
Sequence-Based Prediction for Protein Solvent Accessibility


Abstract
1. Introduction
2. Results and Discussion
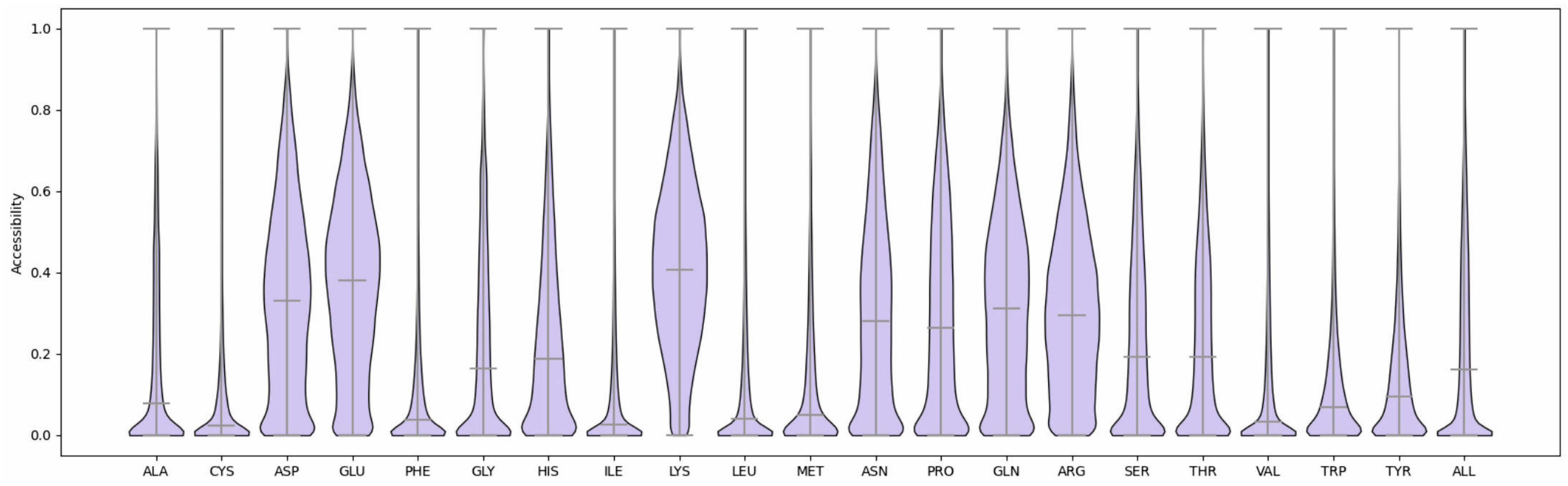
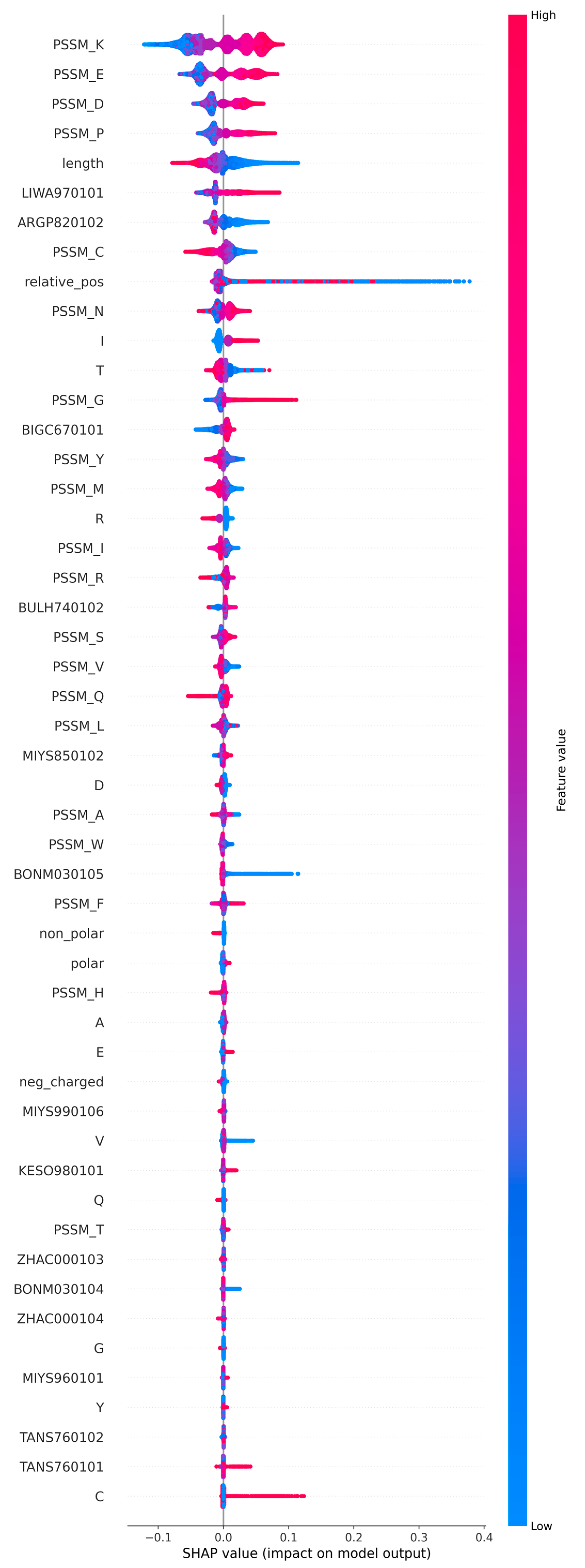
2.1. Feature Selection and Method Training
2.2. Comparison to Other Tools
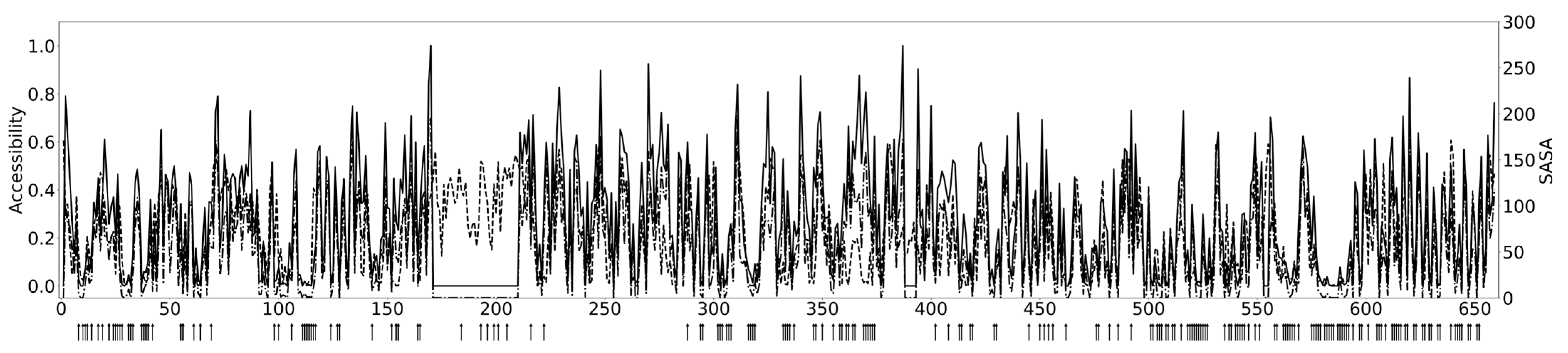
2.3. Example Case
2.4. SolAcc Server
3. Material and Methods
3.1. Data
3.2. Features
3.3. Machine Learning Algorithms
3.4. Feature Selection
3.5. Performance Assessment
4. Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Savojardo, C.; Manfredi, M.; Martelli, P.L.; Casadio, R. Solvent accessibility of residues undergoing pathogenic variations in humans: From protein structures to protein sequences. Front. Mol. Biosci. 2020, 7, 626363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, L.; Lü, Q. Protein solvent-accessibility prediction by a stacked deep bidirectional recurrent neural network. Biomolecules 2018, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Faraggi, E.; Zhang, T.; Yang, Y.; Kurgan, L.; Zhou, Y. SPINE X: Improving protein secondary structure prediction by multistep learning coupled with prediction of solvent accessible surface area and backbone torsion angles. J. Comput. Chem. 2012, 33, 259–267. [Google Scholar] [CrossRef]
- Manfredi, M.; Savojardo, C.; Martelli, P.L.; Casadio, R. DeepREx-WS: A web server for characterising protein-solvent interaction starting from sequence. Comput. Struct. Biotechnol. J. 2021, 19, 5791–5799. [Google Scholar] [CrossRef]
- Fan, X.Q.; Hu, J.; Jia, N.X.; Yu, D.J.; Zhang, G.J. Improved protein relative solvent accessibility prediction using deep multi-view feature learning framework. Anal. Biochem. 2021, 631, 114358. [Google Scholar] [CrossRef]
- Manfredi, M.; Savojardo, C.; Martelli, P.L.; Casadio, R. E-pRSA: Embeddings Improve the Prediction of Residue Relative Solvent Accessibility in Protein Sequence. J. Mol. Biol. 2024, 436, 168494. [Google Scholar] [CrossRef]
- Kaleel, M.; Torrisi, M.; Mooney, C.; Pollastri, G. PaleAle 5.0: Prediction of protein relative solvent accessibility by deep learning. Amino Acids 2019, 51, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, W.; Liu, S.; Xu, J. RaptorX-Property: A web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Yang, Y.; Zhou, Y. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci. Rep. 2015, 5, 11476. [Google Scholar] [CrossRef]
- Singh, J.; Paliwal, K.; Litfin, T.; Singh, J.; Zhou, Y. Reaching alignment-profile-based accuracy in predicting protein secondary and tertiary structural properties without alignment. Sci. Rep. 2022, 12, 7607. [Google Scholar] [CrossRef] [PubMed]
- Urban, G.; Magnan, C.N.; Baldi, P. SSpro/ACCpro 6: Almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, deep learning and structural similarity. Bioinformatics 2022, 38, 2064–2065. [Google Scholar] [CrossRef]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins 2019, 87, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Liu, D.; Huang, R.; Chen, Z.; Deng, L. PredRSA: A gradient boosted regression trees approach for predicting protein solvent accessibility. BMC Bioinform. 2016, 17 (Suppl. 1), 8. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L., Jr. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef]
- Høie, M.H.; Kiehl, E.N.; Petersen, B.; Nielsen, M.; Winther, O.; Nielsen, H.; Hallgren, J.; Marcatili, P. NetSurfP-3.0: Accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic Acids Res. 2022, 50, W510–W515. [Google Scholar] [CrossRef]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Singh, J.; Yang, Y.; Zhou, Y. Single-sequence-based prediction of protein secondary structures and solvent accessibility by deep whole-sequence learning. J. Comput. Chem. 2018, 39, 2210–2216. [Google Scholar] [CrossRef]
- Mitternacht, S. FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Res 2016, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- Hyvönen, M.; Saraste, M. Structure of the PH domain and Btk motif from Bruton’s tyrosine kinase: Molecular explanations for X-linked agammaglobulinaemia. EMBO J. 1997, 16, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Hansson, H.; Mattsson, P.T.; Allard, P.; Haapaniemi, P.; Vihinen, M.; Smith, C.I.; Hard, T. Solution structure of the SH3 domain from Bruton’s tyrosine kinase. Biochemistry 1998, 37, 2912–2924. [Google Scholar] [CrossRef]
- Huang, K.C.; Cheng, H.T.; Pai, M.T.; Tzeng, S.R.; Cheng, J.W. Solution structure and phosphopeptide binding of the SH2 domain from the human Bruton’s tyrosine kinase. J. Biomol. NMR 2006, 36, 73–78. [Google Scholar] [CrossRef]
- Bender, A.T.; Gardberg, A.; Pereira, A.; Johnson, T.; Wu, Y.; Grenningloh, R.; Head, J.; Morandi, F.; Haselmayer, P.; Liu-Bujalski, L. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Mol. Pharmacol. 2017, 91, 208–219. [Google Scholar] [CrossRef]
- Schaafsma, G.P.; Väliaho, J.; Wang, Q.; Berglöf, A.; Zain, R.; Smith, C.I.E.; Vihinen, M. BTKbase, Bruton tyrosine kinase variant database in X-linked agammaglobulinemia—Looking back and ahead. Hum. Mutat. 2023, 2023, 5797541. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Kawashima, S.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28, 374. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. LightGBM: A Highly Efficient Gradient Boosting Decision Tree; Neural Information Processing Systems: La Jolla, CA, USA, 2017. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Sequences | Residues | |
|---|---|---|
| Training set | 6000 | 1,402,211 |
| Blind test set | 500 | 118,180 |
| Validation set | 500 | 121,276 |
| Total | 7000 | 1,641,667 |
| RF | XGBoost | LGBM | MLP | ResNet | LSTM | |
|---|---|---|---|---|---|---|
| MAE | 0.123 | 0.126 | 0.120 | 0.120 | 0.128 | 0.109 |
| PCC | 0.679 | 0.651 | 0.690 | 0.696 | 0.674 | 0.729 |
| MSE | 0.027 | 0.029 | 0.026 | 0.026 | 0.028 | 0.023 |
| RMSE | 0.163 | 0.169 | 0.161 | 0.160 | 0.166 | 0.153 |
| MSLE | 0.016 | 0.017 | 0.015 | 0.015 | 0.017 | 0.013 |
| R2 | 0.457 | 0.421 | 0.475 | 0.482 | 0.439 | 0.525 |
| MAE | PCC | MSE | RMSE | MSLE | R2 | |
|---|---|---|---|---|---|---|
| 10 | 0.115 | 0.705 | 0.025 | 0.158 | 0.015 | 0.496 |
| 20 | 0.113 | 0.710 | 0.025 | 0.157 | 0.014 | 0.501 |
| 30 | 0.113 | 0.715 | 0.024 | 0.156 | 0.014 | 0.508 |
| 40 | 0.112 | 0.721 | 0.024 | 0.154 | 0.014 | 0.516 |
| 50 | 0.107 | 0.740 | 0.023 | 0.153 | 0.013 | 0.526 |
| 60 | 0.114 | 0.715 | 0.024 | 0.154 | 0.014 | 0.510 |
| 70 | 0.113 | 0.712 | 0.025 | 0.157 | 0.014 | 0.508 |
| 80 | 0.113 | 0.711 | 0.025 | 0.157 | 0.014 | 0.500 |
| 90 | 0.112 | 0.720 | 0.024 | 0.155 | 0.014 | 0.515 |
| 100 | 0.112 | 0.714 | 0.025 | 0.157 | 0.014 | 0.503 |
| 200 | 0.110 | 0.728 | 0.023 | 0.153 | 0.014 | 0.527 |
| 300 | 0.110 | 0.729 | 0.023 | 0.152 | 0.014 | 0.530 |
| 400 | 0.109 | 0.734 | 0.023 | 0.151 | 0.013 | 0.538 |
| 643 | 0.109 | 0.729 | 0.023 | 0.153 | 0.014 | 0.525 |
| MAE | PCC | MSE | RMSE | MSLE | R2 | |
|---|---|---|---|---|---|---|
| E-pRSA | 0.116 | 0.779 | 0.024 | 0.155 | 0.015 | 0.513 |
| SPIDER3 | 0.167 | 0.607 | 0.043 | 0.208 | 0.024 | 0.360 |
| NetSufP-3.0 | 0.127 | 0.763 | 0.031 | 0.177 | 0.017 | 0.538 |
| SolAcc | 0.104 | 0.750 | 0.022 | 0.148 | 0.013 | 0.555 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Chen, M.; Liu, C.; Vihinen, M. Sequence-Based Prediction for Protein Solvent Accessibility. Int. J. Mol. Sci. 2025, 26, 5604. https://doi.org/10.3390/ijms26125604
Yang Y, Chen M, Liu C, Vihinen M. Sequence-Based Prediction for Protein Solvent Accessibility. International Journal of Molecular Sciences. 2025; 26(12):5604. https://doi.org/10.3390/ijms26125604
Chicago/Turabian StyleYang, Yang, Mengqi Chen, Congrui Liu, and Mauno Vihinen. 2025. "Sequence-Based Prediction for Protein Solvent Accessibility" International Journal of Molecular Sciences 26, no. 12: 5604. https://doi.org/10.3390/ijms26125604
APA StyleYang, Y., Chen, M., Liu, C., & Vihinen, M. (2025). Sequence-Based Prediction for Protein Solvent Accessibility. International Journal of Molecular Sciences, 26(12), 5604. https://doi.org/10.3390/ijms26125604