
Plasma Proteome Profiling Reveals Inflammation Markers and Tafamidis Effects in V30M Transthyretin Polyneuropathy

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
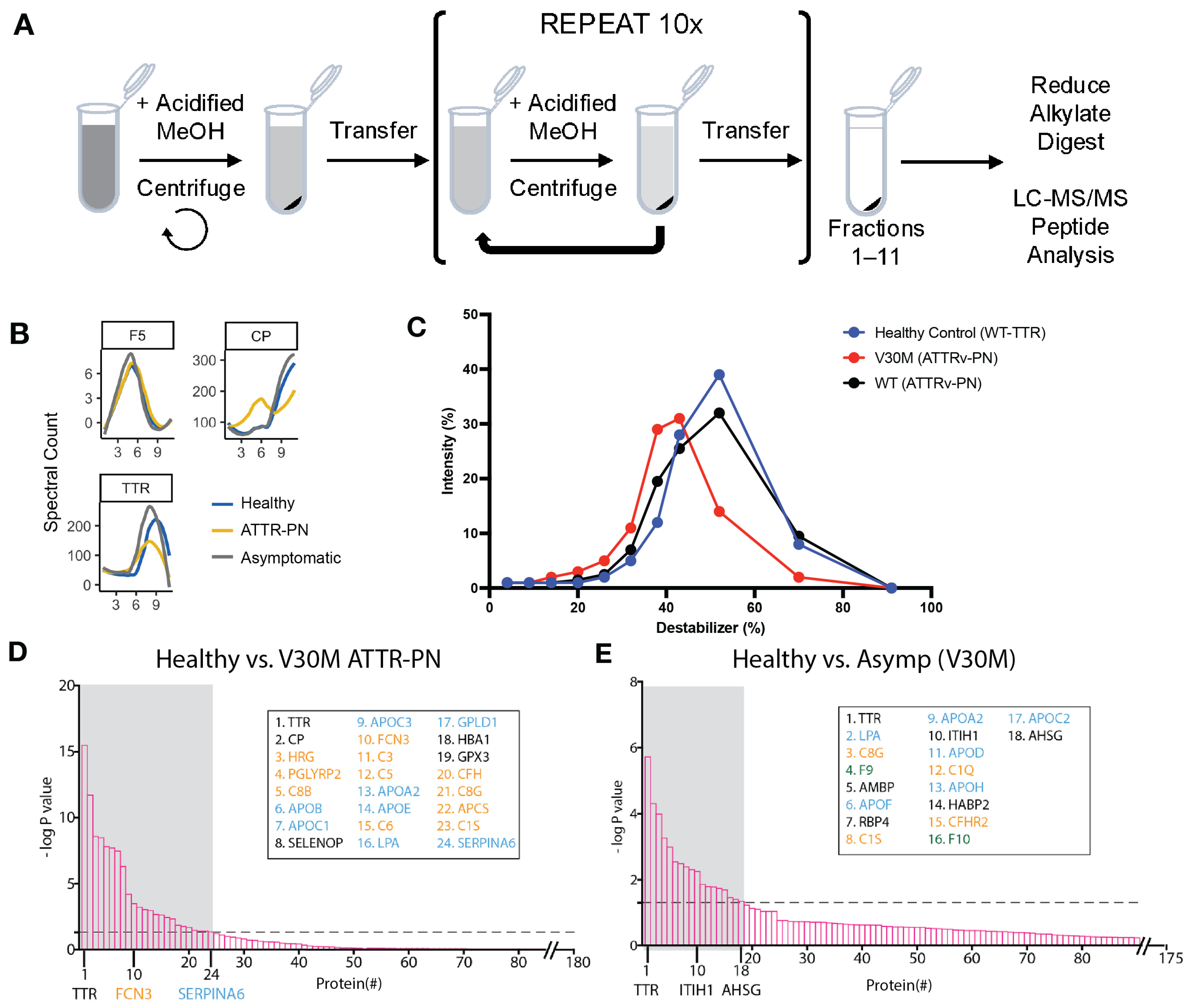
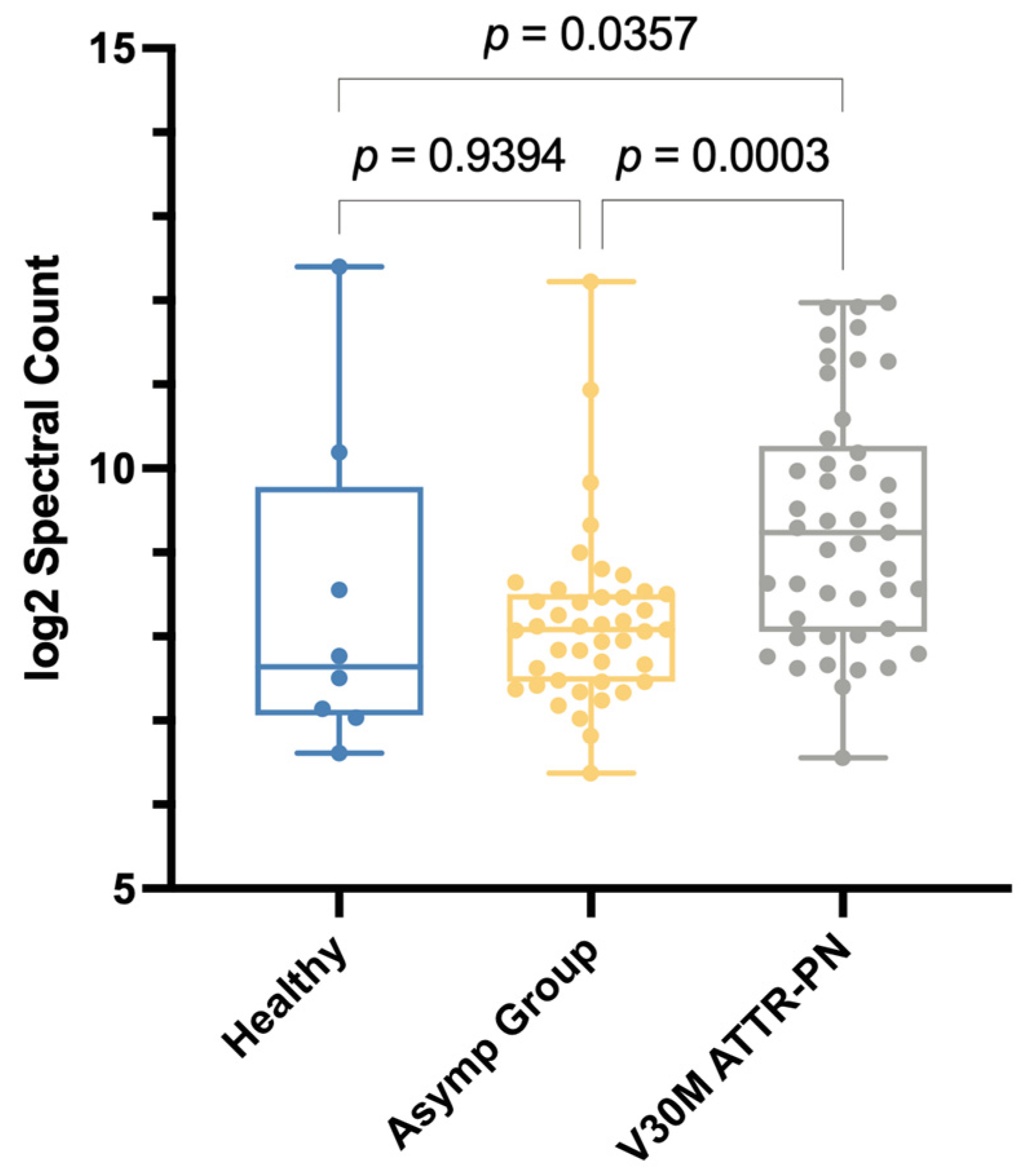
2.1. Plasma Proteome Profiling of Heterozygotic V30M ATTRv-PN Patients and Heterozygotic Asymptomatic V30M Carriers Versus Healthy WT TTR Controls Revealed Changes in Fractionation Profiles of TTR, Apolipoproteins, and Complement Proteins
2.2. V30M ATTRv-PN Disease-Specific Signature of Complement Pathway Activation and Apolipoprotein Alterations
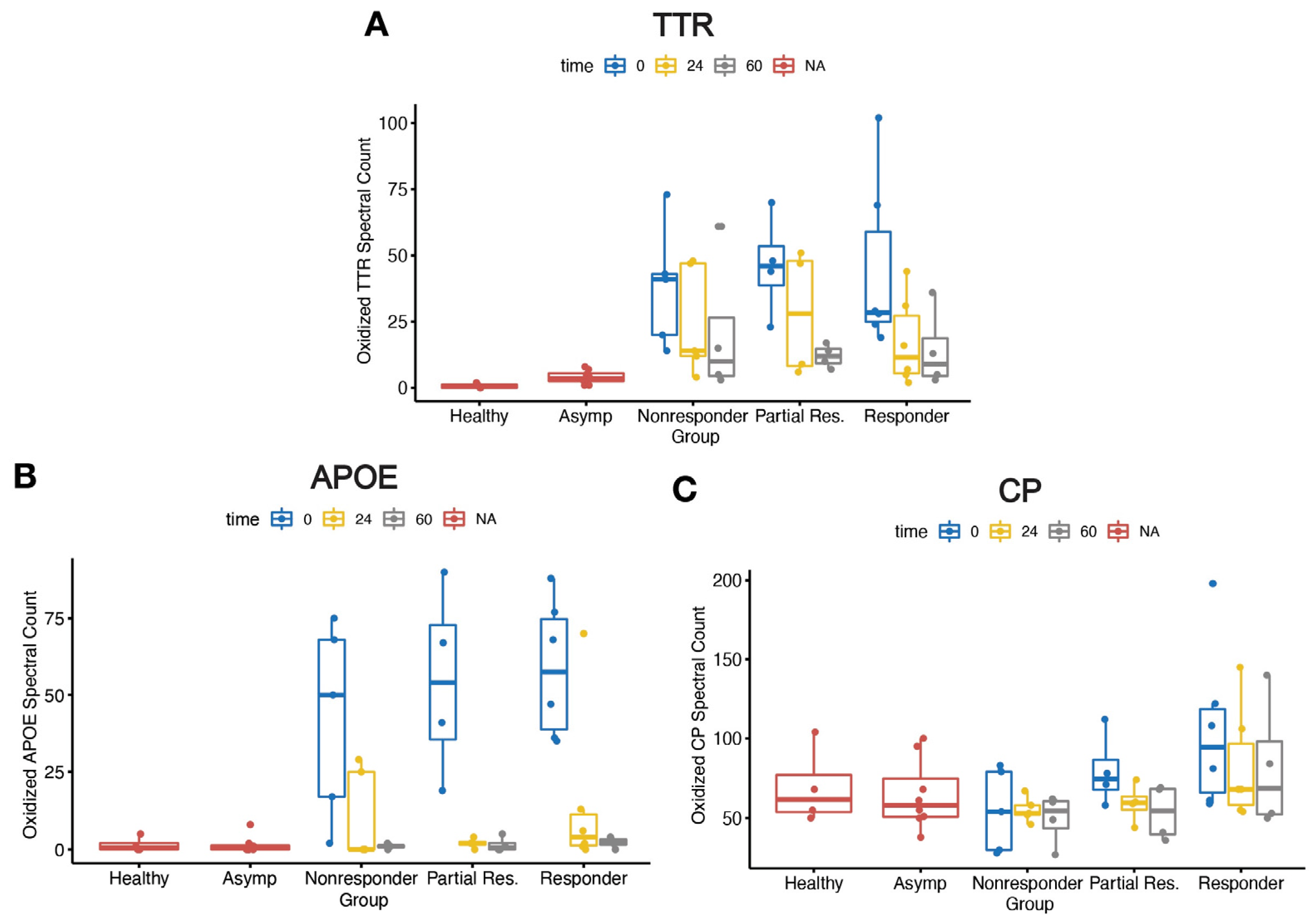
2.3. Elevated Oxidative Modifications
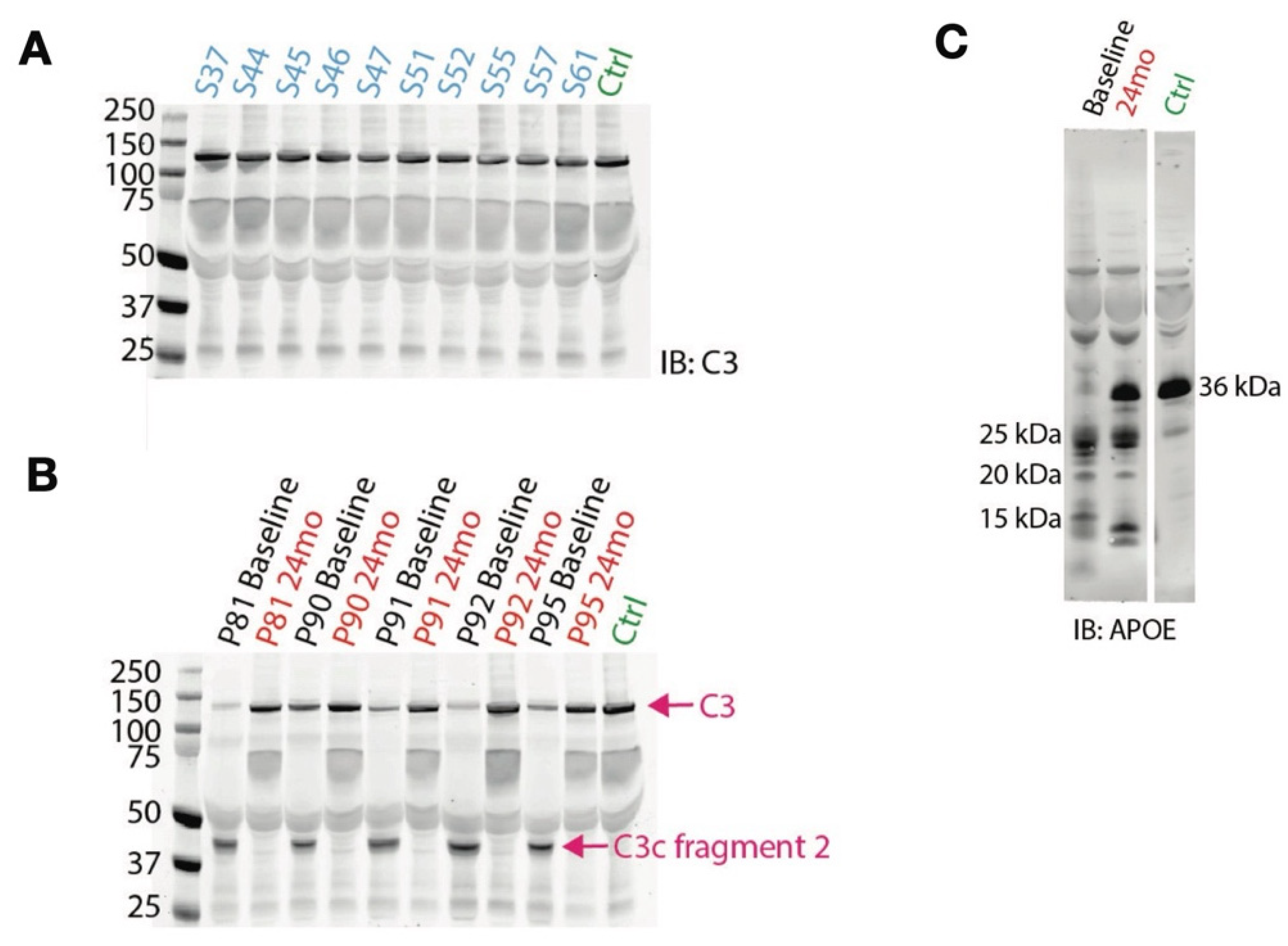
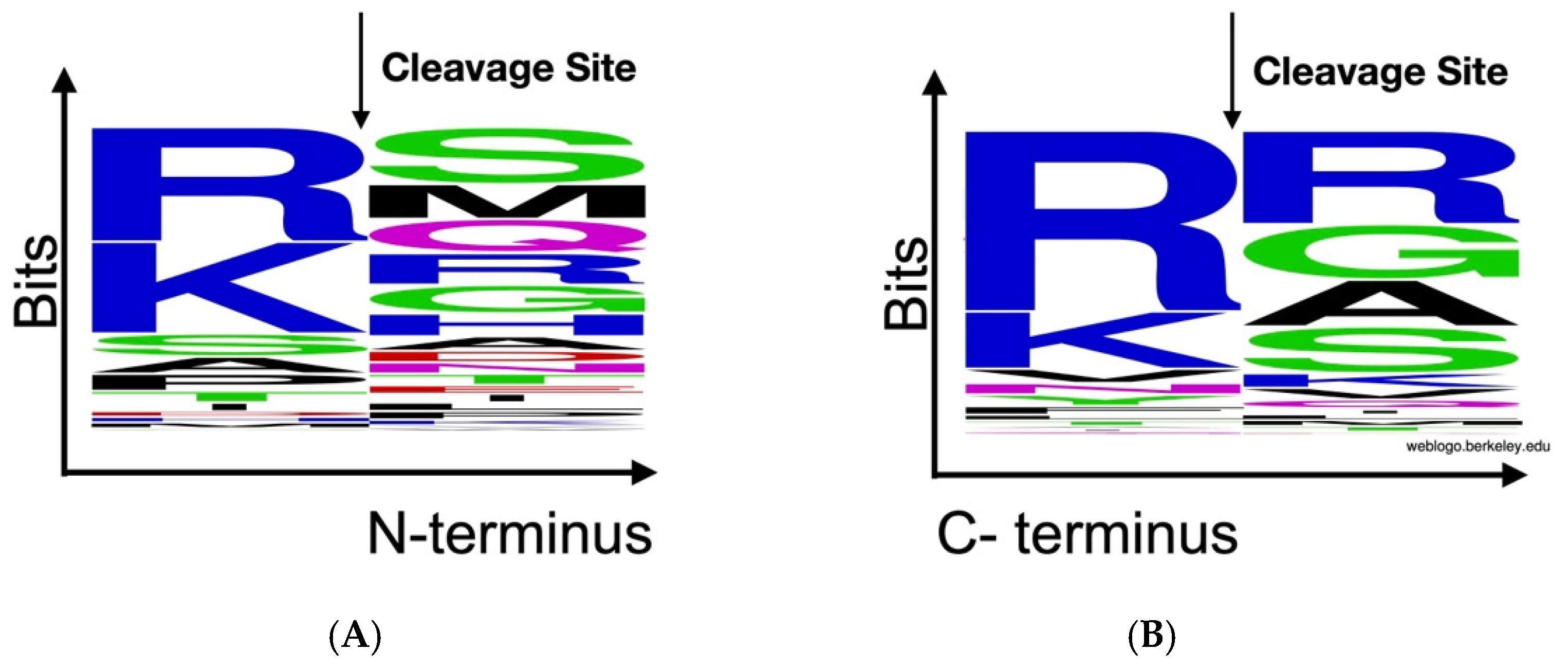
2.4. Peptidomics Analysis Reveals Aberrant Proteolytic Activity and a Signature of Inflammation
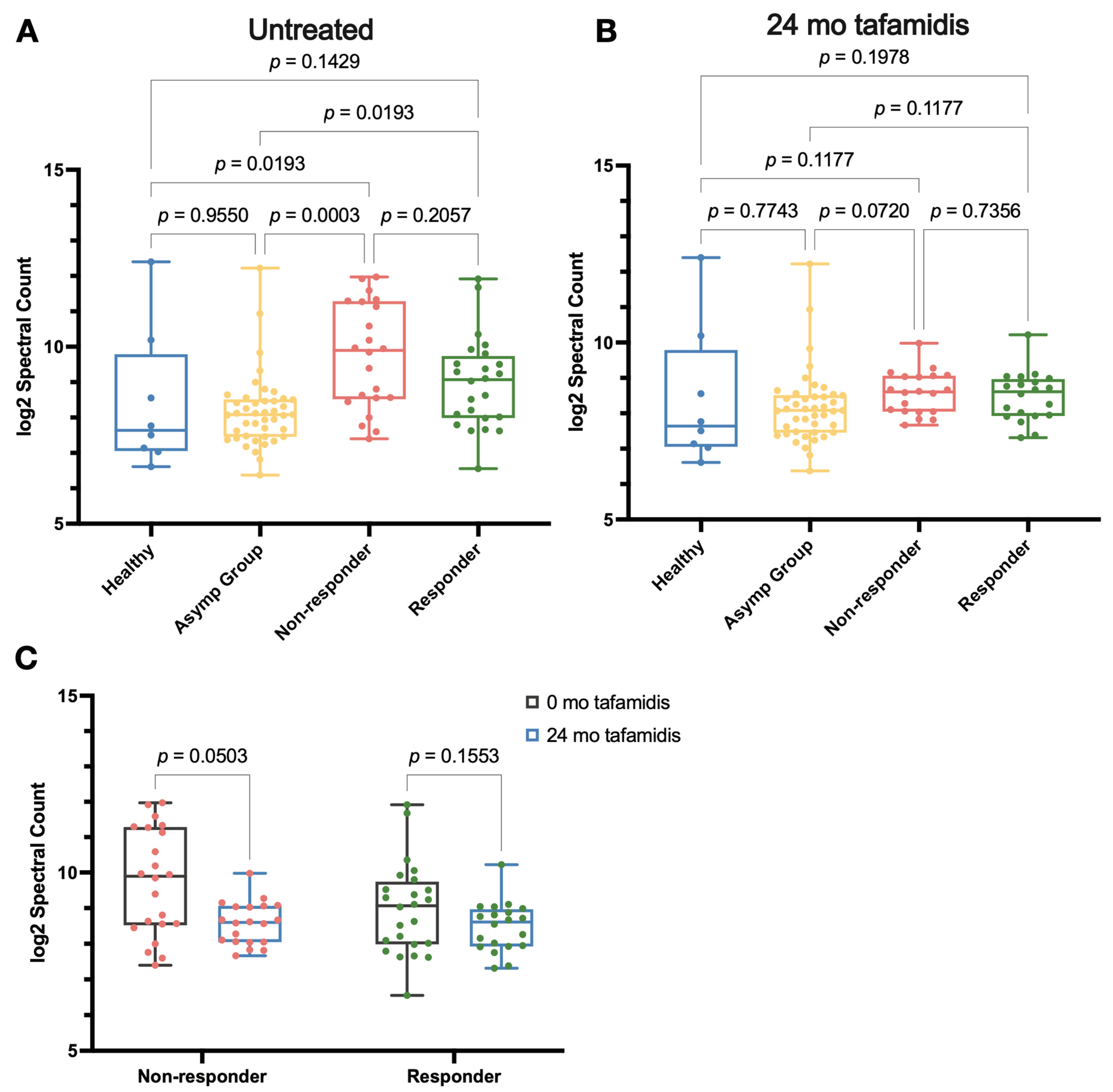
2.5. Effects of Tafamidis Treatment on Proteolysis and Oxidative Modification in the Proteome
3. Discussion
3.1. DiffPOP Is an Effective Method for Clinical Biomarker Discovery
3.2. Inflammation Is a Key Feature of ATTRv-PN Pathological Progression
3.3. Proteome Regulation by Tafamidis
4. Materials and Methods
4.1. Study Design
4.2. Differential Precipitation of Proteins (DiffPOP)
4.3. Proteomics Sample Preparation and Liquid Chromatography Coupled to MS/MS Analysis
4.4. DiffPOP Data Processing for TargetSeeker-MS
4.5. DiffPOP Data Processing with Skyline
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. SDS-PAGE and Immunoblotting
4.8. Peptidomics Sample Preparation
5. Conclusions
6. Limitation of the Study and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATTR | Transthyretin Amyloidosis |
| ATTRv-PN | Transthyretin Amyloid Polyneuropathy |
| APOE | Apolipoprotein E |
| C3 | Complement Component 3 |
| C3a | Complement Component 3a |
| C3b | Complement Component 3b |
| C3c | Complement Component 3c |
| C5 | Complement Component 5 |
| C6 | Complement Component 6 |
| CP | Ceruloplasmin |
| LPA | Lipoprotein A |
| TTR | Transthyretin |
| V30M | Valine to methionine mutation at position 30 in TTR |
Appendix A
References
- Poli, L.; Labella, B.; Cotti Piccinelli, S.; Caria, F.; Risi, B.; Damioli, S.; Padovani, A.; Filosto, M. Hereditary transthyretin amyloidosis: A comprehensive review with a focus on peripheral neuropathy. Front. Neurol. 2023, 14, 1242815. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington Cruz, M.; Plante-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceicao, I.M.; Schmidt, H.H.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.J.; Birken, S.; Costa, P.P.; Goodman, D.S. Family studies of the genetic abnormality in transthyretin (prealbumin) in Portuguese patients with familial amyloidotic polyneuropathy. Ann. N. Y. Acad. Sci. 1984, 435, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Ines, M.; Conceicao, I.; Soares, M.; de Carvalho, M.; Costa, J. Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology 2018, 91, e1999–e2009. [Google Scholar] [CrossRef]
- Waddington-Cruz, M.; Wixner, J.; Amass, L.; Kiszko, J.; Chapman, D.; Ando, Y.; the THAOS investigators. Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol. Ther. 2021, 10, 753–766. [Google Scholar] [CrossRef]
- Bulawa, C.E.; Connelly, S.; Devit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Judge, D.P.; Cappelli, F.; Fontana, M.; Garcia-Pavia, P.; Gibbs, S.; Grogan, M.; Hanna, M.; Hoffman, J.; Masri, A.; et al. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2024, 390, 132–142. [Google Scholar] [CrossRef]
- Judge, D.P.; Gillmore, J.D.; Alexander, K.M.; Ambardekar, A.V.; Cappelli, F.; Fontana, M.; Garcia-Pavia, P.; Grodin, J.L.; Grogan, M.; Hanna, M.; et al. Long-Term Efficacy and Safety of Acoramidis in ATTR-CM: Initial Report From the Open-Label Extension of the ATTRibute-CM Trial. Circulation 2024, 151, 601–611. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Plante-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Coelho, T.; Marques, W., Jr.; Dasgupta, N.R.; Chao, C.C.; Parman, Y.; Franca, M.C., Jr.; Guo, Y.C.; Wixner, J.; Ro, L.S.; Calandra, C.R.; et al. Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy. JAMA 2023, 330, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Urits, I.; Swanson, D.; Swett, M.C.; Patel, A.; Berardino, K.; Amgalan, A.; Berger, A.A.; Kassem, H.; Kaye, A.D.; Viswanath, O. A Review of Patisiran (ONPATTRO(R)) for the Treatment of Polyneuropathy in People with Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2020, 9, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Plante-Bordeneuve, V.; Berk, J.L.; Gonzalez-Duarte, A.; Gillmore, J.D.; Low, S.C.; Sekijima, Y.; et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: A randomized clinical trial. Amyloid 2023, 30, 18–26. [Google Scholar] [CrossRef]
- Fontana, M.; Berk, J.L.; Gillmore, J.D.; Witteles, R.M.; Grogan, M.; Drachman, B.; Damy, T.; Garcia-Pavia, P.; Taubel, J.; Solomon, S.D.; et al. Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy. N. Engl. J. Med. 2025, 392, 33–44. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef]
- Coelho, T.; Maia, L.F.; da Silva, A.M.; Cruz, M.W.; Plante-Bordeneuve, V.; Suhr, O.B.; Conceicao, I.; Schmidt, H.H.; Trigo, P.; Kelly, J.W.; et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol. 2013, 260, 2802–2814. [Google Scholar] [CrossRef]
- Cortese, A.; Vita, G.; Luigetti, M.; Russo, M.; Bisogni, G.; Sabatelli, M.; Manganelli, F.; Santoro, L.; Cavallaro, T.; Fabrizi, G.M.; et al. Monitoring effectiveness and safety of Tafamidis in transthyretin amyloidosis in Italy: A longitudinal multicenter study in a non-endemic area. J. Neurol. 2016, 263, 916–924. [Google Scholar] [CrossRef]
- Lozeron, P.; Theaudin, M.; Mincheva, Z.; Ducot, B.; Lacroix, C.; Adams, D.; French Network for FAP (CORNAMYL). Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur. J. Neurol. 2013, 20, 1539–1545. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V.; Gorram, F.; Salhi, H.; Nordine, T.; Ayache, S.S.; Le Corvoisier, P.; Azoulay, D.; Feray, C.; Damy, T.; Lefaucheur, J.P. Long-term treatment of transthyretin familial amyloid polyneuropathy with tafamidis: A clinical and neurophysiological study. J. Neurol. 2017, 264, 268–276. [Google Scholar] [CrossRef]
- Mundayat, R.; Stewart, M.; Alvir, J.; Short, S.; Ong, M.L.; Keohane, D.; Rill, D.; Sultan, M.B. Positive Effectiveness of Tafamidis in Delaying Disease Progression in Transthyretin Familial Amyloid Polyneuropathy up to 2 Years: An Analysis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol. Ther. 2018, 7, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, C.; Mesgazardeh, J.S.; Anselmo, J.; Fernandes, J.; Novais, M.; Rodrigues, C.; Brighty, G.J.; Powers, D.L.; Powers, E.T.; Coelho, T.; et al. Predictive model of response to tafamidis in hereditary ATTR polyneuropathy. JCI Insight 2019, 4, e126526. [Google Scholar] [CrossRef]
- Buxbaum, J.N. Treatment of hereditary and acquired forms of transthyretin amyloidosis in the era of personalized medicine: The role of randomized controlled trials. Amyloid 2019, 26, 55–65. [Google Scholar] [CrossRef]
- Schonhoft, J.D.; Monteiro, C.; Plate, L.; Eisele, Y.S.; Kelly, J.M.; Boland, D.; Parker, C.G.; Cravatt, B.F.; Teruya, S.; Helmke, S.; et al. Peptide probes detect misfolded transthyretin oligomers in plasma of hereditary amyloidosis patients. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Monteiro, C.; Mesgarzadeh, J.S.; Anselmo, J.; Fernandes, J.; Novais, M.; Rodrigues, C.; Powers, D.L.; Powers, E.T.; Coelho, T.; Kelly, J.W. Tafamidis polyneuropathy amelioration requires modest increases in transthyretin stability even though increases in plasma native TTR and decreases in non-native TTR do not predict response. Amyloid 2023, 30, 81–95. [Google Scholar] [CrossRef]
- Jiang, X.; Labaudiniere, R.; Buxbaum, J.N.; Monteiro, C.; Novais, M.; Coelho, T.; Kelly, J.W. A circulating, disease-specific, mechanism-linked biomarker for ATTR polyneuropathy diagnosis and response to therapy prediction. Proc. Natl. Acad. Sci. USA 2021, 118, e2016072118. [Google Scholar] [CrossRef]
- Buxbaum, J.; Anan, I.; Suhr, O. Serum transthyretin levels in Swedish TTR V30M carriers. Amyloid 2010, 17, 83–85. [Google Scholar] [CrossRef]
- Schneider, F.; Hammarstrom, P.; Kelly, J.W. Transthyretin slowly exchanges subunits under physiological conditions: A convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 2001, 10, 1606–1613. [Google Scholar] [CrossRef]
- Nylander, P.O.; Beckman, L.; Holmgren, G.; Steen, L. Association of C3 and C4A complement types with familial amyloidotic polyneuropathy. Hum. Hered. 1990, 40, 272–277. [Google Scholar] [CrossRef]
- Dardiotis, E.; Koutsou, P.; Zamba-Papanicolaou, E.; Vonta, I.; Hadjivassiliou, M.; Hadjigeorgiou, G.; Cariolou, M.; Christodoulou, K.; Kyriakides, T. Complement C1Q polymorphisms modulate onset in familial amyloidotic polyneuropathy TTR Val30Met. J. Neurol. Sci. 2009, 284, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Lambris, J.D. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001, 180, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Reis, E.S.; Mastellos, D.C.; Gros, P.; Lambris, J.D. Complement component C3-The “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 2016, 274, 33–58. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Damotte, V.; van der Lee, S.J.; Chouraki, V.; Grenier-Boley, B.; Simino, J.; Adams, H.; Tosto, G.; White, C.; Terzikhan, N.; Cruchaga, C.; et al. Plasma amyloid beta levels are driven by genetic variants near APOE, BACE1, APP, PSEN2: A genome-wide association study in over 12,000 non-demented participants. Alzheimers Dement. 2021, 17, 1663–1674. [Google Scholar] [CrossRef]
- Zhao, L.; Buxbaum, J.N.; Reixach, N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 2013, 52, 1913–1926. [Google Scholar] [CrossRef]
- Orzechowski, A.; Cywinska, A.; Rostagno, A.A.; Rizzi, F.M. Oxidative Stress, Chronic Inflammation, and Amyloidoses. Oxidative Med. Cell. Longev. 2019, 2019, 6024975. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Scaloni, A.; Giustarini, D.; Cavarra, E.; Tell, G.; Lungarella, G.; Colombo, R.; Rossi, R.; Milzani, A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox proteomics. Mass Spectrom. Rev. 2005, 24, 55–99. [Google Scholar] [CrossRef]
- da Costa, G.; Ribeiro-Silva, C.; Ribeiro, R.; Gilberto, S.; Gomes, R.A.; Ferreira, A.; Mateus, E.; Barroso, E.; Coelho, A.V.; Freire, A.P.; et al. Transthyretin Amyloidosis: Chaperone Concentration Changes and Increased Proteolysis in the Pathway to Disease. PLoS ONE 2015, 10, e0125392. [Google Scholar] [CrossRef]
- Srivastava, A.R.; Kumar, S.; Agarwal, G.G.; Ranjan, P. Blunt abdominal injury: Serum ALT—A marker of liver injury and a guide to assessment of its severity. Injury 2007, 38, 1069–1074. [Google Scholar] [CrossRef]
- Gonzalez-Ortiz, F.; Kac, P.R.; Brum, W.S.; Zetterberg, H.; Blennow, K.; Karikari, T.K. Plasma phospho-tau in Alzheimer’s disease: Towards diagnostic and therapeutic trial applications. Mol. Neurodegener. 2023, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Pang, Y.; Fu, X.; Ren, Z.; Jia, L. Plasma biomarkers predict Alzheimer’s disease before clinical onset in Chinese cohorts. Nat. Commun. 2023, 14, 6747. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arslan, B.; Jonaitis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol. 2024, 81, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Gerszten, R.E.; Accurso, F.; Bernard, G.R.; Caprioli, R.M.; Klee, E.W.; Klee, G.G.; Kullo, I.; Laguna, T.A.; Roth, F.P.; Sabatine, M.; et al. Challenges in translating plasma proteomics from bench to bedside: Update from the NHLBI Clinical Proteomics Programs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L16–L22. [Google Scholar] [CrossRef]
- Pinto, A.F.M.; Diedrich, J.K.; Moresco, J.J.; Yates, J.R., 3rd. Differential Precipitation of Proteins: A Simple Protein Fractionation Strategy to Gain Biological Insights with Proteomics. J. Am. Soc. Mass Spectrom. 2023, 34, 2025–2033. [Google Scholar] [CrossRef]
- Xu, M.; Moresco, J.J.; Chang, M.; Mukim, A.; Smith, D.; Diedrich, J.K.; Yates, J.R., 3rd; Jones, K.A. SHMT2 and the BRCC36/BRISC deubiquitinase regulate HIV-1 Tat K63-ubiquitylation and destruction by autophagy. PLoS Pathog. 2018, 14, e1007071. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Chan, G.G.; Koch, C.M.; Connors, L.H. Serum Proteomic Variability Associated with Clinical Phenotype in Familial Transthyretin Amyloidosis (ATTRm). J. Proteome Res. 2017, 16, 4104–4112. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Bandeira-Melo, C.; Chimelli, L.; Waddington-Cruz, M.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. Inflammatory profiling of patients with familial amyloid polyneuropathy. BMC Neurol. 2019, 19, 146. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.P. Does Ceruloplasmin Defend Against Neurodegenerative Diseases? Curr. Neuropharmacol. 2019, 17, 539–549. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef]
- Fernandez Lopez, M.T.; Guillin Amarelle, C.; Mato Mato, J.A. Severe hypocupremia and familial amyloid polyneuropathy. Nutr. Hosp. 2020, 37, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Fruchart-Gaillard, C.; Mourier, G.; Savko, M.; Nencetti, S.; Orlandini, E.; Servent, D.; Stura, E.A.; Shepard, W. Copper mediated amyloid-beta binding to Transthyretin. Sci. Rep. 2018, 8, 13744. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, A.; Terazawa-Watanabe, M.; Kazumi, T.; Fukuo, K. Associations of decreased serum transthyretin with elevated high-sensitivity CRP, serum copper and decreased hemoglobin in ambulatory elderly women. Asia Pac. J. Clin. Nutr. 2015, 24, 83–89. [Google Scholar] [CrossRef]
- Keene, C.D.; Cudaback, E.; Li, X.; Montine, K.S.; Montine, T.J. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer’s disease. Curr. Opin. Neurobiol. 2011, 21, 920–928. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Giau, V.V.; Bagyinszky, E.; An, S.S.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef]
- Fernandez-Calle, R.; Konings, S.C.; Frontinan-Rubio, J.; Garcia-Revilla, J.; Camprubi-Ferrer, L.; Svensson, M.; Martinson, I.; Boza-Serrano, A.; Venero, J.L.; Nielsen, H.M.; et al. APOE in the bullseye of neurodegenerative diseases: Impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol. Neurodegener. 2022, 17, 62. [Google Scholar] [CrossRef]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef]
- Bartolacci, C.; Andreani, C.; El-Gammal, Y.; Scaglioni, P.P. Lipid Metabolism Regulates Oxidative Stress and Ferroptosis in RAS-Driven Cancers: A Perspective on Cancer Progression and Therapy. Front. Mol. Biosci. 2021, 8, 706650. [Google Scholar] [CrossRef]
- Endale, H.T.; Tesfaye, W.; Mengstie, T.A. ROS induced lipid peroxidation and their role in ferroptosis. Front. Cell Dev. Biol. 2023, 11, 1226044. [Google Scholar] [CrossRef]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nugroho, K.; Lin, C.-y.; Monteiro, C.; Coelho, T.; Moresco, J.J.; Pinto, A.F.M.; Powers, E.T.; Yates, J.R., III; Diedrich, J.K.; Kelly, J.W. Plasma Proteome Profiling Reveals Inflammation Markers and Tafamidis Effects in V30M Transthyretin Polyneuropathy. Int. J. Mol. Sci. 2025, 26, 5534. https://doi.org/10.3390/ijms26125534
Nugroho K, Lin C-y, Monteiro C, Coelho T, Moresco JJ, Pinto AFM, Powers ET, Yates JR III, Diedrich JK, Kelly JW. Plasma Proteome Profiling Reveals Inflammation Markers and Tafamidis Effects in V30M Transthyretin Polyneuropathy. International Journal of Molecular Sciences. 2025; 26(12):5534. https://doi.org/10.3390/ijms26125534
Chicago/Turabian StyleNugroho, Karina, Chung-yon Lin, Cecilia Monteiro, Teresa Coelho, James J. Moresco, Antonio F. M. Pinto, Evan T. Powers, John R. Yates, III, Jolene K. Diedrich, and Jeffery W. Kelly. 2025. "Plasma Proteome Profiling Reveals Inflammation Markers and Tafamidis Effects in V30M Transthyretin Polyneuropathy" International Journal of Molecular Sciences 26, no. 12: 5534. https://doi.org/10.3390/ijms26125534
APA StyleNugroho, K., Lin, C.-y., Monteiro, C., Coelho, T., Moresco, J. J., Pinto, A. F. M., Powers, E. T., Yates, J. R., III, Diedrich, J. K., & Kelly, J. W. (2025). Plasma Proteome Profiling Reveals Inflammation Markers and Tafamidis Effects in V30M Transthyretin Polyneuropathy. International Journal of Molecular Sciences, 26(12), 5534. https://doi.org/10.3390/ijms26125534