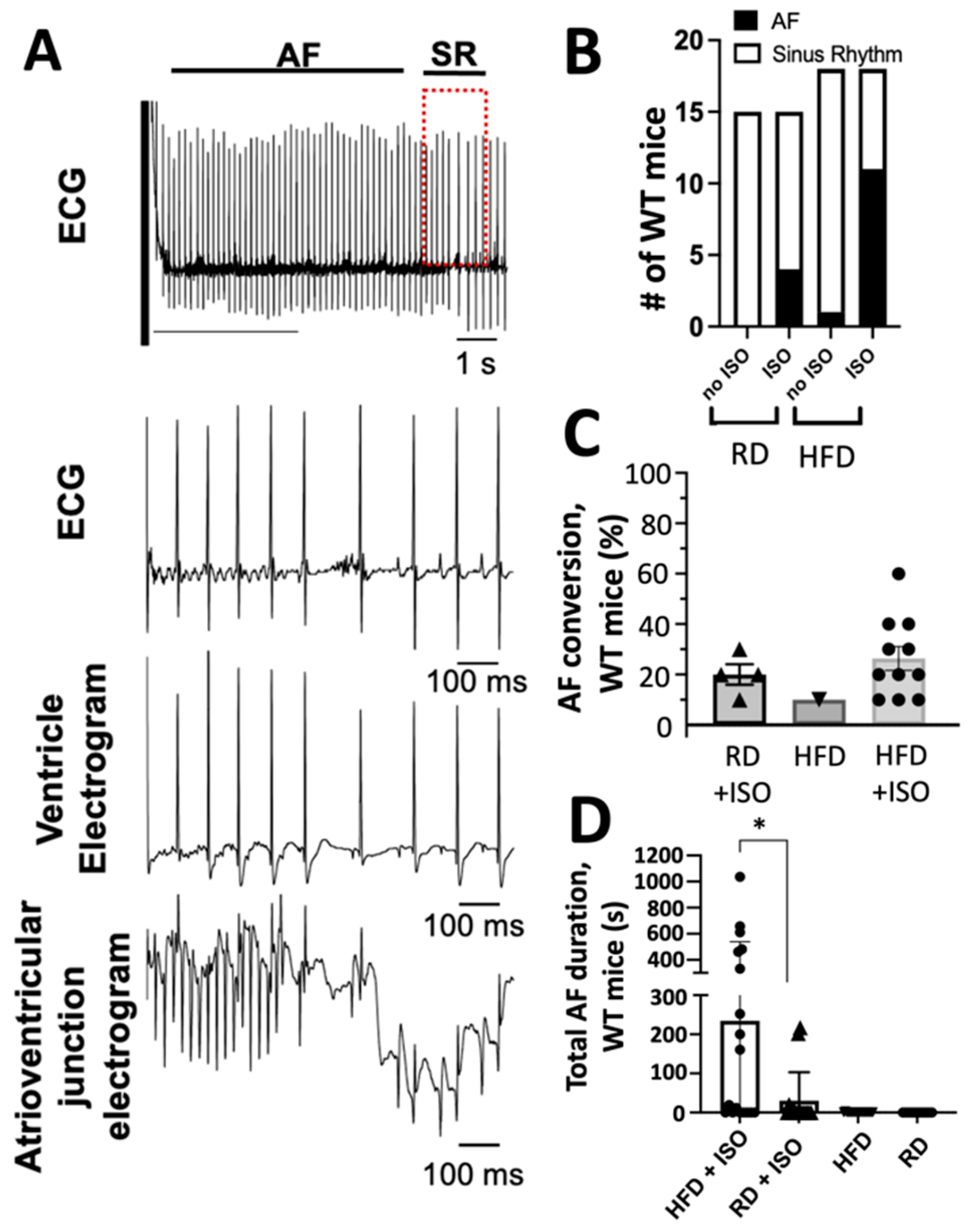
2.2. Diet-Induced Obesity and Acute Sympathetic Activation Synergize During Intracardiac Tachypacing to Induce Atrial Fibrillation
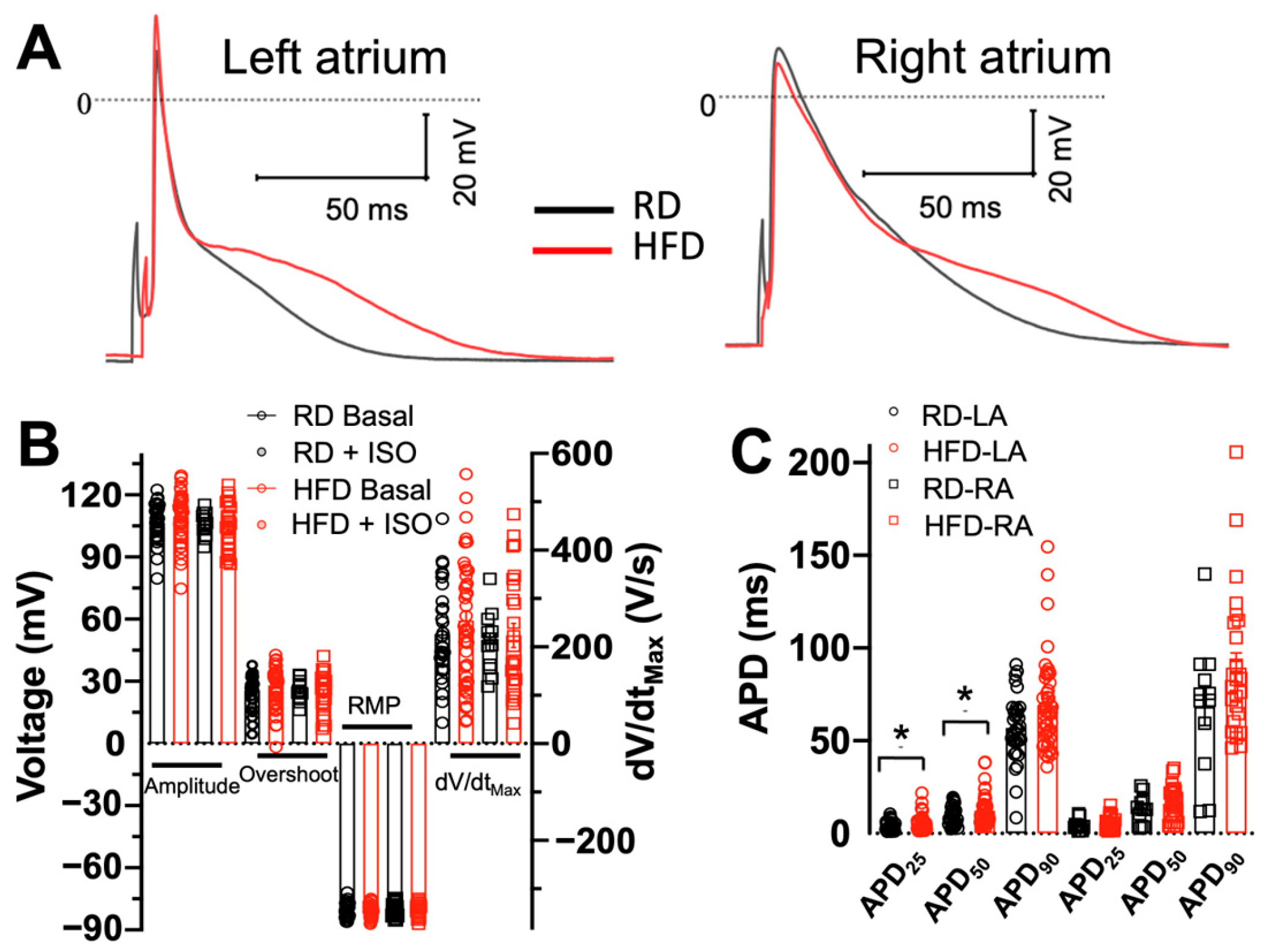
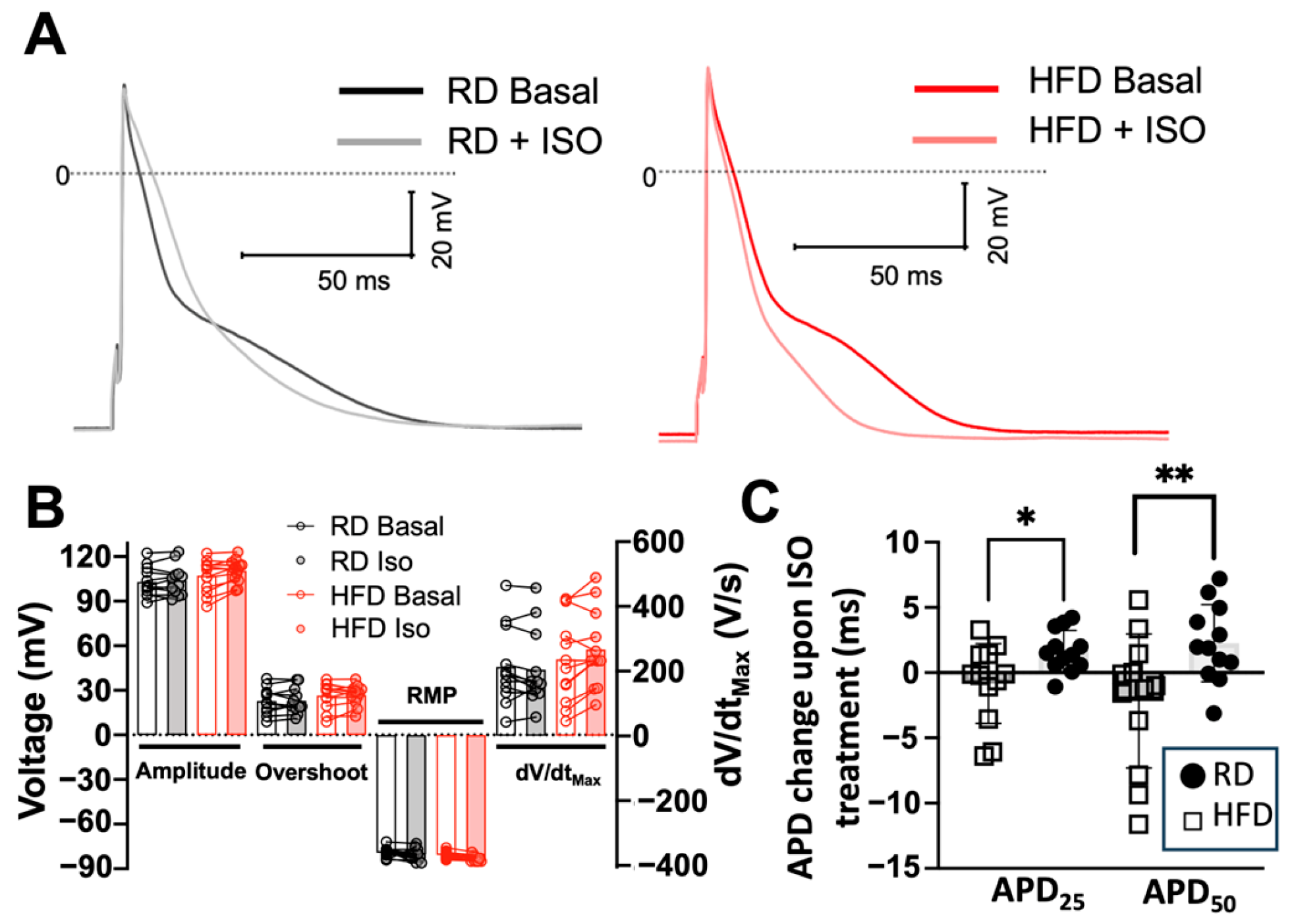
2.3. Effects of Diet-Induced Obesity on the Action Potential Characteristics of Atrial Myocytes
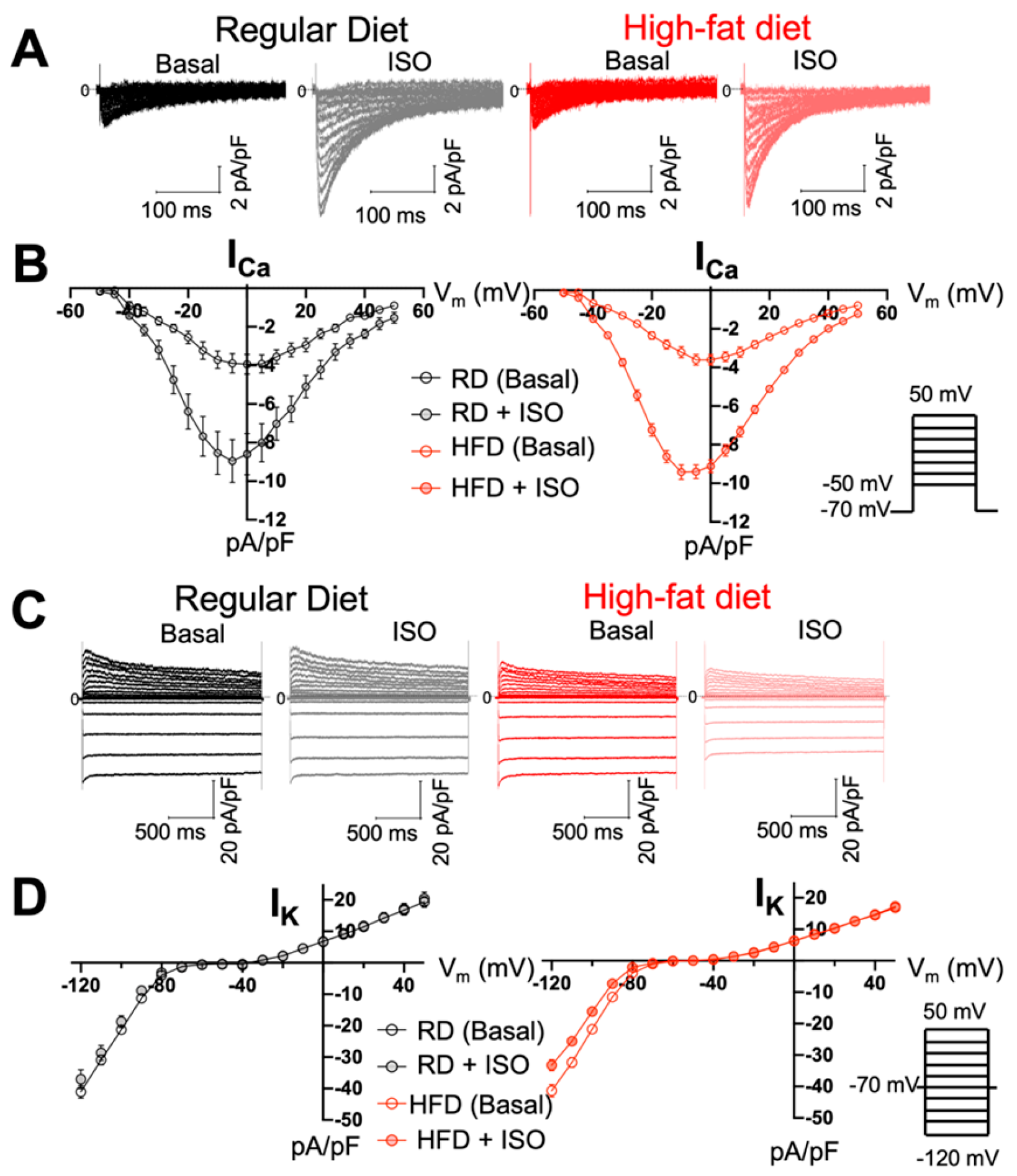
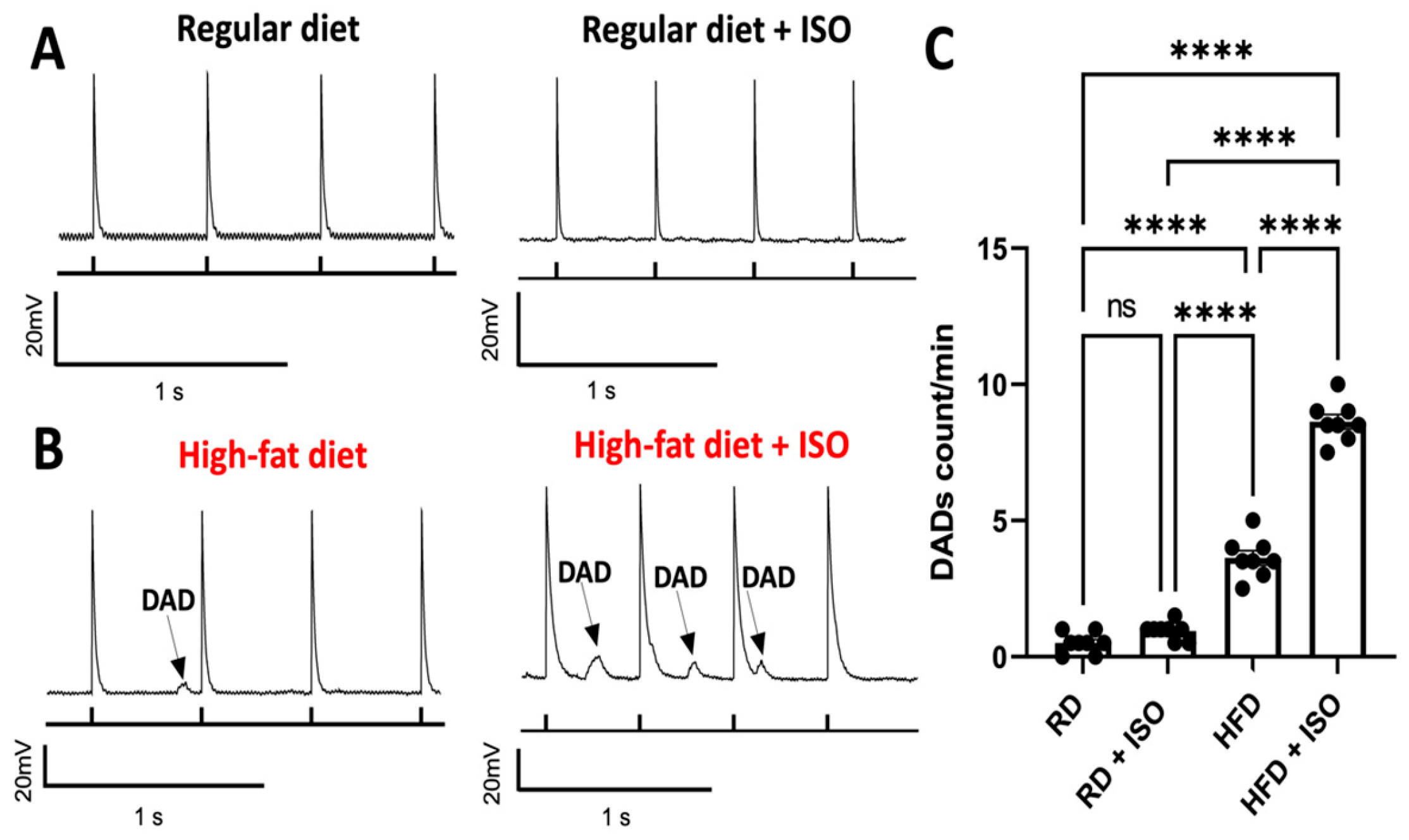
2.4. Diet-Induced Obesity Does Not Alter ICa and IK Densities but Produces Delayed Afterdepolarization Events in Atrial Myocytes
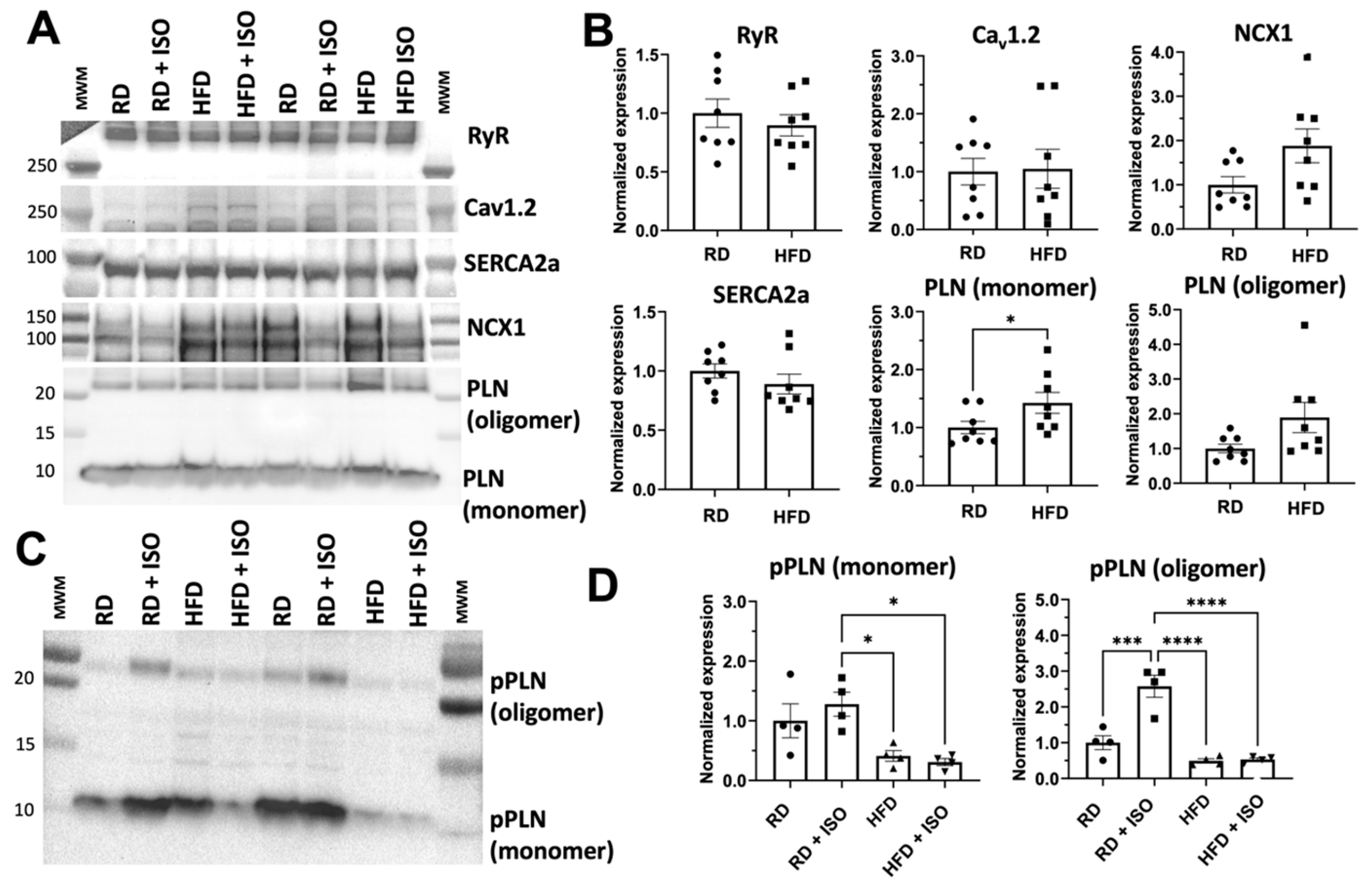
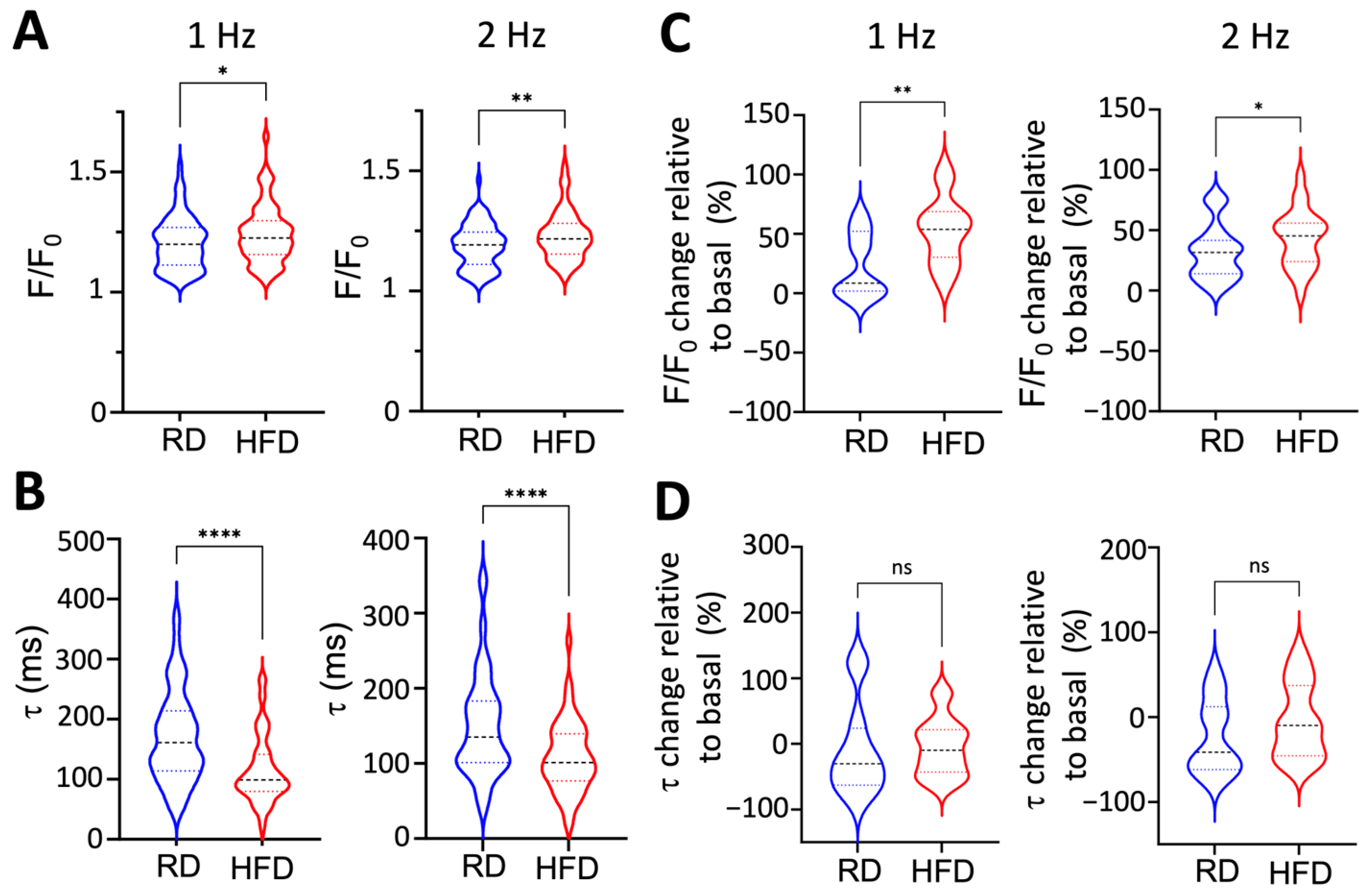
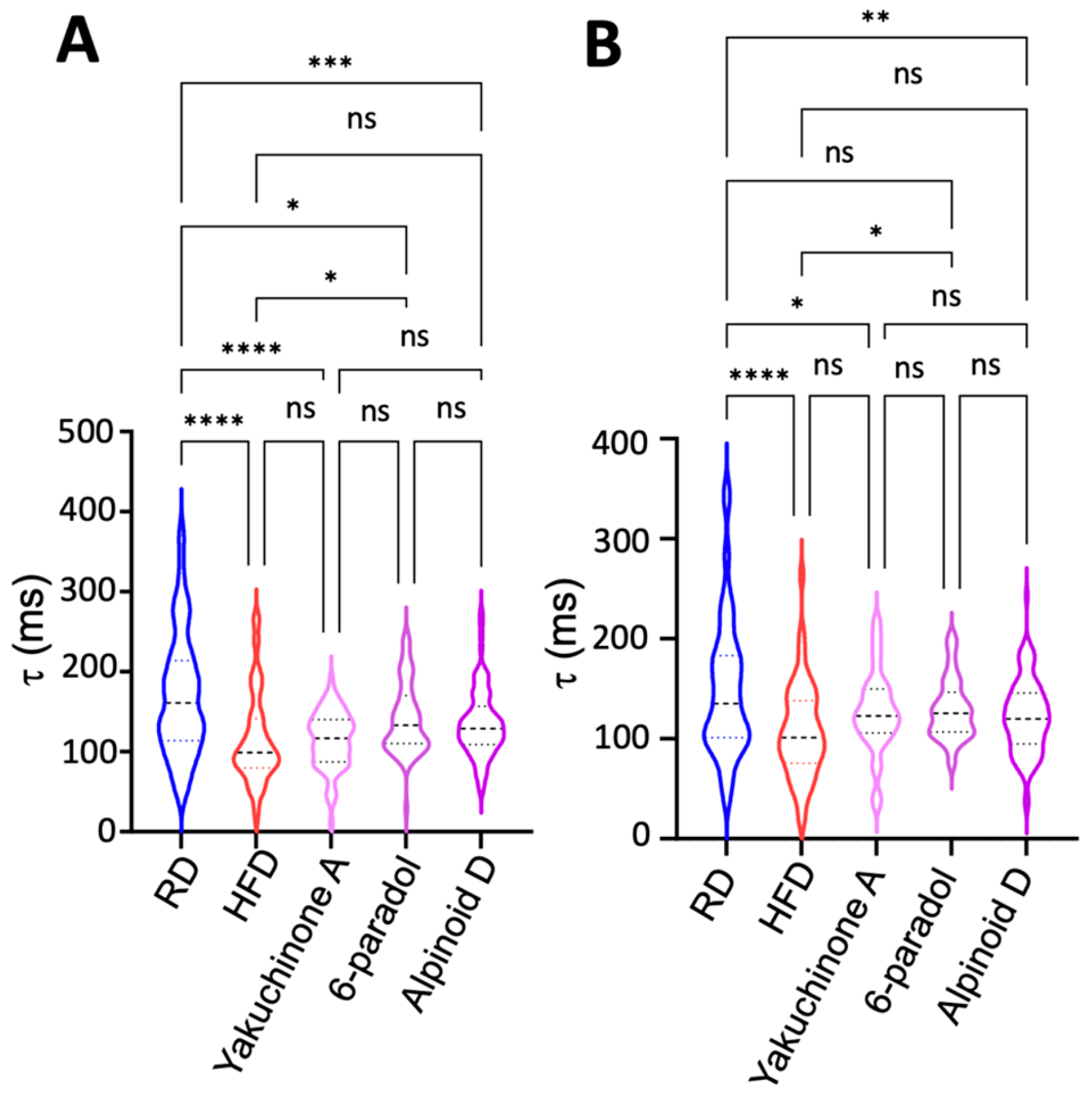
2.5. Obesity Induces Paradoxical Dysregulation of SERCA2a in Atrial Myocytes
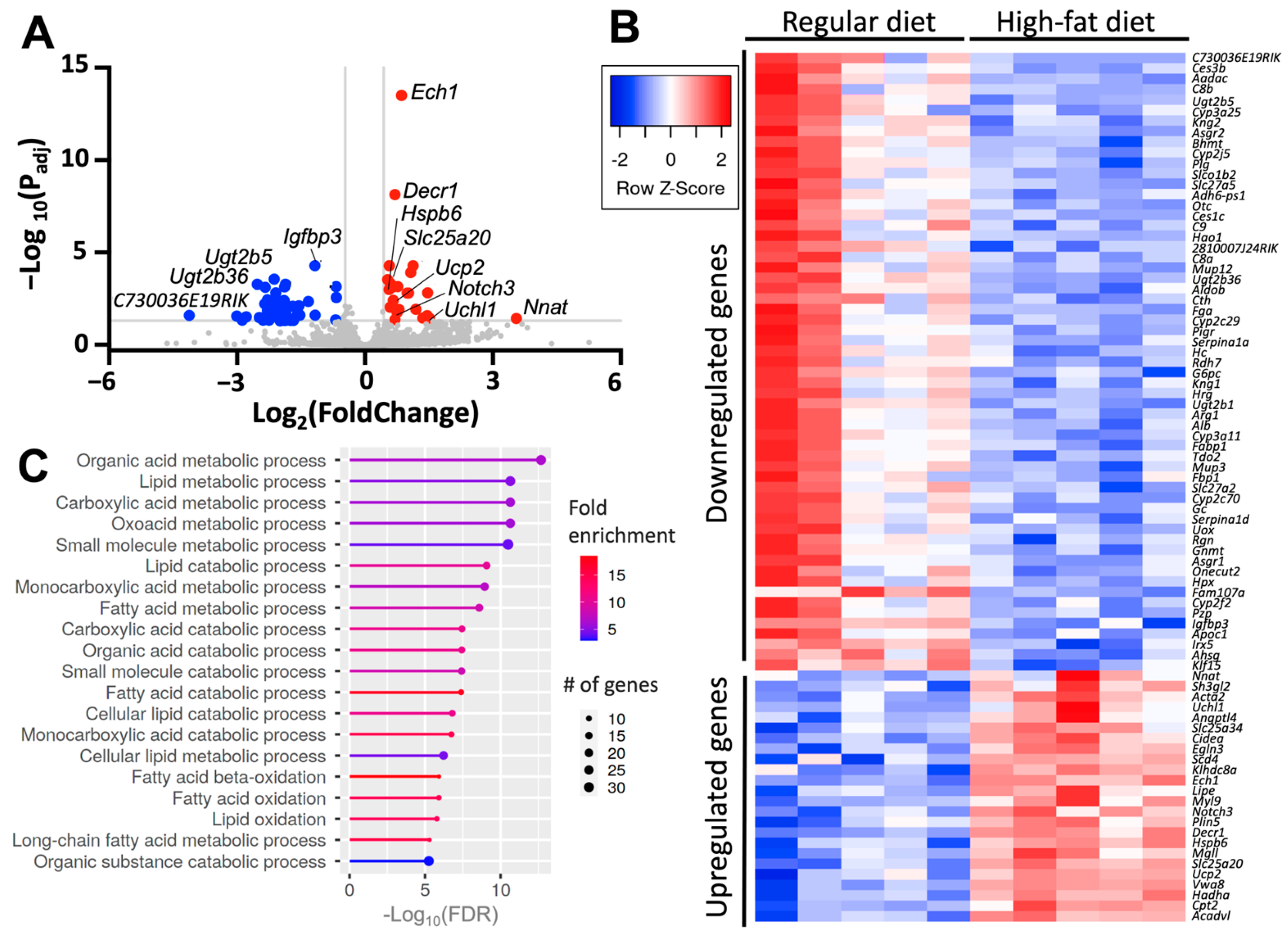
2.6. Diet-Induced Atrial Upregulates Genes Involved in Metabolism and Stress
3. Discussion
Study Limitations
4. Materials and Methods
4.1. High-Fat Diet-Induced Obesity Mice Model
4.2. Enzyme-Linked Immunosorbent Assay (ELISA)
4.3. Surface Electrocardiographic Recording
4.4. Intracardiac Recordings
4.5. Electrophysiology Studies of Isolated Atrial Myocytes
4.6. Western Blot Analysis
4.7. Single-Cell Ca2+ Imaging
4.8. RNA Sequencing and Differential Gene Expression
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 15 April 2025).
- Finkelstein, E.A.; Khavjou, O.A.; Thompson, H.; Trogdon, J.G.; Pan, L.; Sherry, B.; Dietz, W. Obesity and severe obesity forecasts through 2030. Am. J. Prev. Med. 2012, 42, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Despres, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef]
- Dublin, S.; French, B.; Glazer, N.L.; Wiggins, K.L.; Lumley, T.; Psaty, B.M.; Smith, N.L.; Heckbert, S.R. Risk of new-onset atrial fibrillation in relation to body mass index. Arch. Intern. Med. 2006, 166, 2322–2328. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.S.M.; Barnes, M.E.; Miyasaka, Y.; Cha, S.S.; Bailey, K.R.; Verzosa, G.C.; Seward, J.B.; Gersh, B.J. Obesity as a risk factor for the progression of paroxysmal to permanent atrial fibrillation: A longitudinal cohort study of 21 years. Eur. Heart J. 2008, 29, 2227–2233. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.V.; Anumonwo, J.; Jalife, J. Atrial Fibrillation Susceptibility in Obesity. An Excess Adiposity and Fibrosis Complicity? Circ. Res. 2016, 118, 1468–1471. [Google Scholar] [CrossRef]
- Limpitikul, W.B.; Das, S. Obesity-Related Atrial Fibrillation: Cardiac Manifestation of a Systemic Disease. J. Cardiovasc. Dev. Dis. 2023, 10, 323. [Google Scholar] [CrossRef]
- Brundel, B.; Ai, X.; Hills, M.T.; Kuipers, M.F.; Lip, G.Y.H.; de Groot, N.M.S. Atrial fibrillation. Nat. Rev. Dis. Primers 2022, 8, 21. [Google Scholar] [CrossRef]
- Shu, H.; Cheng, J.; Li, N.; Zhang, Z.; Nie, J.; Peng, Y.; Wang, Y.; Wang, D.W.; Zhou, N. Obesity and atrial fibrillation: A narrative review from arrhythmogenic mechanisms to clinical significance. Cardiovasc. Diabetol. 2023, 22, 192. [Google Scholar] [CrossRef]
- Wang, T.J.; Parise, H.; Levy, D.; D’Agostino, R.B., Sr.; Wolf, P.A.; Vasan, R.S.; Benjamin, E.J. Obesity and the risk of new-onset atrial fibrillation. JAMA 2004, 292, 2471–2477. [Google Scholar] [CrossRef]
- Abed, H.S.; Samuel, C.S.; Lau, D.H.; Kelly, D.J.; Royce, S.G.; Alasady, M.; Mahajan, R.; Kuklik, P.; Zhang, Y.; Brooks, A.G.; et al. Obesity results in progressive atrial structural and electrical remodeling: Implications for atrial fibrillation. Heart Rhythm 2012, 10, 90–100. [Google Scholar] [CrossRef]
- Pouwels, S.; Topal, B.; Knook, M.T.; Celik, A.; Sundbom, M.; Ribeiro, R.; Parmar, C.; Ugale, S. Interaction of obesity and atrial fibrillation: An overview of pathophysiology and clinical management. Expert Rev. Cardiovasc. Ther. 2019, 17, 209–223. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.D.; Hong, L.; Sridhar, A.; Menon, A.; Perike, S.; Zhang, M.; Da Silva, I.B.; Yan, J.; Bonini, M.G.; Ai, X.; et al. Ion Channel and Structural Remodeling in Obesity-Mediated Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2020, 13, e008296. [Google Scholar] [CrossRef]
- Scott, L., Jr.; Fender, A.C.; Saljic, A.; Li, L.; Chen, X.; Wang, X.; Linz, D.; Lang, J.; Hohl, M.; Twomey, D.; et al. NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias. Cardiovasc. Res. 2021, 117, 1746–1759. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Liao, J.K. A Mouse Model of Diet-Induced Obesity and Insulin Resistance. In mTOR: Methods and Protocols; Weichhart, T., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 421–433. [Google Scholar]
- Hancock, C.R.; Han, D.H.; Chen, M.; Terada, S.; Yasuda, T.; Wright, D.C.; Holloszy, J.O. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 7815–7820. [Google Scholar] [CrossRef]
- Borst, S.E.; Conover, C.F. High-fat diet induces increased tissue expression of TNF-alpha. Life Sci. 2005, 77, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Eder, K.; Baffy, N.; Falus, A.; Fulop, A.K. The major inflammatory mediator interleukin-6 and obesity. Inflamm. Res. 2009, 58, 727–736. [Google Scholar] [CrossRef]
- Ripplinger, C.M.; Glukhov, A.V.; Kay, M.W.; Boukens, B.J.; Chiamvimonvat, N.; Delisle, B.P.; Fabritz, L.; Hund, T.J.; Knollmann, B.C.; Li, N.; et al. Guidelines for assessment of cardiac electrophysiology and arrhythmias in small animals. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H1137–H1166. [Google Scholar] [CrossRef]
- Calvo, C.J.; Lozano, W.M.; Arias-Mutis, Ó.J.; Such-Miquel, L.; Such, L.; Genovés, P.; Guill, A.; Millet, J.; Chorro, F.J.; Alberola, A.; et al. Modifications of short-term intrinsic pacemaker variability in diet-induced metabolic syndrome: A study on isolated rabbit heart. J. Physiol. Biochem. 2019, 75, 173–183. [Google Scholar] [CrossRef]
- Albarado-Ibañez, A.; Arroyo-Carmona, R.E.; Sánchez-Hernández, R.; Ramos-Ortiz, G.; Frank, A.; García-Gudiño, D.; Torres-Jácome, J. The Role of the Autonomic Nervous System on Cardiac Rhythm during the Evolution of Diabetes Mellitus Using Heart Rate Variability as a Biomarker. J. Diabetes Res. 2019, 2019, 5157024. [Google Scholar] [CrossRef]
- Duan, J.; Wang, Q.; Zhang, B.; Liu, C.; Li, C.; Wang, L. Accurate detection of atrial fibrillation events with R-R intervals from ECG signals. PLoS ONE 2022, 17, e0271596. [Google Scholar] [CrossRef]
- Roden, D.M.; Balser, J.R.; George, A.L., Jr.; Anderson, M.E. Cardiac Ion Channels. Annu. Rev. Physiol. 2002, 64, 431–475. [Google Scholar] [CrossRef]
- Denham, N.C.; Pearman, C.M.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front. Physiol. 2018, 9, 1380. [Google Scholar] [CrossRef] [PubMed]
- Kistamás, K.; Veress, R.; Horváth, B.; Bányász, T.; Nánási, P.P.; Eisner, D.A. Calcium Handling Defects and Cardiac Arrhythmia Syndromes. Front. Pharmacol. 2020, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.Y.; Bai, J.; Gladding, P.A.; Fedorov, V.V.; Zhao, J. Afterdepolarizations and abnormal calcium handling in atrial myocytes with modulated SERCA uptake: A sensitivity analysis of calcium handling channels. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2020, 378, 20190557. [Google Scholar] [CrossRef]
- Singh, D.R.; Dalton, M.P.; Cho, E.E.; Pribadi, M.P.; Zak, T.J.; Seflova, J.; Makarewich, C.A.; Olson, E.N.; Robia, S.L. Newly Discovered Micropeptide Regulators of SERCA Form Oligomers but Bind to the Pump as Monomers. J. Mol. Biol. 2019, 431, 4429–4443. [Google Scholar] [CrossRef] [PubMed]
- Glaves, J.P.; Primeau, J.O.; Espinoza-Fonseca, L.M.; Lemieux, M.J.; Young, H.S. The Phospholamban Pentamer Alters Function of the Sarcoplasmic Reticulum Calcium Pump SERCA. Biophys. J. 2019, 116, 633–647. [Google Scholar] [CrossRef]
- Liu, A.Y.; Aguayo-Ortiz, R.; Guerrero-Serna, G.; Wang, N.; Blin, M.G.; Goldstein, D.R.; Espinoza-Fonseca, L.M. Homologous cardiac calcium pump regulators phospholamban and sarcolipin adopt distinct oligomeric states in the membrane. Comput. Struct. Biotechnol. J. 2022, 20, 380–384. [Google Scholar] [CrossRef]
- Funk, F.; Kronenbitter, A.; Hackert, K.; Oebbeke, M.; Klebe, G.; Barth, M.; Koch, D.; Schmitt, J.P. Phospholamban pentamerization increases sensitivity and dynamic range of cardiac relaxation. Cardiovasc. Res. 2023, 119, 1568–1582. [Google Scholar] [CrossRef]
- Kimura, Y.; Kurzydlowski, K.; Tada, M.; MacLennan, D.H. Phospholamban inhibitory function is activated by depolymerization. J. Biol. Chem. 1997, 272, 15061–15064. [Google Scholar] [CrossRef]
- Zvaritch, E.; Backx, P.H.; Jirik, F.; Kimura, Y.; de Leon, S.; Schmidt, A.G.; Hoit, B.D.; Lester, J.W.; Kranias, E.G.; MacLennan, D.H. The transgenic expression of highly inhibitory monomeric forms of phospholamban in mouse heart impairs cardiac contractility. J. Biol. Chem. 2000, 275, 14985–14991. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef]
- Li, L.; Chu, G.; Kranias, E.G.; Bers, D.M. Cardiac myocyte calcium transport in phospholamban knockout mouse: Relaxation and endogenous CaMKII effects. Am. J. Physiol. 1998, 274, H1335–H1347. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cortes, C.; Fernandez-de Gortari, E.; Aguayo-Ortiz, R.; Seflova, J.; Ard, A.; Clasby, M.; Anumonwo, J.; Espinoza-Fonseca, L.M. Machine Learning-Driven Discovery of Structurally Related Natural Products as Activators of the Cardiac Calcium Pump SERCA2a. ChemMedChem 2025, 20, e202400913. [Google Scholar] [CrossRef]
- Drobysheva, A.; Ahmad, M.; White, R.; Wang, H.W.; Leenen, F.H. Cardiac sympathetic innervation and PGP9.5 expression by cardiomyocytes after myocardial infarction: Effects of central MR blockade. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1817–H1829. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Aeschbacher, S.; Bossard, M.; Hochgruber, T.; Zimmermann, A.J.; Kaufmann, B.A.; Pumpol, K.; Rickenbacker, P.; Pare, G.; Conen, D. Whole Blood Gene Expression Differentiates between Atrial Fibrillation and Sinus Rhythm after Cardioversion. PLoS ONE 2016, 11, e0157550. [Google Scholar] [CrossRef]
- Larbig, R.; Reda, S.; Paar, V.; Trost, A.; Leitner, J.; Weichselbaumer, S.; Motloch, K.A.; Wernly, B.; Arrer, A.; Strauss, B.; et al. Through modulation of cardiac Ca(2+) handling, UCP2 affects cardiac electrophysiology and influences the susceptibility for Ca(2+)-mediated arrhythmias. Exp. Physiol. 2017, 102, 650–662. [Google Scholar] [CrossRef]
- Turner, J.D.; Gaspers, L.D.; Wang, G.; Thomas, A.P. Uncoupling protein-2 modulates myocardial excitation-contraction coupling. Circ. Res. 2010, 106, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Ma, S.; Tse, G.; Wong, W.T.; Huang, Y. Uncoupling Protein 2 in Cardiovascular Health and Disease. Front. Physiol. 2018, 9, 1060. [Google Scholar] [CrossRef]
- Yap, J.Q.; Seflova, J.; Sweazey, R.; Artigas, P.; Robia, S.L. FXYD proteins and sodium pump regulatory mechanisms. J. Gen. Physiol. 2021, 153, e202012633. [Google Scholar] [CrossRef]
- Ke, L.; Meijering, R.A.; Hoogstra-Berends, F.; Mackovicova, K.; Vos, M.J.; Van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS ONE 2011, 6, e20395. [Google Scholar] [CrossRef]
- Shi, J.; Xiao, P.; Liu, X.; Chen, Y.; Xu, Y.; Fan, J.; Yin, Y. Notch3 Modulates Cardiac Fibroblast Proliferation, Apoptosis, and Fibroblast to Myofibroblast Transition via Negative Regulation of the RhoA/ROCK/Hif1alpha Axis. Front. Physiol. 2020, 11, 669. [Google Scholar] [CrossRef] [PubMed]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef]
- Gorski, P.A.; Jang, S.P.; Jeong, D.; Lee, A.; Lee, P.; Oh, J.G.; Chepurko, V.; Yang, D.K.; Kwak, T.H.; Eom, S.H.; et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca(2+)-ATPase in Heart Failure. Circ. Res. 2019, 124, e63–e80. [Google Scholar] [CrossRef]
- Braun, J.L.; Geromella, M.S.; Hamstra, S.I.; Fajardo, V.A. Neuronatin regulates whole-body metabolism: Is thermogenesis involved? FASEB Bioadv. 2020, 2, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.L.; Teng, A.C.T.; Geromella, M.S.; Ryan, C.R.; Fenech, R.K.; MacPherson, R.E.K.; Gramolini, A.O.; Fajardo, V.A. Neuronatin promotes SERCA uncoupling and its expression is altered in skeletal muscles of high-fat diet-fed mice. FEBS Lett. 2021, 595, 2756–2767. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.Y.; Guo, L.; Zhang, B.; Chen, S.; Wu, X.R.; Liu, X.D.; Xu, X.Y.; Li, B.Y.; Chen, S.; Xu, N.J.; et al. Aberrant miR-339-5p/neuronatin signaling causes prodromal neuronal calcium dyshomeostasis in mutant presenilin mice. J. Clin. Investig. 2022, 132, e149160. [Google Scholar] [CrossRef]
- Wondmkun, Y.T. Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications. Diabetes Metab. Syndr. Obes. 2020, 13, 3611–3616. [Google Scholar] [CrossRef]
- Chan, Y.H.; Chang, G.J.; Lai, Y.J.; Chen, W.J.; Chang, S.H.; Hung, L.M.; Kuo, C.T.; Yeh, Y.H. Atrial fibrillation and its arrhythmogenesis associated with insulin resistance. Cardiovasc. Diabetol. 2019, 18, 125. [Google Scholar] [CrossRef]
- Takemoto, Y.; Ramirez, R.J.; Yokokawa, M.; Kaur, K.; Ponce-Balbuena, D.; Sinno, M.C.; Willis, B.C.; Ghanbari, H.; Ennis, S.R.; Guerrero-Serna, G.; et al. Galectin-3 Regulates Atrial Fibrillation Remodeling and Predicts Catheter Ablation Outcomes. JACC Basic Transl. Sci. 2016, 1, 143–154. [Google Scholar] [CrossRef]
- Ablorh, N.A.; Dong, X.; James, Z.M.; Xiong, Q.; Zhang, J.; Thomas, D.D.; Karim, C.B. Synthetic phosphopeptides enable quantitation of the content and function of the four phosphorylation states of phospholamban in cardiac muscle. J. Biol. Chem. 2014, 289, 29397–29405. [Google Scholar] [CrossRef]
- MacGrogan, D.; Munch, J.; de la Pompa, J.L. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 685–704. [Google Scholar] [CrossRef] [PubMed]
- Remes, J.; Van Brakel, T.J.; Bolotin, G.; Garber, C.; De Jong, M.M.; Van Der Veen, F.H.; Maessen, J.G. Persistent atrial fibrillation in a goat model of chronic left atrial overload. J. Thorac. Cardiovasc. Surg. 2008, 136, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Pandey, A.; Lau, D.H.; Alpert, M.A.; Sanders, P. Obesity and Atrial Fibrillation Prevalence, Pathogenesis, and Prognosis: Effects of Weight Loss and Exercise. J. Am. Coll. Cardiol. 2017, 70, 2022–2035. [Google Scholar] [CrossRef]
- Lohr, D.; Thiele, A.; Stahnke, M.; Braun, V.; Smeir, E.; Spranger, J.; Brachs, S.; Klopfleisch, R.; Foryst-Ludwig, A.; Schreiber, L.M.; et al. Assessment of Myocardial Microstructure in a Murine Model of Obesity-Related Cardiac Dysfunction by Diffusion Tensor Magnetic Resonance Imaging at 7T. Front. Cardiovasc. Med. 2022, 9, 839714. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Rudy, Y. Early afterdepolarizations in cardiac myocytes: Mechanism and rate dependence. Biophys. J. 1995, 68, 949–964. [Google Scholar] [CrossRef]
- Johnson, D.M.; Heijman, J.; Bode, E.F.; Greensmith, D.J.; van der Linde, H.; Abi-Gerges, N.; Eisner, D.A.; Trafford, A.W.; Volders, P.G. Diastolic spontaneous calcium release from the sarcoplasmic reticulum increases beat-to-beat variability of repolarization in canine ventricular myocytes after beta-adrenergic stimulation. Circ. Res. 2013, 112, 246–256. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Bers, D.M. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 2004, 14, 61–66. [Google Scholar] [CrossRef]
- Varro, A.; Baczko, I. Cardiac ventricular repolarization reserve: A principle for understanding drug-related proarrhythmic risk. Br. J. Pharmacol. 2011, 164, 14–36. [Google Scholar] [CrossRef]
- Cleary, S.R.; Seflova, J.; Cho, E.E.; Bisht, K.; Khandelia, H.; Espinoza-Fonseca, L.M.; Robia, S.L. Phospholamban inhibits the cardiac calcium pump by interrupting an allosteric activation pathway. J. Biol. Chem. 2024, 300, 107267. [Google Scholar] [CrossRef]
- Lima-Leopoldo, A.P.; Leopoldo, A.S.; da Silva, D.C.; do Nascimento, A.F.; de Campos, D.H.; Luvizotto, R.A.; de Deus, A.F.; Freire, P.P.; Medeiros, A.; Okoshi, K.; et al. Long-term obesity promotes alterations in diastolic function induced by reduction of phospholamban phosphorylation at serine-16 without affecting calcium handling. J. Appl. Physiol. 2014, 117, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Lugenbiel, P.; Wenz, F.; Govorov, K.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Atrial fibrillation complicated by heart failure induces distinct remodeling of calcium cycling proteins. PLoS ONE 2015, 10, e0116395. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Heijman, J.; Wang, Q.; Chiang, D.Y.; Li, N.; Karck, M.; Wehrens, X.H.T.; Nattel, S.; Dobrev, D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014, 129, 145–156. [Google Scholar] [CrossRef]
- Greiser, M.; Neuberger, H.R.; Harks, E.; El-Armouche, A.; Boknik, P.; de Haan, S.; Verheyen, F.; Verheule, S.; Schmitz, W.; Ravens, U.; et al. Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J. Mol. Cell. Cardiol. 2009, 46, 385–394. [Google Scholar] [CrossRef]
- Fernandez-Tenorio, M.; Niggli, E. Stabilization of Ca(2+) signaling in cardiac muscle by stimulation of SERCA. J. Mol. Cell. Cardiol. 2018, 119, 87–95. [Google Scholar] [CrossRef]
- Tarifa, C.; Vallmitjana, A.; Jimenez-Sabado, V.; Marchena, M.; Llach, A.; Herraiz-Martinez, A.; Godoy-Marin, H.; Nolla-Colomer, C.; Ginel, A.; Vinolas, X.; et al. Spatial Distribution of Calcium Sparks Determines Their Ability to Induce Afterdepolarizations in Human Atrial Myocytes. JACC Basic Transl. Sci. 2023, 8, 1–15. [Google Scholar] [CrossRef]
- Dobrev, D.; Wehrens, X.H. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ. Res. 2014, 114, 1311–1319; discussion 1319. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Papa, A.; Katchman, A.N.; Zakharov, S.I.; Roybal, D.; Hennessey, J.A.; Kushner, J.; Yang, L.; Chen, B.X.; Kushnir, A.; et al. Mechanism of adrenergic Ca(V)1.2 stimulation revealed by proximity proteomics. Nature 2020, 577, 695–700. [Google Scholar] [CrossRef]
- Kaplan, A.D.; Boyman, L.; Ward, C.W.; Lederer, W.J.; Greiser, M. Ryanodine Receptor Stabilization Therapy Suppresses Ca2+-Based Arrhythmias in a Novel Model of Metabolic HFpEF. bioRxiv 2024. [Google Scholar] [CrossRef]
- Rudolph, A.; Stengel, A.; Suhs, M.; Schaper, S.; Wolk, E.; Rose, M.; Hofmann, T. Circulating Neuronatin Levels Are Positively Associated with BMI and Body Fat Mass but Not with Psychological Parameters. Nutrients 2023, 15, 3657. [Google Scholar] [CrossRef] [PubMed]
- Mzhavia, N.; Yu, S.; Ikeda, S.; Chu, T.T.; Goldberg, I.; Dansky, H.M. Neuronatin: A new inflammation gene expressed on the aortic endothelium of diabetic mice. Diabetes 2008, 57, 2774–2783. [Google Scholar] [CrossRef]
- Tsai, W.C.; Hung, T.C.; Kusayama, T.; Han, S.; Fishbein, M.C.; Chen, L.S.; Chen, P.S. Autonomic Modulation of Atrial Fibrillation. JACC Basic Transl. Sci. 2023, 8, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cortes, C.; Velasco-Saavedra, M.A.; Fernandez-de Gortari, E.; Guerrero-Serna, G.; Aguayo-Ortiz, R.; Espinoza-Fonseca, L.M. A novel machine learning-based screening identifies statins as inhibitors of the calcium pump SERCA. J. Biol. Chem. 2023, 299, 104681. [Google Scholar] [CrossRef] [PubMed]
- Lomax, A.E.; Kondo, C.S.; Giles, W.R. Comparison of time- and voltage-dependent K+ currents in myocytes from left and right atria of adult mice. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1837–H1848. [Google Scholar] [CrossRef]
- Kurata, H.T. Emerging complexities of lipid regulation of potassium channels. J. Gen. Physiol. 2016, 148, 201–205. [Google Scholar] [CrossRef]
- Takano, M.; Kuratomi, S. Regulation of cardiac inwardly rectifying potassium channels by membrane lipid metabolism. Prog. Biophys. Mol. Biol. 2003, 81, 67–79. [Google Scholar] [CrossRef]
- van der Cruijsen, E.A.; Nand, D.; Weingarth, M.; Prokofyev, A.; Hornig, S.; Cukkemane, A.A.; Bonvin, A.M.; Becker, S.; Hulse, R.E.; Perozo, E.; et al. Importance of lipid-pore loop interface for potassium channel structure and function. Proc. Natl. Acad. Sci. USA 2013, 110, 13008–13013. [Google Scholar] [CrossRef]
- Abriel, H. Roles and regulation of the cardiac sodium channel Na v 1.5: Recent insights from experimental studies. Cardiovasc. Res. 2007, 76, 381–389. [Google Scholar] [CrossRef]
- Dewal, R.S.; Greer-Short, A.; Lane, C.; Nirengi, S.; Manzano, P.A.; Hernandez-Saavedra, D.; Wright, K.R.; Nassal, D.; Baer, L.A.; Mohler, P.J.; et al. Phospho-ablation of cardiac sodium channel Na(v)1.5 mitigates susceptibility to atrial fibrillation and improves glucose homeostasis under conditions of diet-induced obesity. Int. J. Obes. 2021, 45, 795–807. [Google Scholar] [CrossRef]
- Pei, Z.; Xiao, Y.; Meng, J.; Hudmon, A.; Cummins, T.R. Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation. Nat. Commun. 2016, 7, 12035. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, M.; Traaseth, N.J.; Veglia, G. Activating and deactivating roles of lipid bilayers on the Ca(2+)-ATPase/phospholamban complex. Biochemistry 2011, 50, 10367–10374. [Google Scholar] [CrossRef]
- Li, J.; James, Z.M.; Dong, X.; Karim, C.B.; Thomas, D.D. Structural and functional dynamics of an integral membrane protein complex modulated by lipid headgroup charge. J. Mol. Biol. 2012, 418, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Griffin, C.; McKernan, K.; Eter, L.; Abrishami, S.; Singer, K. Female adipose tissue has improved adaptability and metabolic health compared to males in aged obesity. Aging 2020, 12, 1725–1746. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314. [Google Scholar] [CrossRef]
- Filgueiras-Rama, D.; Vasilijevic, J.; Jalife, J.; Noujaim, S.F.; Alfonso, J.M.; Nicolas-Avila, J.A.; Gutierrez, C.; Zamarreno, N.; Hidalgo, A.; Bernabe, A.; et al. Human influenza A virus causes myocardial and cardiac-specific conduction system infections associated with early inflammation and premature death. Cardiovasc. Res. 2021, 117, 876–889. [Google Scholar] [CrossRef]
- Bao, Y.; Willis, B.C.; Frasier, C.R.; Lopez-Santiago, L.F.; Lin, X.; Ramos-Mondragon, R.; Auerbach, D.S.; Chen, C.; Wang, Z.; Anumonwo, J.; et al. Scn2b Deletion in Mice Results in Ventricular and Atrial Arrhythmias. Circ. Arrhythm. Electrophysiol. 2016, 9, e003923. [Google Scholar] [CrossRef]
- Musa, H.; Marcou, C.A.; Herron, T.J.; Makara, M.A.; Tester, D.J.; O’Connell, R.P.; Rosinski, B.; Guerrero-Serna, G.; Milstein, M.L.; Monteiro da Rocha, A.; et al. Abnormal myocardial expression of SAP97 is associated with arrhythmogenic risk. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1357–H1370. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Balbuena, D.; Guerrero-Serna, G.; Valdivia, C.R.; Caballero, R.; Diez-Guerra, F.J.; Jimenez-Vazquez, E.N.; Ramirez, R.J.; Monteiro da Rocha, A.; Herron, T.J.; Campbell, K.F.; et al. Cardiac Kir2.1 and NaV1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 2018, 122, 1501–1516. [Google Scholar] [CrossRef]
- Cerrone, M.; Noujaim, S.F.; Tolkacheva, E.G.; Talkachou, A.; O’Connell, R.; Berenfeld, O.; Anumonwo, J.; Pandit, S.V.; Vikstrom, K.; Napolitano, C.; et al. Arrhythmogenic Mechanisms in a Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2007, 101, 1039–1048. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2011, 39, D52–D57. [Google Scholar] [CrossRef] [PubMed]
- Draghici, S.; Khatri, P.; Tarca, A.L.; Amin, K.; Done, A.; Voichita, C.; Georgescu, C.; Romero, R. A systems biology approach for pathway level analysis. Genome Res. 2007, 17, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2019, 36, 2628–2629. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regular Diet | High-FAT Diet | p-Value | |
|---|---|---|---|
| LV ejection fraction (%) | 44.3 ± 2.6 (N = 10) | 44.2 ± 2.3 (N = 10) | >0.9 |
| LV mass (mg) | 102.4 ± 4.5 (N = 10) | 109.2 ± 3.0 (N = 10) | 0.22 |
| LA volume index (mL/m2) | 4.0 ± 0.3 (N = 10) | 3.5 ± 0.3 (N = 10) | 0.25 |
| LA length (mm) | 1.9 ± 0.1 (N = 10) | 1.9 ± 0.1 (N = 10) | >0.9 |
| Systolic pressure (mmHg) | 122.2 ± 3.4 (N = 4) | 111.5 ± 4.8 (N = 5) | 0.13 |
| Diastolic pressure (mmHg) | 105.3 ± 1.5 (N = 4) | 104.2 ± 3.5 (N = 5) | 0.8 |
| Regular Diet | High-Fat Diet | p-Value | |
|---|---|---|---|
| P-wave duration (ms) | 8.9 ± 0.2 (N = 16) | 9.1 ± 0.1 (N = 18) | 0.4 |
| PR interval (ms) | 37 ± 0.7 (N = 16) | 38 ± 0.7 (N = 18) | 0.4 |
| QRS duration (ms) | 11 ± 0.17 (N = 16) | 11 ± 0.13 (N = 18) | 0.3 |
| RR interval (ms) | 142 ± 2.8 (N = 16) | 153 ± 2.4 (N = 18) | 0.004 |
| Regular Diet, Basal (N = 10) | High-Fat Diet, Basal (N = 9) | p-Value | Regular Diet, ISO (N = 10) | High-Fat Diet, ISO (N = 9) | p-Value | |
|---|---|---|---|---|---|---|
| P-wave duration (ms) | 8.5 ± 0.4 | 8.6 ± 0.4 | 0.8 | 8.1 ± 0.6 | 8.8 ± 0.3 | 0.3 |
| PR interval (ms) | 37 ± 1.7 | 35 ± 0.9 | 0.3 | 35 ± 1.8 | 30 ± 1.6 | 0.07 |
| QRS duration (ms) | 11 ± 0.4 | 10 ± 0.1 | 0.1 | 11 ± 0.5 | 10 ± 0.1 | 0.1 |
| RR interval (ms) | 124.5 ± 3.7 | 125.6 ± 3.1 | 0.8 | 102 ± 1.3 | 91 ± 1.4 | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponce-Balbuena, D.; Tyrrell, D.J.; Cruz-Cortés, C.; Guerrero-Serna, G.; Monteiro Da Rocha, A.; Herron, T.J.; Song, J.; Raza, D.S.; Anumonwo, J.; Goldstein, D.R.; et al. Paradoxical SERCA2a Dysregulation Contributes to Atrial Fibrillation in a Model of Diet-Induced Obesity. Int. J. Mol. Sci. 2025, 26, 5603. https://doi.org/10.3390/ijms26125603
Ponce-Balbuena D, Tyrrell DJ, Cruz-Cortés C, Guerrero-Serna G, Monteiro Da Rocha A, Herron TJ, Song J, Raza DS, Anumonwo J, Goldstein DR, et al. Paradoxical SERCA2a Dysregulation Contributes to Atrial Fibrillation in a Model of Diet-Induced Obesity. International Journal of Molecular Sciences. 2025; 26(12):5603. https://doi.org/10.3390/ijms26125603
Chicago/Turabian StylePonce-Balbuena, Daniela, Daniel J. Tyrrell, Carlos Cruz-Cortés, Guadalupe Guerrero-Serna, Andre Monteiro Da Rocha, Todd J. Herron, Jianrui Song, Danyal S. Raza, Justus Anumonwo, Daniel R. Goldstein, and et al. 2025. "Paradoxical SERCA2a Dysregulation Contributes to Atrial Fibrillation in a Model of Diet-Induced Obesity" International Journal of Molecular Sciences 26, no. 12: 5603. https://doi.org/10.3390/ijms26125603
APA StylePonce-Balbuena, D., Tyrrell, D. J., Cruz-Cortés, C., Guerrero-Serna, G., Monteiro Da Rocha, A., Herron, T. J., Song, J., Raza, D. S., Anumonwo, J., Goldstein, D. R., & Espinoza-Fonseca, L. M. (2025). Paradoxical SERCA2a Dysregulation Contributes to Atrial Fibrillation in a Model of Diet-Induced Obesity. International Journal of Molecular Sciences, 26(12), 5603. https://doi.org/10.3390/ijms26125603