
SARS-Cov-2 Replication in a Blood–Brain Barrier Model Established with Human Brain Microvascular Endothelial Cells Induces Permeability and Disables ACE2-Dependent Regulation of Bradykinin B1 Receptor

 ,
,  , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
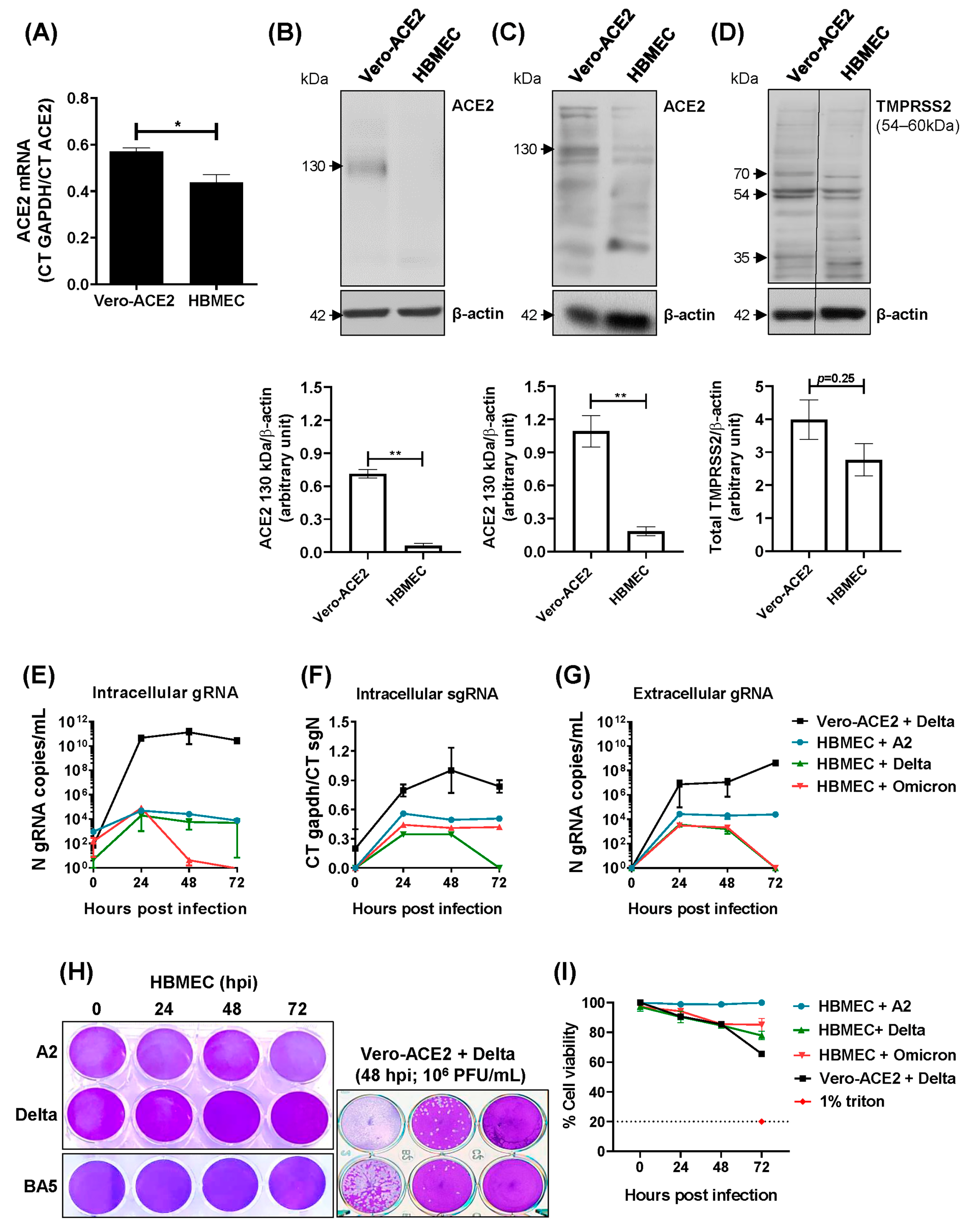
2.1. Transformed HBMECs Do Not Sustain a SARS-CoV-2 Productive Infection
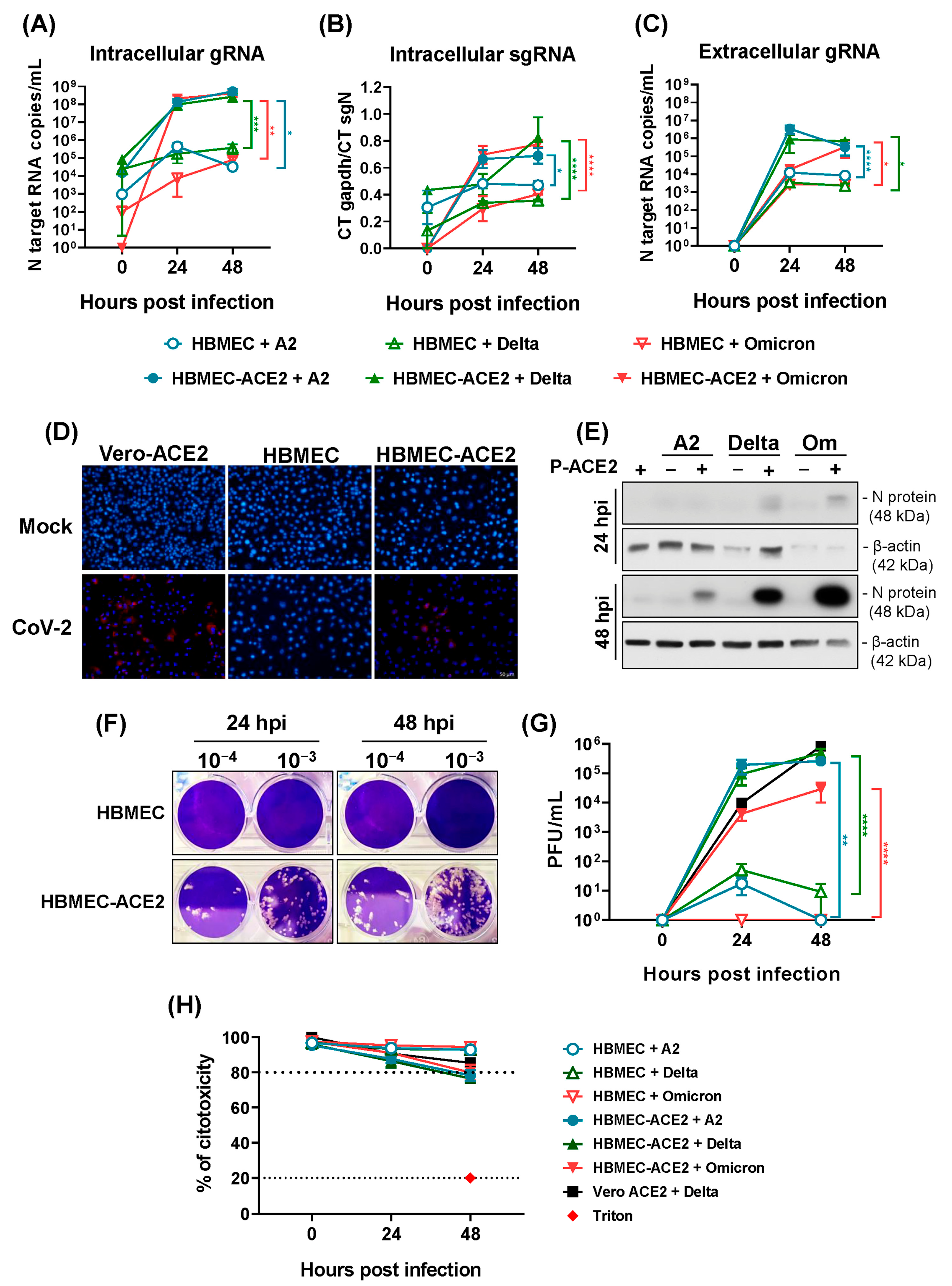
2.2. HBMECs Overexpressing ACE2 Are Permissive to SARS-CoV-2 Replication
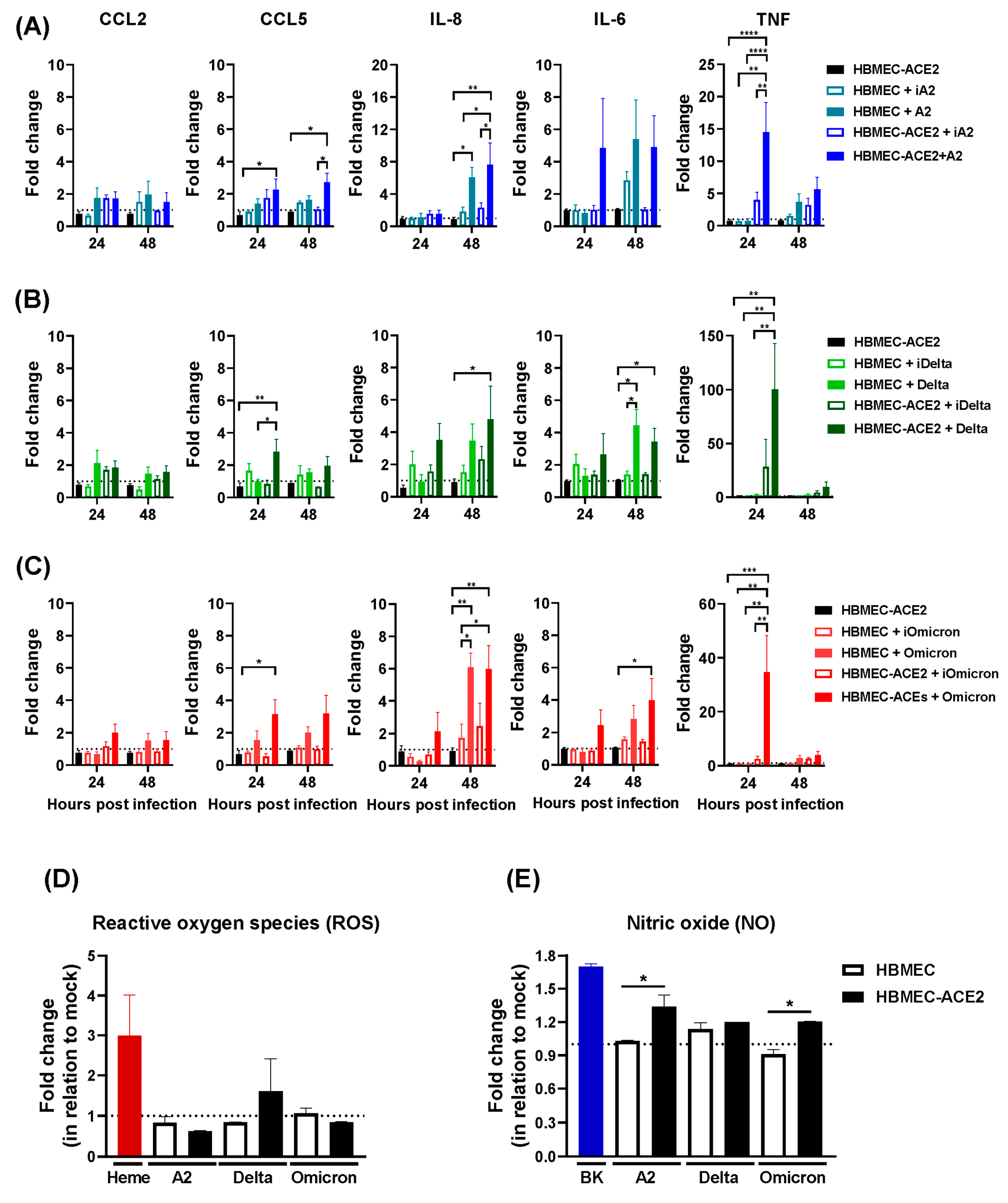
2.3. Production of Inflammatory Mediators by HBMEC-ACE2 Infected with SARS-CoV-2
2.4. Infection of HBMEC-ACE2 with SARS-CoV-2 Induces Endothelial Permeability, with Virus and Mononuclear Cells Crossing
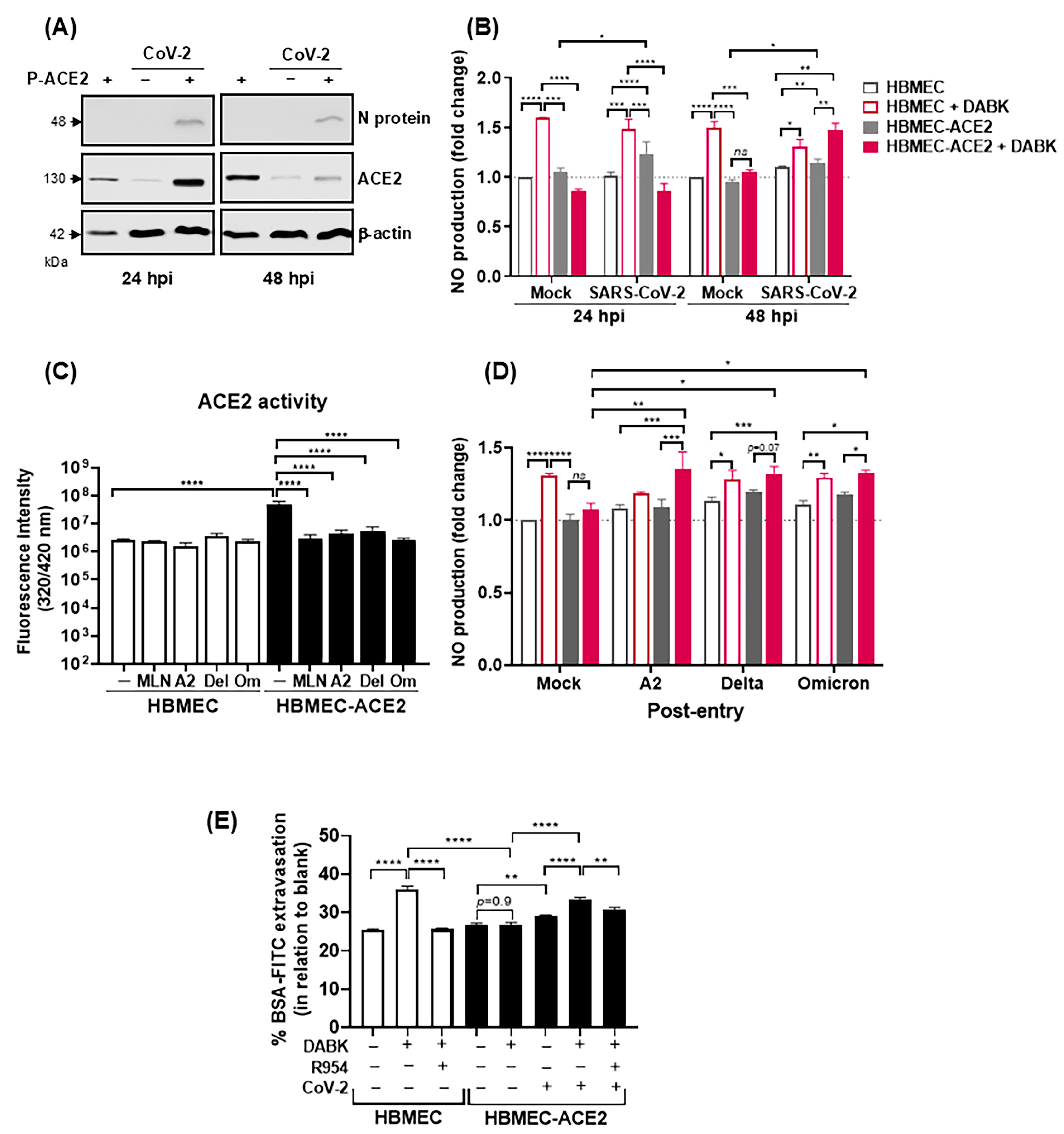
2.5. SARS-CoV-2 Infection Modulates ACE2-Dependent Regulation of DABK/B1R Pathway
3. Discussion
4. Materials and Methods
4.1. Cells and Viruses
4.2. Pseudovirus Construction and Transduction in HBMECs
4.3. HBMECs Infection with SARS-CoV-2
4.4. Analysis of Virus Replication, hACE2 and Cytokine Expression by RT-qPCR
4.5. Evaluation of Protein Expression by Western Blotting
4.6. Cell Viability Assay
4.7. Evaluation of dsRNA Staining by Immunofluorescence
4.8. Quantification of Nitric Oxide (NO) and Reactive Oxygen Species (ROS)
4.9. Assessment of ACE2 Enzymatic Activity
4.10. Ethical Statement and Isolation of PBMC and Human Primary Monocytes
4.11. Analysis of Monocyte Adhesion
4.12. Evaluation of Endothelial Permeability and of Virus and PBMC Extravasation
4.13. Analysis of Angiotensin II Degradation by ELISA
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 15, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 28, 1054–1062. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Perlman, S. COVID-19: Inflammatory Profile. Annu. Rev. Med. 2022, 27, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Mangalmurti, N.; Hunter, C.A. Cytokine Storms: Understanding COVID-19. Immunity 2020, 14, 19–25. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 10, 1330–1341. [Google Scholar] [CrossRef]
- Alfaro, E.; Díaz-García, E.; García-Tovar, S.; Zamarrón, E.; Mangas, A.; Galera, R.; Nanwani-Nanwani, K.; Pérez-de-Diego, R.; López-Collazo, E.; García-Río, F.; et al. Impaired Kallikrein-Kinin System in COVID-19 Patients’ Severity. Front. Immunol. 2022, 13, 909342. [Google Scholar] [CrossRef]
- Lipcsey, M.; Persson, B.; Eriksson, O.; Blom, A.M.; Fromell, K.; Hultström, M.; Huber-Lang, M.; Ekdahl, K.N.; Frithiof, R.; Nilsson, B. The Outcome of Critically Ill COVID-19 Patients Is Linked to Thromboinflammation Dominated by the Kallikrein/Kinin System. Front. Immunol. 2021, 12, 627579. [Google Scholar] [CrossRef]
- Englert, H.; Rangaswamy, C.; Deppermann, C.; Sperhake, J.P.; Krisp, C.; Schreier, D.; Gordon, E.; Konrath, S.; Haddad, M.; Pula, G.; et al. Defective NET clearance contributes to sustained FXII activation in COVID-19-associated pulmonary thrombo-inflammation. eBioMedicine 2021, 67, 103382. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Vicentino, A.R.R.; Fraga-Junior, V.D.S.; Palazzo, M.; Tasmo, N.R.A.; Rodrigues, D.A.S.; Barroso, S.P.C.; Ferreira, S.N.; Neves-Borges, A.C.; Allonso, D.; Fantappié, M.R.; et al. High mobility group box 1, ATP, lipid mediators, and tissue factor are elevated in COVID-19 patients: HMGB1 as a biomarker of worst prognosis. Clin. Transl. Sci. 2023, 16, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 4, 283. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Thorne, L.G.; Reuschl, A.K.; Zuliani-Alvarez, L.; Whelan, M.V.X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G.J. SARS-CoV-2 sensing by RIG-I and MDA5 links epithelial infection to macrophage inflammation. EMBO J. 2021, 2, e107826. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Edvinsson, Å.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef]
- Simoneau, C.R.; Ott, M. Modeling multi-organ infection by SARS-CoV-2 using stem cell technology. Cell Stem Cell 2020, 27, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Valyaeva, A.A.; Zharikova, A.A.; Sheval, E.V. SARS-CoV-2 cellular tropism and direct multiorgan failure in COVID-19 patients: Bioinformatic predictions, experimental observations, and open questions. Cell Biol. Int. 2023, 47, 308–326. [Google Scholar] [CrossRef]
- Greene, C.; Connolly, R.; Brennan, D.; Laffan, A.; O’Keeffe, E.; Zaporojan, L.; O’Callaghan, J.; Thomson, B.; Connolly, E.; Argue, R.; et al. Blood-brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nat. Neurosci. 2024, 27, 421–432. [Google Scholar] [CrossRef]
- Stein, S.R.; Ramelli, S.C.; Grazioli, A.; Chung, J.Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 2022, 612, 758–763. [Google Scholar] [CrossRef]
- Mustafá, Y.M.; Meuren, L.M.; Coelho, S.V.A.; De Arruda, L.B. Pathways exploited by flaviviruses to counteract the blood-brain barrier and invade the central nervous system. Front. Microbiol. 2019, 10, 525. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef] [PubMed]
- Nascimento Conde, J.; Schutt, W.R.; Gorbunova, E.E.; Mackow, E.R. Recombinant ACE2 Expression Is Required for SARS-CoV-2 To Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. mBio 2020, 11, e03185-20. [Google Scholar] [CrossRef]
- Yang, R.C.; Huang, K.; Zhang, H.P.; Li, L.; Zhang, Y.F.; Tan, C.; Chen, H.C.; Jin, M.L.; Wang, X.R. SARS-CoV-2 productively infects human brain microvascular endothelial cells. J. Neuroinflamm. 2022, 19, 149. [Google Scholar] [CrossRef]
- Motta, C.S.; Torices, S.; da Rosa, B.G.; Marcos, A.C.; Alvarez-Rosa, L.; Siqueira, M.; Moreno-Rodriguez, T.; Matos, A.D.R.; Caetano, B.C.; Martins, J.S.C.C.; et al. Human Brain Microvascular Endothelial Cells Exposure to SARS-CoV-2 Leads to Inflammatory Activation through NF-κB Non-Canonical Pathway and Mitochondrial Remodeling. Viruses 2023, 15, 745. [Google Scholar] [CrossRef]
- Kaneko, N.; Satta, S.; Komuro, Y.; Muthukrishnan, S.D.; Kakarla, V.; Guo, L.; An, J.; Elahi, F.; Kornblum, H.I.; Liebeskind, D.S.; et al. Flow-Mediated Susceptibility and Molecular Response of Cerebral Endothelia to SARS-CoV-2 Infection. Stroke 2021, 52, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Guang, C.; Phillips, R.D.; Jiang, B.; Milani, F. Three key proteases—Angiotensin-I-converting enzyme (ACE), ACE2 and renin—Within and beyond the renin-angiotensin system. Arch. Cardiovasc. Dis. 2012, 105, 373–385. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Rhaleb, N.E.; Dion, S.; Drapeau, G. New selective bradykinin receptor antagonists and bradykinin B2 receptor characterization. Trends Pharmacol. Sci. 1990, 11, 156–161. [Google Scholar] [CrossRef]
- Marceau, F.; Bachelard, H.; Bouthillier, J.; Fortin, J.P.; Morissette, G.; Bawolak, M.T.; Charest-Morin, X.; Gera, L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 2020, 82, 106305. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Calo, G.; Gobeil, F. Kallikrein–Kinin System. eLS 2001. [Google Scholar] [CrossRef]
- Moreau, M.E.; Bawolak, M.T.; Morissette, G.; Adam, A.; Marceau, F. Role of Nuclear Factor-κB and Protein Kinase C Signaling in the Expression of the Kinin B1 Receptor in Human Vascular Smooth Muscle Cells. Mol. Pharmacol. 2006, 71, 949–956. [Google Scholar] [CrossRef]
- Regoli, D.; Rizzi, A.; Calo, G.; Nsa Allogho, S.; Gobeil, F. B1 and B2 kinin receptors in various species. Immunopharmacology 1997, 36, 143–147. [Google Scholar] [CrossRef]
- Kuhr, F.; Lowry, J.; Zhang, Y.; Brovkovych, V.; Skidgel, R.A. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 2010, 44, 145–154. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef] [PubMed]
- Martens, C.P.; Van Mol, P.; Wauters, J.; Wauters, E.; Gangnus, T.; Noppen, B.; Callewaert, H.; Feyen, J.H.M.; Liesenborghs, L.; Heylen, E.; et al. Dysregulation of the kallikrein-kinin system in bronchoalveolar lavage fluid of patients with severe COVID-19. eBioMedicine 2022, 83, 104195. [Google Scholar] [CrossRef] [PubMed]
- Edinger, F.; Edinger, S.; Schmidt, G.; Koch, C.; Sander, M.; Schneck, E. The Role of the Kinin System and the Effect of Des-Arginine9-Bradykinin on Coagulation and Platelet Function in Critically Ill COVID-19 Patients: A Secondary Analysis of a Prospective Observational Study. Int. J. Mol. Sci. 2024, 25, 2342. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, Q.; Fox, D.M.; Gao, C.; Stanley, S.A.; Luo, K. SARS-CoV-2 down-regulates ACE2 through lysosomal degradation. Mol. Biol. Cell 2022, 33, ar147. [Google Scholar] [CrossRef]
- Rust, N.M.; Papa, M.P.; Scovino, A.M.; da Silva, M.M.; Calzavara-Silva, C.E.; Marques, E.T., Jr.; Peçanha, L.M.; Scharfstein, J.; Arruda, L.B. Bradykinin enhances Sindbis virus infection in human brain microvascular endothelial cells. Virology 2012, 422, 81–91. [Google Scholar] [CrossRef]
- Coelho, S.V.A.; Neris, R.L.S.; Papa, M.P.; Schnellrath, L.C.; Meuren, L.M.; Tschoeke, D.A.; Leomil, L.; Verçoza, B.R.F.; Miranda, M.; Thompson, F.L.; et al. Development of standard methods for Zika virus propagation, titration, and purification. J. Virol. Methods 2017, 246, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Papa, M.P.; Meuren, L.M.; Coelho, S.V.A.; Lucas, C.G.O.; Mustafá, Y.M.; Matassoli, L.F.; Silveira, P.P.; Frost, P.S.; Pezzuto, P.; Ribeiro, M.R.; et al. Zika Virus Infects, Activates, and Crosses Brain Microvascular Endothelial Cells, without Barrier Disruption. Front. Microbiol. 2017, 22, 2557. [Google Scholar] [CrossRef]
- Rowland, R.; Brandariz-Nuñez, A. Analysis of the Role of N-Linked Glycosylation in Cell Surface Expression, Function, and Binding Properties of SARS-CoV-2 Receptor ACE2. Microbiol. Spectr. 2021, 9, e0119921. [Google Scholar] [CrossRef]
- Stocker, N.; Radzikowska, U.; Wawrzyniak, P.; Tan, G.; Huang, M.; Ding, M.; Akdis, C.A.; Sokolowska, M. Regulation of angiotensin-converting enzyme 2 isoforms by type 2 inflammation and viral infection in human airway epithelium. Mucosal Immunol. 2023, 16, 5–16. [Google Scholar] [CrossRef]
- Coelho, S.V.A.; Rust, N.M.; Vellasco, L.; Papa, M.P.; Pereira, A.S.G.; Silva Palazzo, M.F.D.; Juliano, M.A.; Costa, S.M.; Alves, A.M.B.; Cordeiro, M.T.; et al. Contact System Activation in Plasma from Dengue Patients Might Harness Endothelial Virus Replication through the Signaling of Bradykinin Receptors. Pharmaceuticals 2021, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Torices, S.; Cabrera, R.; Stangis, M.; Naranjo, O.; Fattakhov, N.; Teglas, T.; Adesse, D.; Toborek, M. Expression of SARS-CoV-2-related receptors in cells of the neurovascular unit: Implications for HIV-1 infection. J. Neuroinflamm. 2021, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Haverty, R.; McCormack, J.; Evans, C.; Purves, K.; O’Reilly, S.; Gautier, V.; Rochfort, K.; Fabre, A.; Fletcher, N.F. SARS-CoV-2 infects neurons, astrocytes, choroid plexus epithelial cells and pericytes of the human central nervous system in vitro. J. Gen. Virol. 2024, 105, 002009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q.; et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct. Target. Ther. 2021, 6, 337. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Niethamer, T.K.; Stabler, C.T.; Leach, J.P.; Zepp, J.A.; Morley, M.P.; Babu, A.; Zhou, S.; Morrisey, E.E. Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. eLife 2020, 9, e53072. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H.; Schuster, R.; Zille, M.; Müller, K.; Krohn, M.; Körbelin, J.; Zhang, L.; Özorhan, Ü.; et al. The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.S.L.M.; Vendramini, P.H.; Valença, A.G.F.; Brandão-Teles, C.; Zuccoli, G.D.S.; et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2200960119. [Google Scholar] [CrossRef]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Neumann, B.; Schmidbauer, M.L.; Dimitriadis, K.; Otto, S.; Knier, B.; Niesen, W.D.; Hosp, J.A.; Günther, A.; Lindemann, S.; Nagy, G.; et al. Cerebrospinal fluid findings in COVID-19 patients with neurological symptoms. J. Neurol. Sci. 2020, 15, 117090. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Perl, D.P.; Steiner, J.; Pasternack, N.; Li, W.; Maric, D.; Safavi, F.; Horkayne-Szakaly, I.; Jones, R.; Stram, M.N.; et al. Neurovascular injury with complement activation and inflammation in COVID-19. Brain 2022, 145, 2555–2568. [Google Scholar] [CrossRef] [PubMed]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Savitt, A.G.; Manimala, S.; White, T.; Fandaros, M.; Yin, W.; Duan, H.; Xu, X.; Geisbrecht, B.V.; Rubenstein, D.A.; Kaplan, A.P.; et al. SARS-CoV-2 Exacerbates COVID-19 Pathology Through Activation of the Complement and Kinin Systems. Front. Immunol. 2021, 12, 767347. [Google Scholar] [CrossRef]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9, e59177. [Google Scholar] [CrossRef]
- Bocci, M.; Oudenaarden, C.; Sàenz-Sardà, X.; Simrén, J.; Edén, A.; Sjölund, J.; Möller, C.; Gisslén, M.; Zetterberg, H.; Englund, E.; et al. Infection of Brain Pericytes Underlying Neuropathology of COVID-19 Patients. Int. J. Mol. Sci. 2021, 22, 11622. [Google Scholar] [CrossRef]
- Meinhardt, J.; Streit, S.; Dittmayer, C.; Manitius, R.V.; Radbruch, H.; Heppner, F.L. The neurobiology of SARS-CoV-2 infection. Nat. Rev. Neurosci. 2024, 25, 30–42. [Google Scholar] [CrossRef]
- Motta Junior, J.D.S.; Miggiolaro, A.F.R.D.S.; Nagashima, S.; de Paula, C.B.V.; Baena, C.P.; Scharfstein, J.; de Noronha, L. Mast Cells in Alveolar Septa of COVID-19 Patients: A Pathogenic Pathway That May Link Interstitial Edema to Immunothrombosis. Front Immunol 2020, 11, 574862. [Google Scholar] [CrossRef]
- Moreno-Sanchez, D.; Hernandez-Ruiz, L.; Ruiz, F.A.; Docampo, R. Polyphosphate is a novel pro-inflammatory regulator of mast cells and is located in acidocalcisomes. J. Biol. Chem. 2012, 287, 28435–28444. [Google Scholar] [CrossRef]
- Nikolskaia, O.V.; de A Lima, A.P.; Kim, Y.V.; Lonsdale-Eccles, J.D.; Fukuma, T.; Scharfstein, J.; Grab, D.J. Blood-brain barrier traversal by African trypanosomes requires calcium signaling induced by parasite cysteine protease. J. Clin. Investig. 2006, 116, 2739–2747. [Google Scholar] [CrossRef]
- Voloch, C.M.; da Silva Francisco, R., Jr.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C., Jr.; et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95, e00119-21. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, B.B.; Silva, G.P.D.D.; Coelho, S.V.A.; Correa, I.A.; Souza, M.R.M.; Macedo, K.V.G.; Matos, B.M.; Tanuri, A.; Matassoli, F.L.; Costa, L.J.D.; et al. Hydroxypropyl-beta-cyclodextrin (HP-BCD) inhibits SARS-CoV-2 replication and virus-induced inflammatory cytokines. Antiviral Res. 2022, 205, 105373. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; Na, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.B.; Sriramula, S.; Chhabra, K.H.; Xia, H.; Lazartigues, E. Species-specific inhibitor sensitivity of angiotensin-converting enzyme 2 (ACE2) and its implication for ACE2 activity assays. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1293–R1299. [Google Scholar] [CrossRef]
- Gabra, B.H.; Sirois, P. Role of bradykinin B(1) receptors in diabetes-induced hyperalgesia in streptozotocin-treated mice. Eur. J. Pharmacol. 2002, 457, 115–124, Erratum in Eur. J. Pharmacol. 2003, 458, 329. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′–3′) | |
|---|---|---|
| N genomic | Forward | TTACAAACATTGGCCGCAAA |
| N genomic | Reverse | GCGCGACATTCCGAAGAA |
| N subgenomic | Forward | CGATCTCTTGTAGATCTGTTCTCTAAACGAACAAATTAAAT |
| N subgenomic | Reverse | TCTGGTTACTGCCAGTTGCCTCTG |
| Gapdh | Forward | GTGGACCTGACCTGCCGTCT |
| Gapdh | Reverse | GGAGGAGTGGGTGTCGCTGT |
| Ace2 | Forward | GGGATCAGAGATCGGAAGAAGAAA |
| Ace2 | Reverse | AGGAGGTCTGAACATCATCAGT |
| Ccl5 | Forward | CCAGCAGTCGTCTTTGTCAC |
| Ccl5 | Reverse | CTCTGGGTTGGCACACACTT |
| Ccl2 | Forward | CAGCCAGATGCAATCAATGCC |
| Ccl2 | Reverse | TGGAATCCTGAACCCACTTCT |
| Il8 | Forward | CAGCCAAAACTCCACAGTCA |
| Il8 | Reverse | TTGGAGAGCACATAAAAACATCT |
| Il6 | Forward | TGTGAAAGCAGCAAAGAGGCACTG |
| Il6 | Reverse | ACAGCTCTGGCTTGTTCCTCACTA |
| Tnf | Forward | CAGAGGGAAGAGTTCCCCAGGGACC |
| Tnf | Reverse | CCTTGGTCTGGTAGGAGACGGC |
| Gene | Probe (5′–3′) | |
| N | ACACTAGCCATCCTTACTGCGCTTCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, S.V.A.; Souza, G.L.e.; Bezerra, B.B.; Lima, L.R.; Correa, I.A.; de Almeida, D.V.; Silva-Aguiar, R.P.d.; Pinheiro, A.A.S.; Sirois, P.; Caruso-Neves, C.; et al. SARS-Cov-2 Replication in a Blood–Brain Barrier Model Established with Human Brain Microvascular Endothelial Cells Induces Permeability and Disables ACE2-Dependent Regulation of Bradykinin B1 Receptor. Int. J. Mol. Sci. 2025, 26, 5540. https://doi.org/10.3390/ijms26125540
Coelho SVA, Souza GLe, Bezerra BB, Lima LR, Correa IA, de Almeida DV, Silva-Aguiar RPd, Pinheiro AAS, Sirois P, Caruso-Neves C, et al. SARS-Cov-2 Replication in a Blood–Brain Barrier Model Established with Human Brain Microvascular Endothelial Cells Induces Permeability and Disables ACE2-Dependent Regulation of Bradykinin B1 Receptor. International Journal of Molecular Sciences. 2025; 26(12):5540. https://doi.org/10.3390/ijms26125540
Chicago/Turabian StyleCoelho, Sharton Vinicius Antunes, Gabriela Lisboa e Souza, Bruno Braz Bezerra, Luan Rocha Lima, Isadora Alonso Correa, Dalziza Victalina de Almeida, Rodrigo Pacheco da Silva-Aguiar, Ana Acácia S. Pinheiro, Pierre Sirois, Celso Caruso-Neves, and et al. 2025. "SARS-Cov-2 Replication in a Blood–Brain Barrier Model Established with Human Brain Microvascular Endothelial Cells Induces Permeability and Disables ACE2-Dependent Regulation of Bradykinin B1 Receptor" International Journal of Molecular Sciences 26, no. 12: 5540. https://doi.org/10.3390/ijms26125540
APA StyleCoelho, S. V. A., Souza, G. L. e., Bezerra, B. B., Lima, L. R., Correa, I. A., de Almeida, D. V., Silva-Aguiar, R. P. d., Pinheiro, A. A. S., Sirois, P., Caruso-Neves, C., Costa, L. J. d., Scharfstein, J., & Arruda, L. B. d. (2025). SARS-Cov-2 Replication in a Blood–Brain Barrier Model Established with Human Brain Microvascular Endothelial Cells Induces Permeability and Disables ACE2-Dependent Regulation of Bradykinin B1 Receptor. International Journal of Molecular Sciences, 26(12), 5540. https://doi.org/10.3390/ijms26125540