
Peptidylarginine Deiminase 4 Deficiency Suppresses Neutrophil Extracellular Trap Formation and Ameliorates Elastase-Induced Emphysema in Mouse Lung

 and
and
Abstract
1. Introduction
2. Results
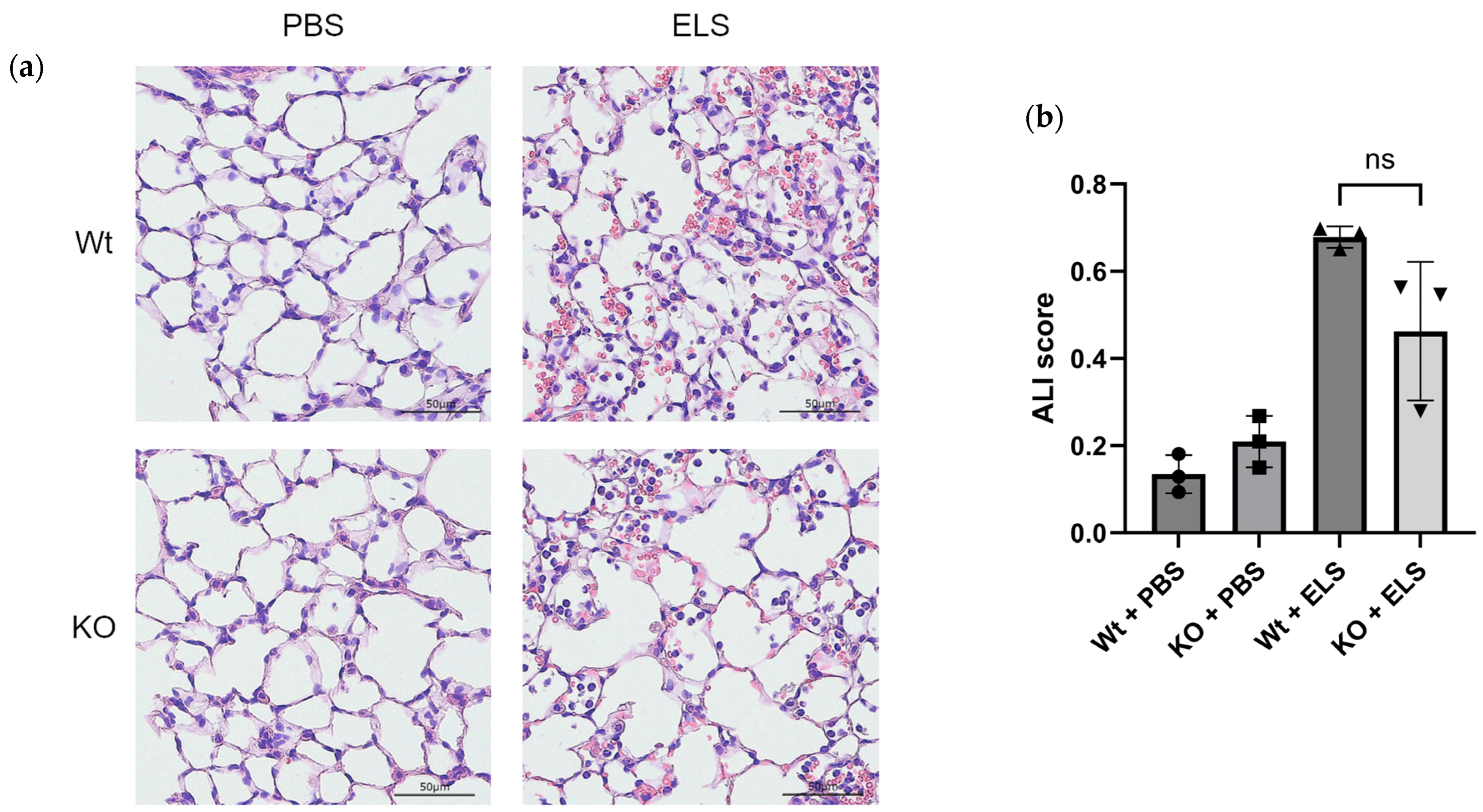
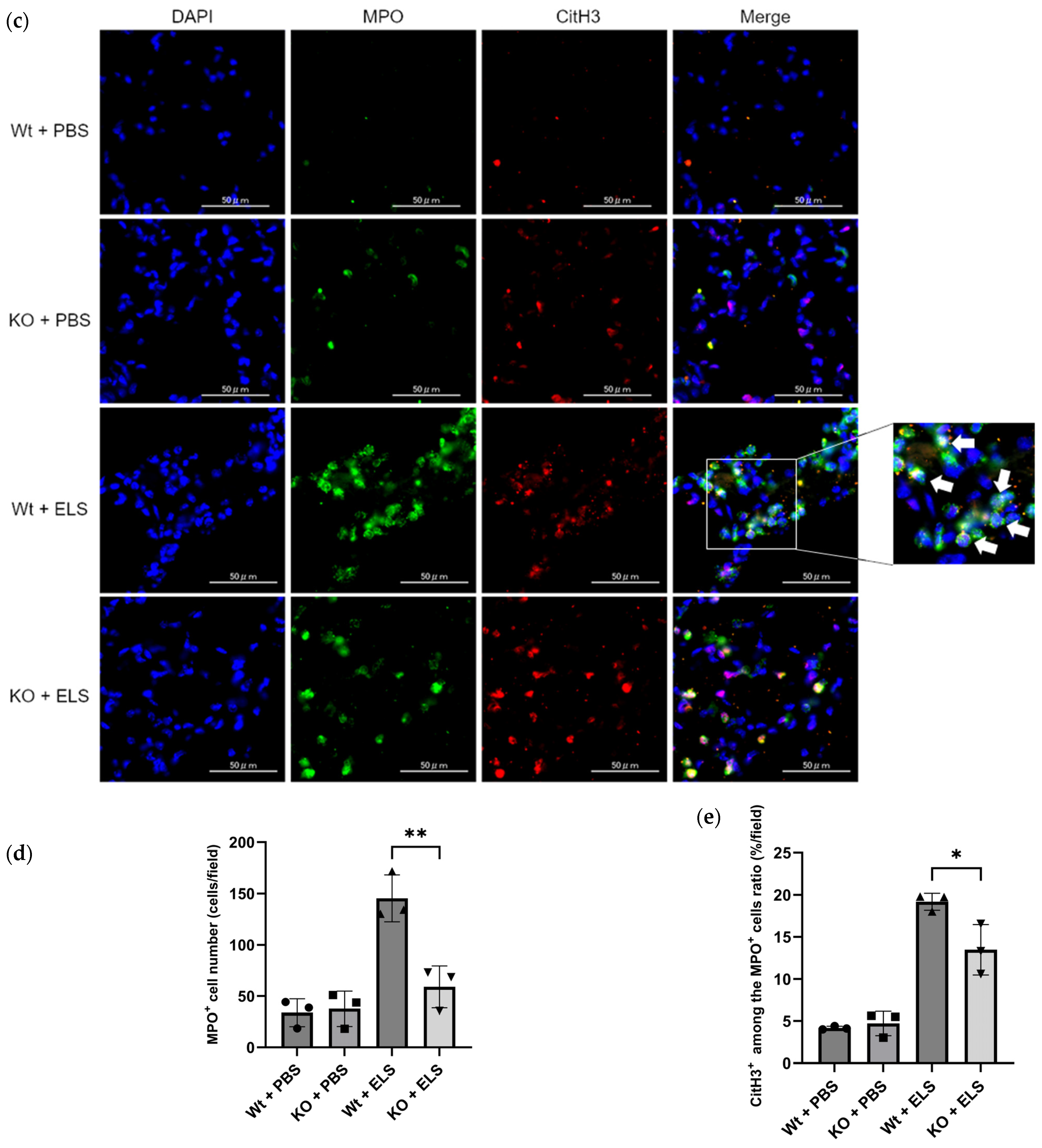
2.1. PAD4 Deficiency Suppresses NET Formation and Tends to Ameliorate ELS-Induced Injury in the Lung on Day 1
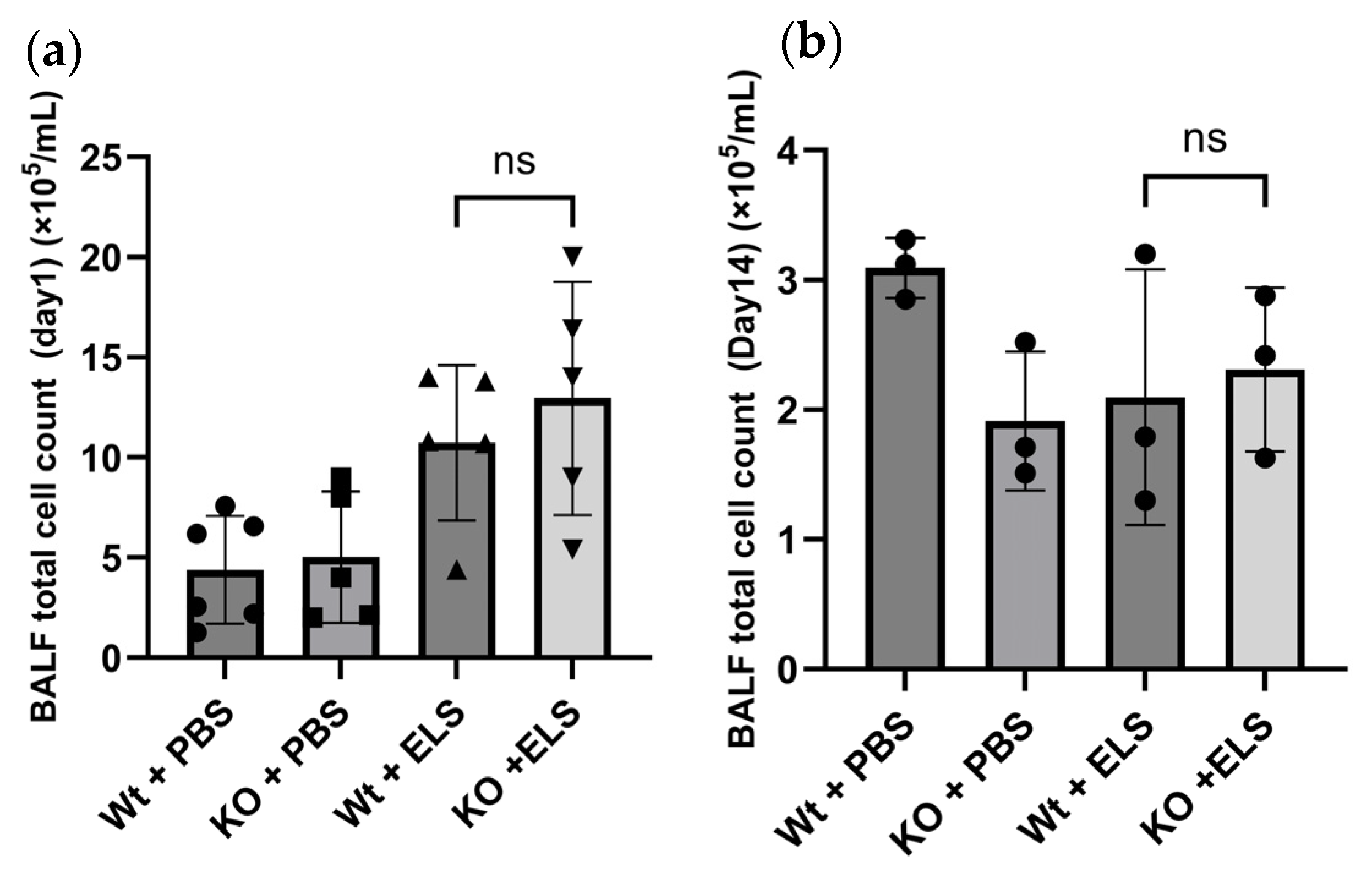
2.2. PAD4 Deficiency Did Not Affect the Cellular Profile of the Bronchoalveolar Lavage Fluid (BALF)
2.3. PAD4 Deficiency Suppresses the Extent of Emphysema
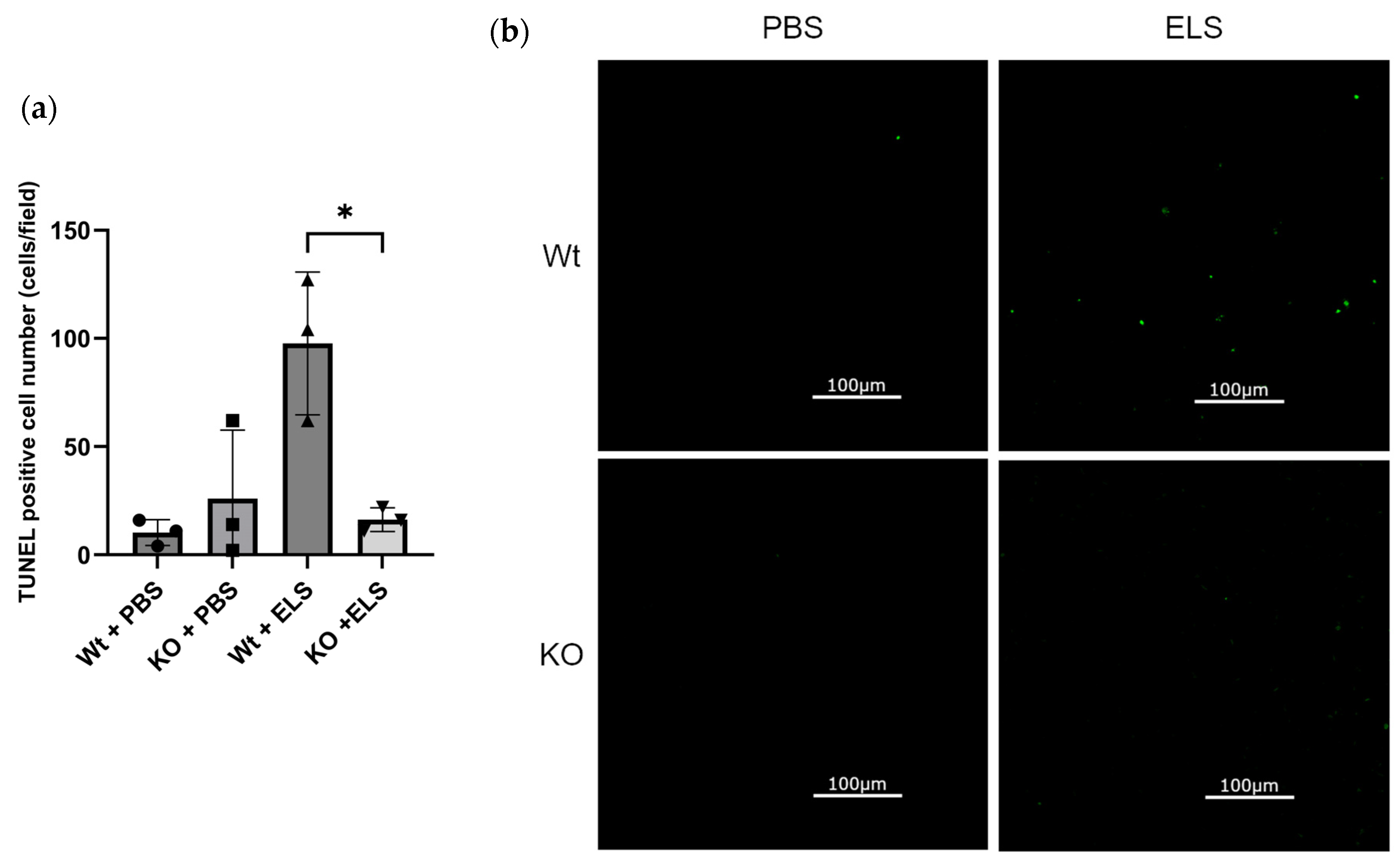
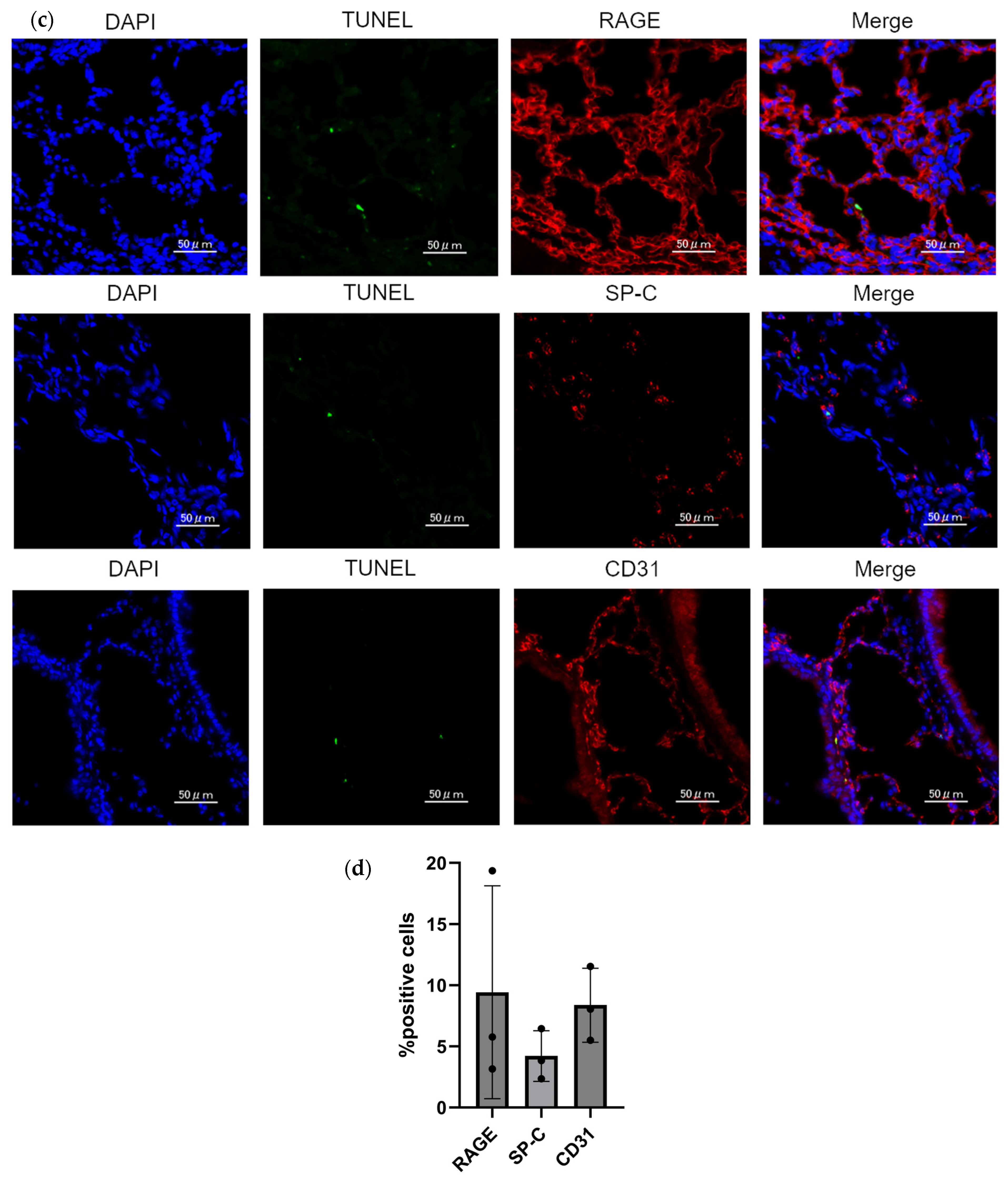
2.4. PAD4 Deficiency Suppresses the Extent of Apoptosis in the Lung
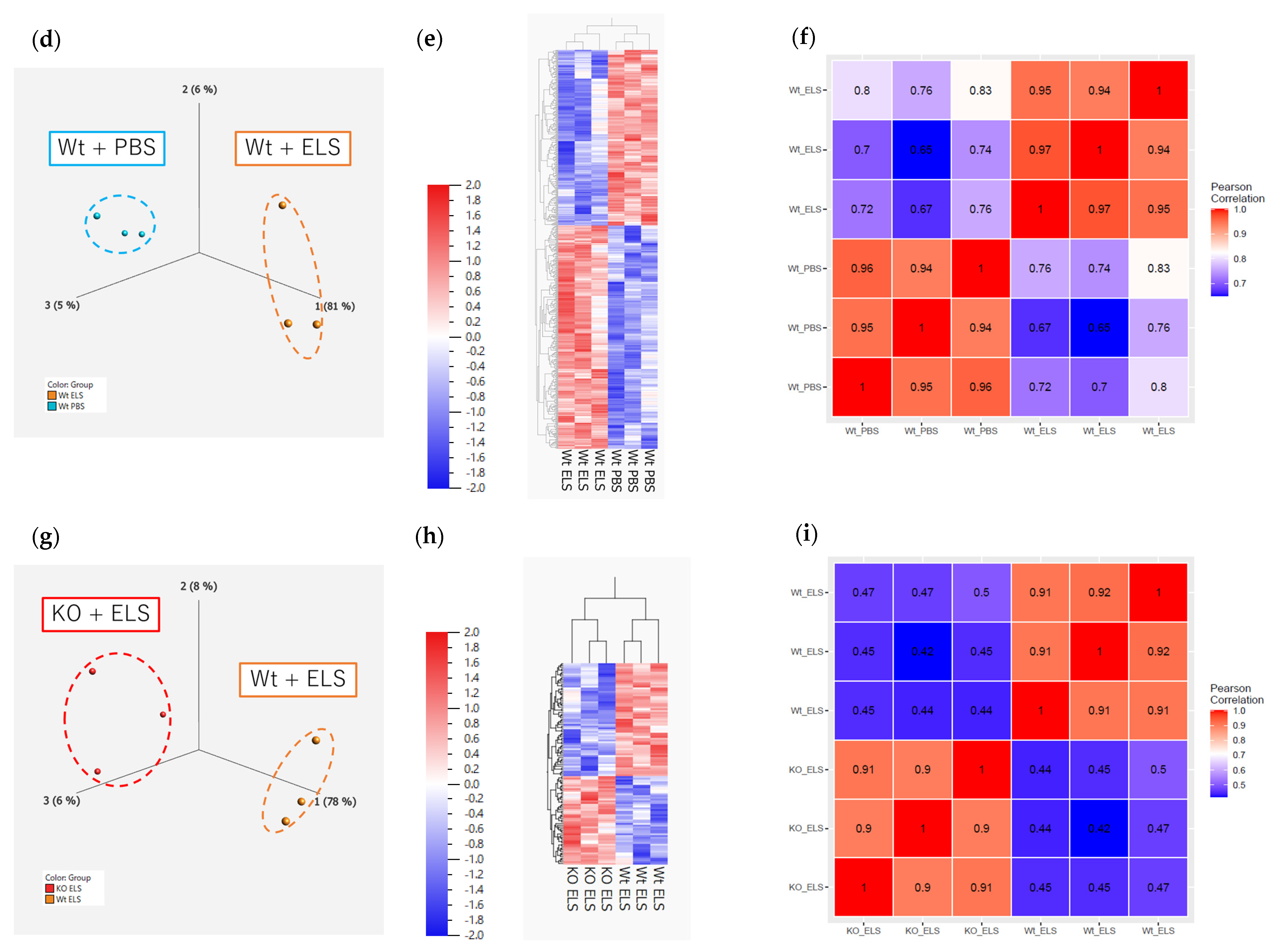
2.5. RNA Sequencing and Real-Time Quantitative PCR Analysis of the Whole Lung
2.5.1. RNA Sequencing on Day 1
2.5.2. RNA Sequencing on Day 14
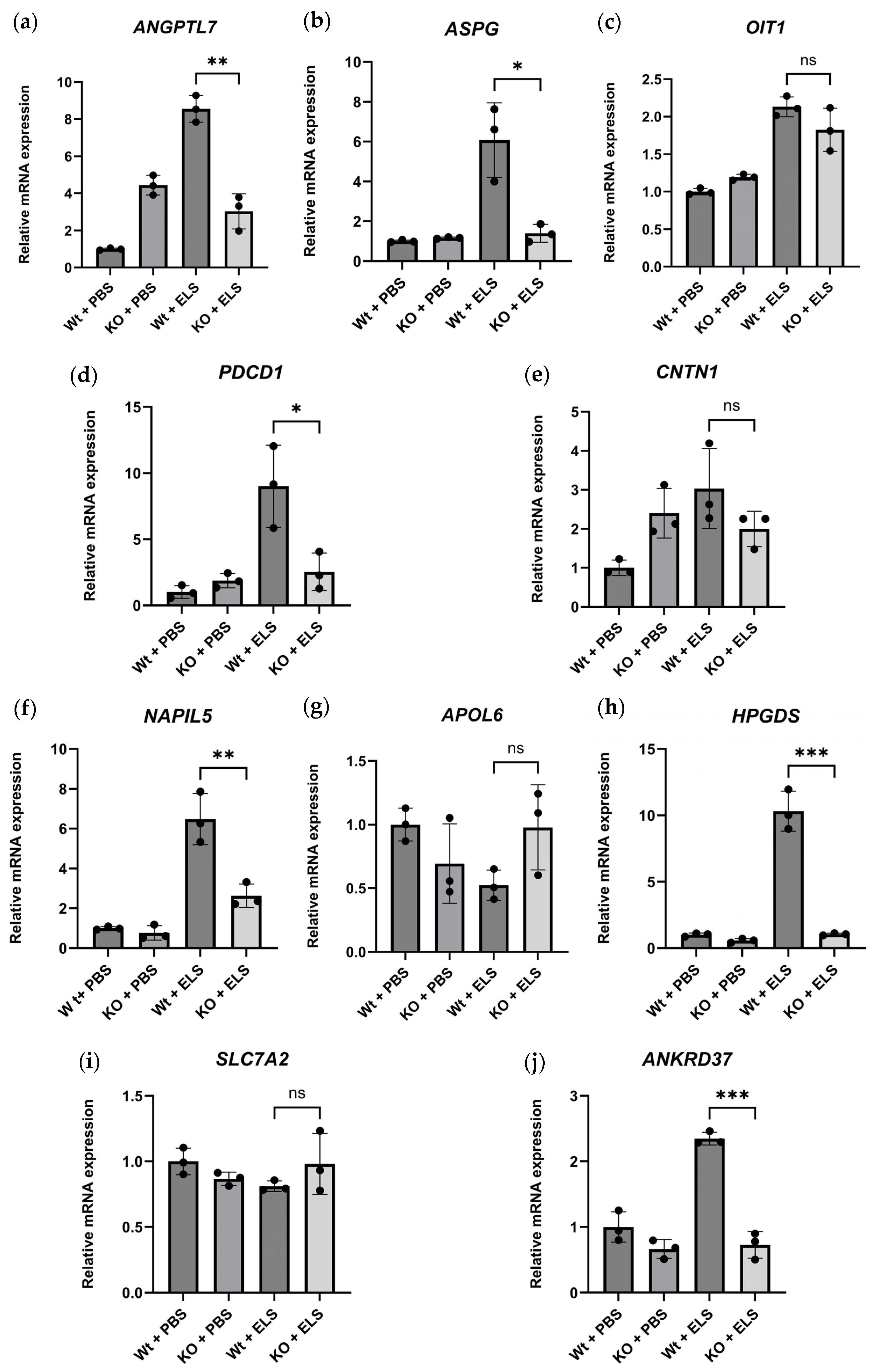
2.5.3. Real-Time Quantitative PCR of the DEGs
3. Discussion
4. Materials and Methods
4.1. Mice and ELS-Induced Emphysema Model
4.2. BALF Analysis
4.3. Evaluating Emphysema Formation
4.4. Detection of NETs in the Lung
4.5. SEM Analysis
4.6. TUNEL Assay
4.7. RNA Sequencing
4.8. Real-Time Quantitative PCR Analysis
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Annotation
References
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Repine, J.E.; Bast, A.; Lankhorst, I.; Oxidative Stress Study Group. Oxidative stress in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1997, 156 Pt 1, 341–357. [Google Scholar] [CrossRef]
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. 2000, 162 Pt 1, 1175–1177. [Google Scholar] [CrossRef]
- Churg, A.; Wright, J.L. Proteases and emphysema. Curr. Opin. Pulm. Med. 2005, 11, 153–159. [Google Scholar] [CrossRef]
- Karadag, F.; Karul, A.B.; Cildag, O.; Yilmaz, M.; Ozcan, H. Biomarkers of systemic inflammation in stable and exacerbation phases of COPD. Lung 2008, 186, 403–409. [Google Scholar] [CrossRef]
- Rutgers, S.R.; Timens, W.; Kaufmann, H.F.; van der Mark, T.W.; Koeter, G.H.; Postma, D.S. Comparison of induced sputum with bronchial wash, bronchoalveolar lavage and bronchial biopsies in COPD. Eur. Respir. J. 2000, 15, 109–115. [Google Scholar] [CrossRef]
- Stanescu, D.; Sanna, A.; Veriter, C.; Kostianev, S.; Calcagni, P.G.; Fabbri, L.M.; Maestrelli, P. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax 1996, 51, 267–271. [Google Scholar] [CrossRef]
- Donaldson, G.C.; Seemungal, T.A.; Patel, I.S.; Bhowmik, A.; Wilkinson, T.M.; Hurst, J.R.; Maccallum, P.K.; Wedzicha, J.A. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest 2005, 128, 1995–2004. [Google Scholar] [CrossRef]
- Amitani, R.; Wilson, R.; Rutman, A.; Read, R.; Ward, C.; Burnett, D.; Stockley, R.A.; Cole, P.J. Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 1991, 4, 26–32. [Google Scholar] [CrossRef]
- Senior, R.M.; Tegner, H.; Kuhn, C.; Ohlsson, K.; Starcher, B.C.; Pierce, J.A. The induction of pulmonary emphysema with human leukocyte elastase. Am. Rev. Respir. Dis. 1977, 116, 469–475. [Google Scholar] [CrossRef]
- Ballieux, B.E.; Hiemstra, P.S.; Klar-Mohamad, N.; Hagen, E.C.; van Es, L.A.; van der Woude, F.J.; Daha, M.R. Detachment and cytolysis of human endothelial cells by proteinase 3. Eur. J. Immunol. 1994, 24, 3211–3215. [Google Scholar] [CrossRef] [PubMed]
- Cheng, O.Z.; Palaniyar, N. NET balancing: A problem in inflammatory lung diseases. Front. Immunol. 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, O.E.; Borregaard, N. Neutrophil extracellular traps—The dark side of neutrophils. J. Clin. Investig. 2016, 126, 1612–1620. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Dwivedi, N.; Neeli, I.; Schall, N.; Wan, H.; Desiderio, D.M.; Csernok, E.; Thompson, P.R.; Dali, H.; Briand, J.P.; Muller, S.; et al. Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity. FASEB J. 2014, 28, 2840–2851. [Google Scholar] [CrossRef]
- Spengler, J.; Lugonja, B.; Ytterberg, A.J.; Zubarev, R.A.; Creese, A.J.; Pearson, M.J.; Grant, M.M.; Milward, M.; Lundberg, K.; Buckley, C.D.; et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol. 2015, 67, 3135–3145. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, A.S.; Martinod, K.; Seidman, M.A.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 2014, 12, 860–870. [Google Scholar] [CrossRef]
- Suzuki, M.; Ikari, J.; Anazawa, R.; Tanaka, N.; Katsumata, Y.; Shimada, A.; Suzuki, E.; Tatsumi, K. PAD4 Deficiency Improves Bleomycin-induced Neutrophil Extracellular Traps and Fibrosis in Mouse Lung. Am. J. Respir. Cell Mol. Biol. 2020, 63, 806–818. [Google Scholar] [CrossRef]
- Wright, T.K.; Gibson, P.G.; Simpson, J.L.; McDonald, V.M.; Wood, L.G.; Baines, K.J. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology 2016, 21, 467–475. [Google Scholar] [CrossRef]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Thompson, P.R. Protein Arginine Deiminases (PADs): Biochemistry and Chemical Biology of Protein Citrullination. Acc. Chem. Res. 2019, 52, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hunt, D.; Herron, M.; McDonnell, J.; Alshuhoumi, R.; McGarvey, L.P.; Fabre, A.; O’Brien, H.; McCarthy, C.; Martin, S.L.; et al. Neutrophil-Derived Peptidyl Arginine Deiminase Activity Contributes to Pulmonary Emphysema by Enhancing Elastin Degradation. J. Immunol. 2024, 213, 75–85. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M. An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef]
- Veras, F.P.; Gomes, G.F.; Silva, B.M.S.; Caetite, D.B.; Almeida, C.; Silva, C.M.S.; Schneider, A.H.; Corneo, E.S.; Bonilha, C.S.; Batah, S.S.; et al. Targeting neutrophils extracellular traps (NETs) reduces multiple organ injury in a COVID-19 mouse model. Respir. Res. 2023, 24, 66. [Google Scholar] [CrossRef] [PubMed]
- Dharwal, V.; Naura, A.S. PARP-1 inhibition ameliorates elastase induced lung inflammation and emphysema in mice. Biochem. Pharmacol. 2018, 150, 24–34. [Google Scholar] [CrossRef]
- Azzouz, D.; Khan, M.A.; Palaniyar, N. ROS induces NETosis by oxidizing DNA and initiating DNA repair. Cell Death Discov. 2021, 7, 113. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Padhy, D.S.; Palit, P.; Ikbal, A.M.A.; Das, N.; Roy, D.K.; Banerjee, S. Selective inhibition of peptidyl-arginine deiminase (PAD): Can it control multiple inflammatory disorders as a promising therapeutic strategy? Inflammopharmacology 2023, 31, 731–744. [Google Scholar] [CrossRef]
- Tian, Y.; Qu, S.; Alam, H.B.; Williams, A.M.; Wu, Z.; Deng, Q.; Pan, B.; Zhou, J.; Liu, B.; Duan, X.; et al. Peptidylarginine deiminase 2 has potential as both a biomarker and therapeutic target of sepsis. JCI Insight 2020, 5, e138873. [Google Scholar] [CrossRef]
- Wu, Z.; Deng, Q.; Pan, B.; Alam, H.B.; Tian, Y.; Bhatti, U.F.; Liu, B.; Mondal, S.; Thompson, P.R.; Li, Y. Inhibition of PAD2 Improves Survival in a Mouse Model of Lethal LPS-Induced Endotoxic Shock. Inflammation 2020, 43, 1436–1445. [Google Scholar] [CrossRef] [PubMed]
- Hemmers, S.; Teijaro, J.R.; Arandjelovic, S.; Mowen, K.A. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 2011, 6, e22043. [Google Scholar] [CrossRef]
- Surolia, R.; Li, F.J.; Wang, Z.; Kashyap, M.; Srivastava, R.K.; Traylor, A.M.; Singh, P.; Dsouza, K.G.; Kim, H.; Pittet, J.F.; et al. NETosis in the pathogenesis of acute lung injury following cutaneous chemical burns. JCI Insight 2021, 6, e147564. [Google Scholar] [CrossRef] [PubMed]
- Alflen, A.; Aranda Lopez, P.; Hartmann, A.K.; Maxeiner, J.; Bosmann, M.; Sharma, A.; Platten, J.; Ries, F.; Beckert, H.; Ruf, W.; et al. Neutrophil extracellular traps impair fungal clearance in a mouse model of invasive pulmonary aspergillosis. Immunobiology 2020, 225, 151867. [Google Scholar] [CrossRef] [PubMed]
- Biron, B.M.; Chung, C.S.; Chen, Y.; Wilson, Z.; Fallon, E.A.; Reichner, J.S.; Ayala, A. PAD4 Deficiency Leads to Decreased Organ Dysfunction and Improved Survival in a Dual Insult Model of Hemorrhagic Shock and Sepsis. J. Immunol. 2018, 200, 1817–1828. [Google Scholar] [CrossRef]
- Fischer, B.M.; Pavlisko, E.; Voynow, J.A. Pathogenic triad in COPD: Oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chronic Obstr. Pulm. Dis. 2011, 6, 413–421. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Wang, K.; Liao, Y.; Li, X.; Wang, R.; Zeng, Z.; Cheng, M.; Gao, L.; Xu, D.; Wen, F.; Wang, T.; et al. Inhibition of neutrophil elastase prevents cigarette smoke exposure-induced formation of neutrophil extracellular traps and improves lung function in a mouse model of chronic obstructive pulmonary disease. Int. Immunopharmacol. 2023, 114, 109537. [Google Scholar] [CrossRef]
- Ahmad, S.; Saleem, M.; Riaz, N.; Lee, Y.S.; Diri, R.; Noor, A.; Almasri, D.; Bagalagel, A.; Elsebai, M.F. The Natural Polypeptides as Significant Elastase Inhibitors. Front. Pharmacol. 2020, 11, 688. [Google Scholar] [CrossRef]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.C.; Parent, J.; Milot, J. Alveolar epithelial and endothelial cell apoptosis in emphysema: What we know and what we need to know. Int. J. Chronic Obstr. Pulm. Dis. 2009, 4, 19–31. [Google Scholar]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Investig. 2000, 106, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Kurian, N.; Cohen, T.S.; Oberg, L.; De Zan, E.; Skogberg, G.; Vollmer, S.; Baturcam, E.; Svanberg, P.; Bonn, B.; Smith, P.D.; et al. Dual Role for a MEK Inhibitor as a Modulator of Inflammation and Host Defense Mechanisms with Potential Therapeutic Application in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 2611–2624. [Google Scholar] [CrossRef]
- Mercer, B.A.; D’Armiento, J.M. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2006, 1, 137–150. [Google Scholar] [CrossRef]
- Qian, F.; Deng, J.; Gantner, B.N.; Flavell, R.A.; Dong, C.; Christman, J.W.; Ye, R.D. Map kinase phosphatase 5 protects against sepsis-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L866–L874. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef]
- Robison, P.; Sussan, T.E.; Chen, H.; Biswal, S.; Schneider, M.F.; Hernandez-Ochoa, E.O. Impaired calcium signaling in muscle fibers from intercostal and foot skeletal muscle in a cigarette smoke-induced mouse model of COPD. Muscle Nerve 2017, 56, 282–291. [Google Scholar] [CrossRef]
- Antunes, M.A.; Rocco, P.R. Elastase-induced pulmonary emphysema: Insights from experimental models. An. Acad. Bras. Cienc. 2011, 83, 1385–1396. [Google Scholar] [CrossRef]
- Sergio, L.P.S.; Lucinda, L.M.F.; Reboredo, M.M.; de Paoli, F.; Fonseca, L.M.C.; Pinheiro, B.V.; Mencalha, A.L.; Fonseca, A.S. Emphysema induced by elastase alters the mRNA relative levels from DNA repair genes in acute lung injury in response to sepsis induced by lipopolysaccharide administration in Wistar rats. Exp. Lung Res. 2018, 44, 79–88. [Google Scholar] [CrossRef]
- Baila, B.; Ohno, Y.; Nagamoto, H.; Kotosai, K.; Yabuuchi, Y.; Funaguchi, N.; Ito, F.; Endo, J.; Mori, H.; Takemura, G.; et al. Tetomilast attenuates elastase-induced pulmonary emphysema through inhibition of oxidative stress in rabbits. Biol. Pharm. Bull. 2012, 35, 494–502. [Google Scholar] [CrossRef]
- Ritzmann, F.; Borchardt, K.; Vella, G.; Chitirala, P.; Angenendt, A.; Herr, C.; Menger, M.D.; Hoth, M.; Lis, A.; Bohle, R.M.; et al. Blockade of PD-1 decreases neutrophilic inflammation and lung damage in experimental COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 320, L958–L968. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, R.; Strange, C. Use of multitarget tyrosine kinase inhibitors to attenuate platelet-derived growth factor signalling in lung disease. Eur. Respir. Rev. 2017, 26, 170061. [Google Scholar] [CrossRef]
- Bostrom, H.; Willetts, K.; Pekny, M.; Leveen, P.; Lindahl, P.; Hedstrand, H.; Pekna, M.; Hellstrom, M.; Gebre-Medhin, S.; Schalling, M.; et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 1996, 85, 863–873. [Google Scholar] [CrossRef]
- Jones, J.H.; Friedrich, E.; Hong, Z.; Minshall, R.D.; Malik, A.B. PV1 in Caveolae Controls Lung Endothelial Permeability. Am. J. Respir. Cell Mol. Biol. 2020, 63, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Q.; Liu, T.; Bai, T.; Zhang, M.; Hu, Y.; Li, J.; Chang, F. Role and mechanism of benzo[a]pyrene in the transformation of chronic obstructive pulmonary disease into lung adenocarcinoma. J. Cancer Res. Clin. Oncol. 2023, 149, 4741–4760. [Google Scholar] [CrossRef] [PubMed]
- Hardavella, G.; Tzortzaki, E.G.; Siozopoulou, V.; Galanis, P.; Vlachaki, E.; Avgousti, M.; Stefanou, D.; Siafakas, N.M. Lymphangiogenesis in COPD: Another link in the pathogenesis of the disease. Respir. Med. 2012, 106, 687–693. [Google Scholar] [CrossRef]
- Vardar-Yagli, N.; Saglam, M.; Savci, S.; Inal-Ince, D.; Calik-Kutukcu, E.; Arikan, H.; Coplu, L. Impact of sleep quality on functional capacity, peripheral muscle strength and quality of life in patients with chronic obstructive pulmonary disease. Expert Rev. Respir. Med. 2015, 9, 233–239. [Google Scholar] [CrossRef]
- Nunes, D.M.; Mota, R.M.; de Pontes Neto, O.L.; Pereira, E.D.; de Bruin, V.M.; de Bruin, P.F. Impaired sleep reduces quality of life in chronic obstructive pulmonary disease. Lung 2009, 187, 159–163. [Google Scholar] [CrossRef]
- Sundar, I.K.; Yao, H.; Sellix, M.T.; Rahman, I. Circadian molecular clock in lung pathophysiology. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1056–L1075. [Google Scholar] [CrossRef]
- Yao, H.; Sundar, I.K.; Huang, Y.; Gerloff, J.; Sellix, M.T.; Sime, P.J.; Rahman, I. Disruption of Sirtuin 1-Mediated Control of Circadian Molecular Clock and Inflammation in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2015, 53, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Markelic, I.; Hlapcic, I.; Ceri, A.; Radic Antolic, M.; Samarzija, M.; Popovic-Grle, S.; Vukic Dugac, A.; Rumora, L. Activation of NLRP3 inflammasome in stable chronic obstructive pulmonary disease. Sci. Rep. 2022, 12, 7544. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, N.; Singh, M.; Kaur, M.; Singh, G.; Narang, A.; Kanwal, A.; Sharma, K.; Singh, B.; Napoli, M.D.; et al. Neutrophil Extracellular Traps and NLRP3 Inflammasome: A Disturbing Duo in Atherosclerosis, Inflammation and Atherothrombosis. Vaccines 2023, 11, 261. [Google Scholar] [CrossRef] [PubMed]
- Munzer, P.; Negro, R.; Fukui, S.; di Meglio, L.; Aymonnier, K.; Chu, L.; Cherpokova, D.; Gutch, S.; Sorvillo, N.; Shi, L.; et al. NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front. Immunol. 2021, 12, 683803. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xu, S.L.; Ge, L.Y.; Zhu, J.; Zheng, T.; Zhu, Z.; Zhou, L. Sialic acid-binding immunoglobulin-like lectin 9 as a potential therapeutic target for chronic obstructive pulmonary disease. Chin. Med. J. 2021, 134, 757–764. [Google Scholar] [CrossRef]
- Zeng, Z.; Li, M.; Wang, M.; Wu, X.; Li, Q.; Ning, Q.; Zhao, J.; Xu, Y.; Xie, J. Increased expression of Siglec-9 in chronic obstructive pulmonary disease. Sci. Rep. 2017, 7, 10116. [Google Scholar] [CrossRef]
- Lee, K.Y.; Feng, P.H.; Ho, S.C.; Chuang, K.J.; Chen, T.T.; Su, C.L.; Liu, W.T.; Chuang, H.C. Inter-alpha-trypsin inhibitor heavy chain 4: A novel biomarker for environmental exposure to particulate air pollution in patients with chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 831–841. [Google Scholar]
- Migulina, N.; Tjin, G.; Faiz, A.; Borghuis, T.; Zhao, F.; Kaper, H.J.; Metzlar, M.; van Dijk, E.; Sharma, P.K.; Timens, W.; et al. Differential roles for lysyl oxidase (like), family members in chronic obstructive pulmonary disease; from gene and protein expression to function. FASEB J. 2022, 36, e22374. [Google Scholar] [CrossRef]
- Barnes, P.J.; Burney, P.G.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Prim. 2015, 1, 15076. [Google Scholar] [CrossRef]
- Yang, J.; Siqueira, M.F.; Behl, Y.; Alikhani, M.; Graves, D.T. The transcription factor ST18 regulates proapoptotic and proinflammatory gene expression in fibroblasts. FASEB J. 2008, 22, 3956–3967. [Google Scholar] [CrossRef]
- Matsuoka, T.; Hirata, M.; Tanaka, H.; Takahashi, Y.; Murata, T.; Kabashima, K.; Sugimoto, Y.; Kobayashi, T.; Ushikubi, F.; Aze, Y.; et al. Prostaglandin D2 as a mediator of allergic asthma. Science 2000, 287, 2013–2017. [Google Scholar] [CrossRef] [PubMed]
- Celejewska-Wojcik, N.; Kania, A.; Gorka, K.; Nastalek, P.; Wojcik, K.; Gielicz, A.; Mastalerz, L.; Sanak, M.; Sladek, K. Eicosanoids and Eosinophilic Inflammation of Airways in Stable COPD. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 1415–1424. [Google Scholar] [CrossRef]
- Stebbins, K.J.; Broadhead, A.R.; Baccei, C.S.; Scott, J.M.; Truong, Y.P.; Coate, H.; Stock, N.S.; Santini, A.M.; Fagan, P.; Prodanovich, P.; et al. Pharmacological blockade of the DP2 receptor inhibits cigarette smoke-induced inflammation, mucus cell metaplasia, and epithelial hyperplasia in the mouse lung. J. Pharmacol. Exp. Ther. 2010, 332, 764–775. [Google Scholar] [CrossRef]
- Aiken, S.G.; Grimes, T.; Munro, S.; Zarganes-Tzitzikas, T.; La Thangue, N.B.; Brennan, P.E. A patent review of peptidylarginine deiminase 4 (PAD4) inhibitors (2014–present). Expert Opin. Ther. Pat. 2025, 35, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Arfman, T.; Wichapong, K.; Reutelingsperger, C.P.M.; Voorberg, J.; Nicolaes, G.A.F. PAD4 takes charge during neutrophil activation: Impact of PAD4 mediated NET formation on immune-mediated disease. J. Thromb. Haemost. 2021, 19, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.E.; Causey, C.P.; Knuckley, B.; Slack-Noyes, J.L.; Thompson, P.R. Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr. Opin. Drug Discov. Dev. 2009, 12, 616–627. [Google Scholar]
- Ghorani, V.; Boskabady, M.H.; Khazdair, M.R.; Kianmeher, M. Experimental animal models for COPD: A methodological review. Tob. Induc. Dis. 2017, 15, 25. [Google Scholar] [CrossRef]
- Ueno, M.; Maeno, T.; Nishimura, S.; Ogata, F.; Masubuchi, H.; Hara, K.; Yamaguchi, K.; Aoki, F.; Suga, T.; Nagai, R.; et al. Alendronate inhalation ameliorates elastase-induced pulmonary emphysema in mice by induction of apoptosis of alveolar macrophages. Nat. Commun. 2015, 6, 6332. [Google Scholar] [CrossRef]
- Shim, Y.M.; Paige, M.; Hanna, H.; Kim, S.H.; Burdick, M.D.; Strieter, R.M. Role of LTB(4) in the pathogenesis of elastase-induced murine pulmonary emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L749–L759. [Google Scholar] [CrossRef]
- Murphy, M.P.; Zieger, M.; Henry, M.; Meleady, P.; Mueller, C.; McElvaney, N.G.; Reeves, E.P. Citrullination, a novel posttranslational modification of elastin, is involved in COPD pathogenesis. Am. J. Physiol. Lung Cell Mol. Physiol. 2024, 327, L600–L606. [Google Scholar] [CrossRef]
- Munoz-Barrutia, A.; Ceresa, M.; Artaechevarria, X.; Montuenga, L.M.; Ortiz-de-Solorzano, C. Quantification of lung damage in an elastase-induced mouse model of emphysema. Int. J. Biomed. Imaging 2012, 2012, 734734. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO: Relevant Terms Were Excerpted | ||
|---|---|---|
| GO Terms | DEGs | p-Value |
| Negative Regulation Of ERK1 And ERK2 Cascade (GO:0070373) | DUSP10 RGS14 | 0.0067 |
| Negative Regulation Of MAP Kinase Activity (GO:0043407) | RGS14 | 0.0076 |
| Release Of Sequestered Calcium Ion Into Cytosol By Sarcoplasmic Reticulum (GO:0014808) | RYR1 | 0.015 |
| Base-Excision Repair, AP Site Formation (GO:0006285) | MUTYH | 0.028 |
| GO: Relevant Terms Were Excerpted | ||
|---|---|---|
| GO Terms | DEGs | p-Value |
| Positive Regulation Of Lymphocyte Apoptotic Process (GO:0070230) | PDCD1 | 0.019 |
| Proteolysis (GO:0006508) | DDI2 HTRA4 CNTN1 ADAMTS12 | 0.022 |
| Regulation Of Platelet-Derived Growth Factor Receptor Signalling Pathway (GO:0010640) | CNTN1 | 0.038 |
| DEGs | Difference (Wt + PBS vs. Wt + ELS) (Fold Change) | Difference (Wt + ELS vs. KO + ELS) (Fold Change) |
|---|---|---|
| 4.06 | −1.96 |
| 2.93 | −2.96 |
| 2.39 | −3.46 |
| 2.74 | −2.56 |
| 3.02 | −1.77 |
| GO: Relevant Terms Were Excerpted | ||
|---|---|---|
| GO Term | DEGs | p-Value |
| Regulation Of Cellular Extravasation (GO:0002691) | PLVAP LYVE1 | 0.0011 |
| Entrainment Of Circadian Clock By Photoperiod (GO:0043153) | USP2 CRY1 | 0.0070 |
| Negative Regulation Of Phosphatidylinositol 3-Kinase Signalling (GO:0014067) | NLRC3 | 0.048 |
| Wikipathways: Relevant Pathways Were Excerpted | ||
| WikiPathway | DEGs | p-value |
| Microglia Pathogen Phagocytosis Pathway (WP3626) | LAT SIGLECE | 0.021 |
| GO: Relevant Terms Were Excerpted | ||
|---|---|---|
| GO Term | DEGs | p-Value |
| Acute Inflammatory Response (GO:0002526) | ITIH4 VNN1 | 0.0043 |
| Extracellular Matrix Organization (GO:0030198) | LOX ELN ADAMTS6 COL19A1 | 0.0095 |
| Positive Regulation Of Cysteine-Type Endopeptidase Activity Involved In Apoptotic Signalling Pathway (GO:2001269) | ST18 | 0.022 |
| Wikipathways: Relevant Pathways Were Excerpted | ||
| WikiPathway | DEGs | p-value |
| Osteoblast Signalling (WP238) | SLC17A2 | 0.037 |
| DEGs | Difference (Wt + PBS vs. Wt + ELS) (Fold Change) | Difference (Wt + ELS vs. KO + ELS) (Fold Change) |
|---|---|---|
| 3.18 | −2.39 |
| 3.19 | −2.25 |
| 3.56 | −1.87 |
| 2.62 | −2.19 |
| 1.25 | −3.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsumata, M.; Ikari, J.; Urano, A.; Suzuki, E.; Kugou, K.; Hasegawa, Y.; Tatsumi, K.; Suzuki, T. Peptidylarginine Deiminase 4 Deficiency Suppresses Neutrophil Extracellular Trap Formation and Ameliorates Elastase-Induced Emphysema in Mouse Lung. Int. J. Mol. Sci. 2025, 26, 5573. https://doi.org/10.3390/ijms26125573
Katsumata M, Ikari J, Urano A, Suzuki E, Kugou K, Hasegawa Y, Tatsumi K, Suzuki T. Peptidylarginine Deiminase 4 Deficiency Suppresses Neutrophil Extracellular Trap Formation and Ameliorates Elastase-Induced Emphysema in Mouse Lung. International Journal of Molecular Sciences. 2025; 26(12):5573. https://doi.org/10.3390/ijms26125573
Chicago/Turabian StyleKatsumata, Megumi, Jun Ikari, Akira Urano, Eiko Suzuki, Kazuto Kugou, Yoshinori Hasegawa, Koichiro Tatsumi, and Takuji Suzuki. 2025. "Peptidylarginine Deiminase 4 Deficiency Suppresses Neutrophil Extracellular Trap Formation and Ameliorates Elastase-Induced Emphysema in Mouse Lung" International Journal of Molecular Sciences 26, no. 12: 5573. https://doi.org/10.3390/ijms26125573
APA StyleKatsumata, M., Ikari, J., Urano, A., Suzuki, E., Kugou, K., Hasegawa, Y., Tatsumi, K., & Suzuki, T. (2025). Peptidylarginine Deiminase 4 Deficiency Suppresses Neutrophil Extracellular Trap Formation and Ameliorates Elastase-Induced Emphysema in Mouse Lung. International Journal of Molecular Sciences, 26(12), 5573. https://doi.org/10.3390/ijms26125573