

Anticancer Potential of Halogen Derivatives of Methyl 6-Acetyl-5-Hydroxy-2-Methyl-1-Benzofuran-3-Carboxylate

 and
and
Abstract
1. Introduction
2. Results and Discussion
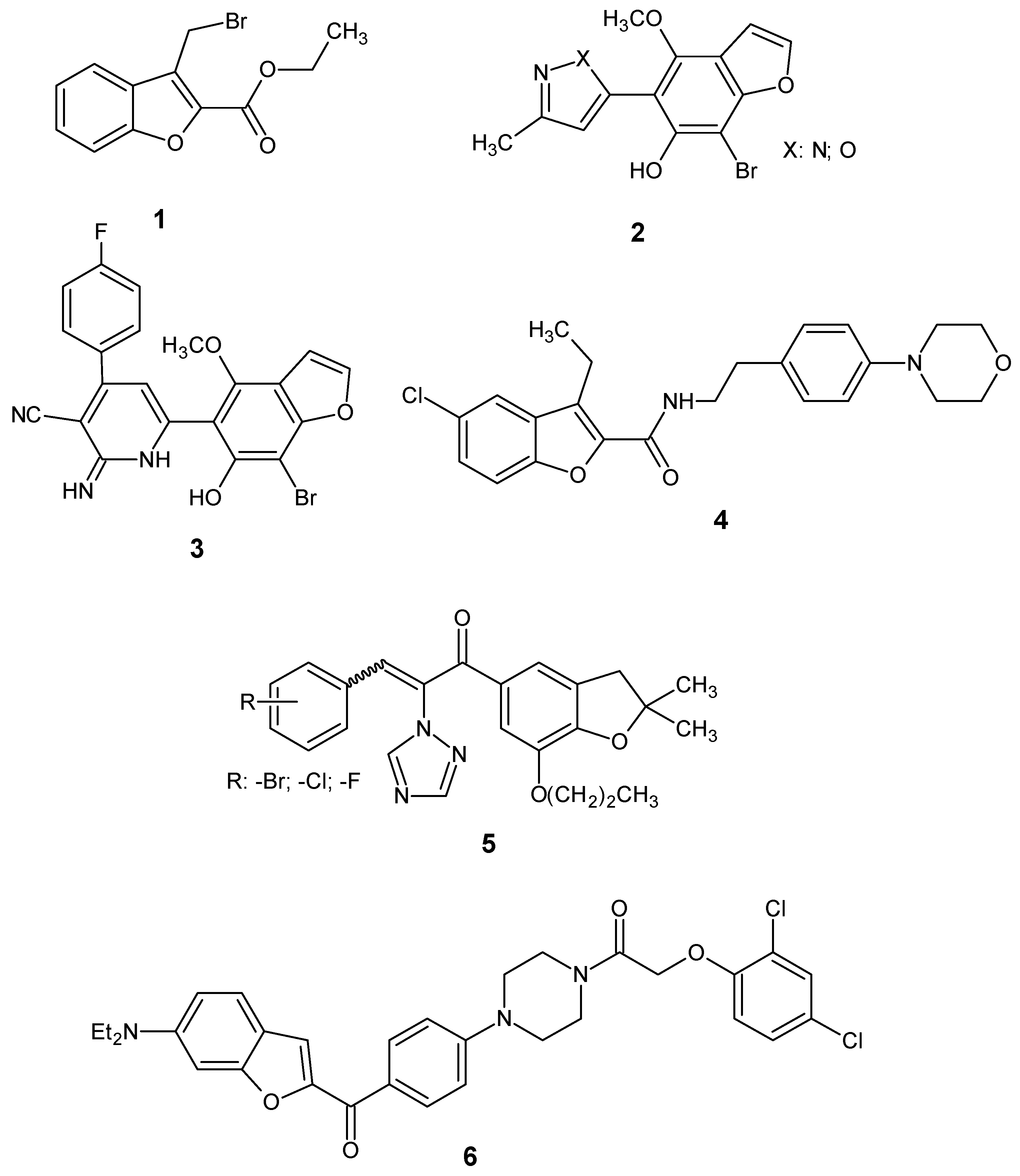
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro Cytotoxic Activity
2.2.2. Antiproliferative Activity
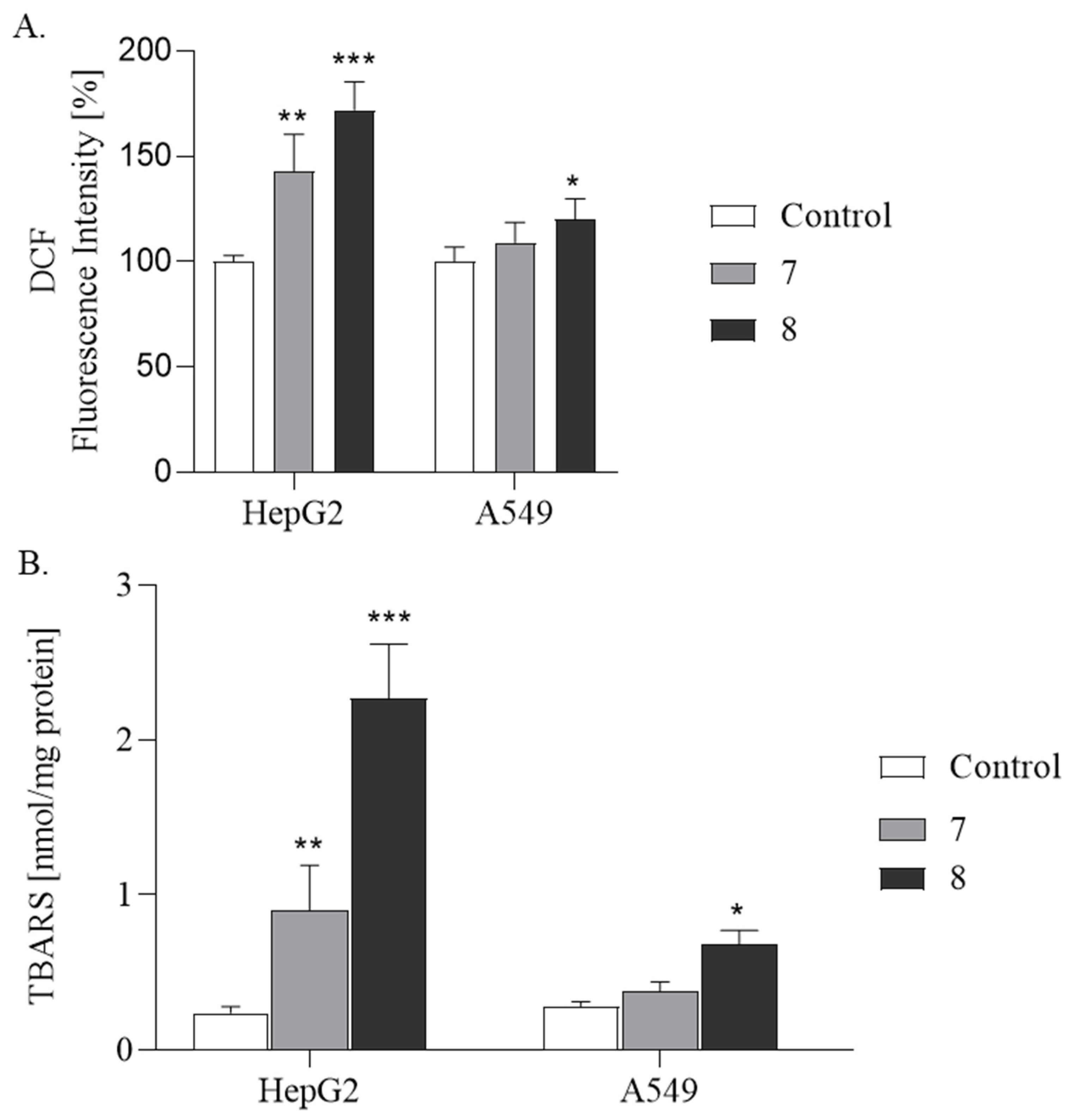
2.2.3. ROS Production and Lipid Peroxidation Studies
2.2.4. Interleukin-6 Assay
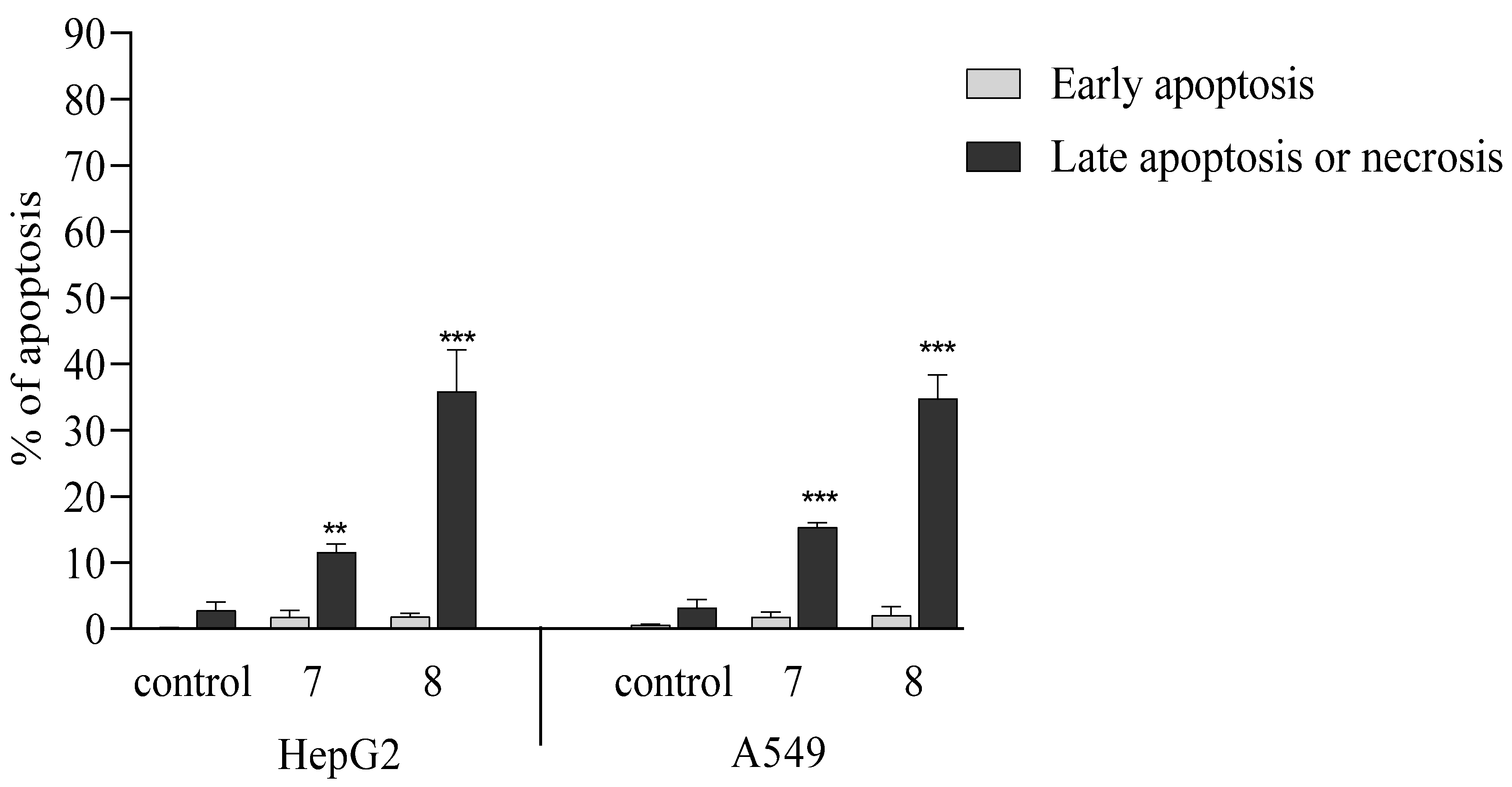
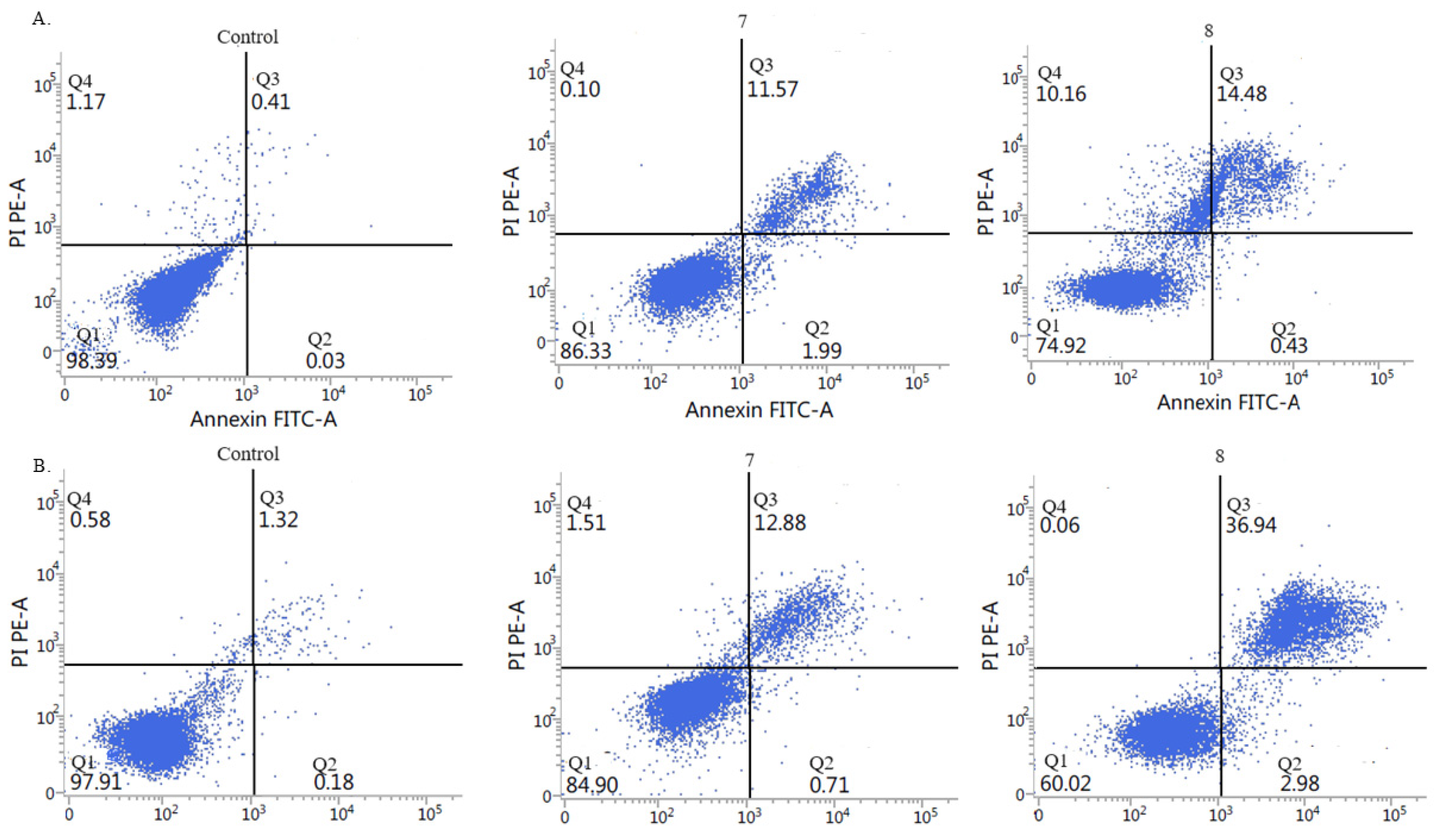
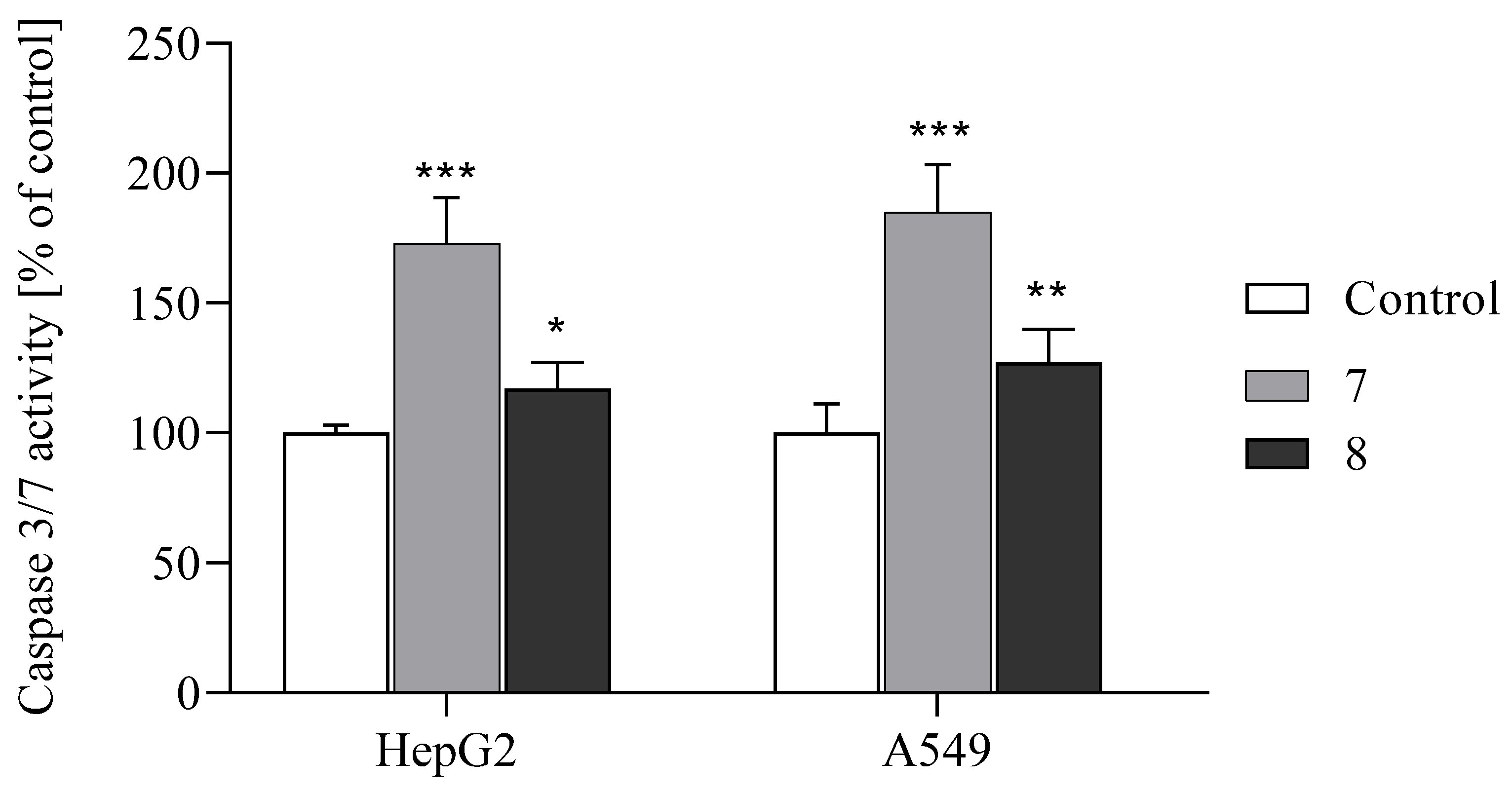
2.2.5. Activation of Apoptosis
2.2.6. Test Benzo [B] Furans Cause G2/M Cell Cycle Arrest
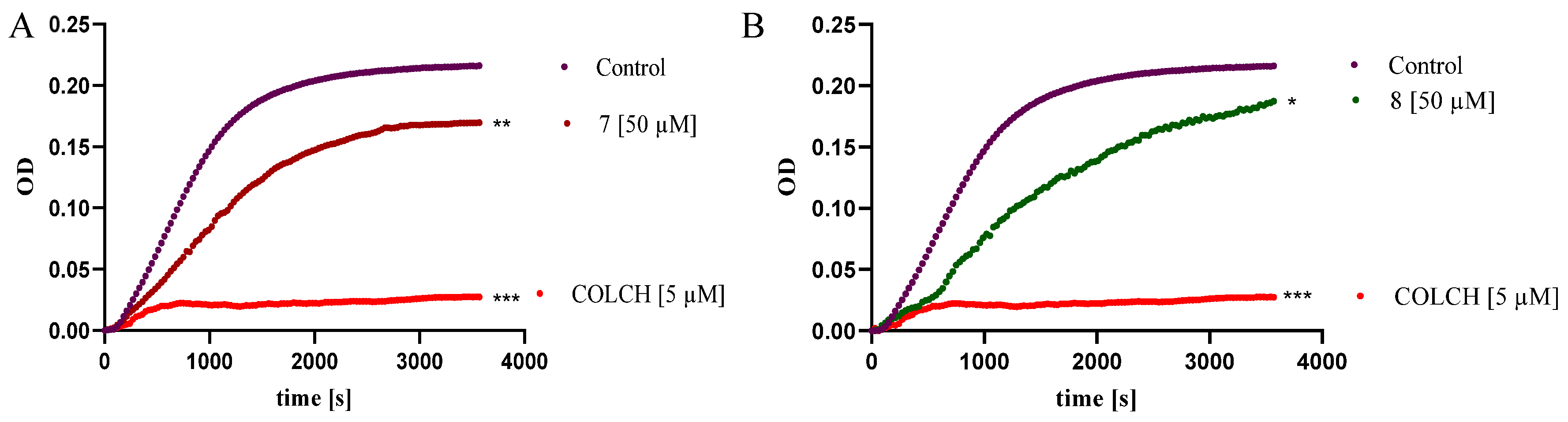
2.2.7. Benzo [B] Furans Inhibit Tubulin Polymerization In Vitro
3. Materials and Methods
3.1. Chemistry
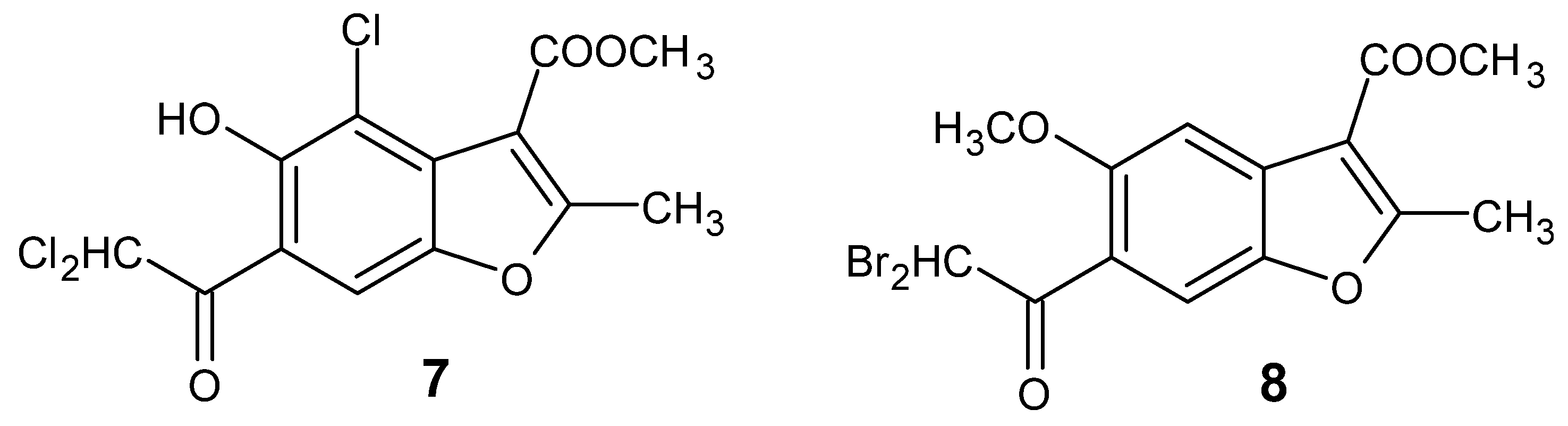
3.1.1. Synthesis of Methyl 4-Chloro-6-(Dichloroacetyl)-5-Hydroxy-2-Methyl-1-Benzofuran-3-Carboxylate (7, Figure 2)
3.1.2. Synthesis of Methyl 6-(Dibromoacetyl)-5-Methoxy-2-Methyl-1-Benzofuran-3-Carboxylate (8, Figure 2)
3.2. Biological Studies
3.2.1. Human Cell Line, MTT Assay
3.2.2. Measurement of Live Cell Number and Viability (%) by Trypan Blue Exclusion Assay
3.2.3. Measurement of Cell Apoptosis by Flow Cytometry Annexin V-FITC Binding Assay
3.2.4. Cell Cycle Analysis
3.2.5. Caspases-3 and -7 Activity Assay
3.2.6. DCFH-DA Assay
3.2.7. TBA Assay
3.2.8. Interleukin-6
3.2.9. Tubulin Polymerization
3.2.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khanam, H.; Shamsuzzaman. Bioactive Benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.H.; Hu, Y.H.; Yang, J.; Liu, T.; Sun, J.; Wang, X.J. Natural source, bioactivity and synthesis of benzofuran derivatives. RSC Adv. 2019, 9, 27510–27540. [Google Scholar] [CrossRef] [PubMed]
- Nevagi, R.J.; Dighe, S.N.; Dighe, S.N. Biological and medicinal significance of benzofuran. Eur. J. Med. Chem. 2015, 97, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Farhat, J.; Alzyoud, L.; Alwahsh, M.; Al-Omari, B. Structure-Activity Relationship of Benzofuran Derivatives with Potential Anticancer Activity. Cancers 2022, 14, 2196. [Google Scholar] [CrossRef]
- Asadi, P.; Khodarahmi, G.; Jahanian-Najafabadi, A.; Saghaie, L.; Hassanzadeh, F. Biologically Active Heterocyclic Hybrids Based on Quinazolinone, Benzofuran and Imidazolium Moieties: Synthesis, Characterization, Cytotoxic and Antibacterial Evaluation. Chem. Biodivers. 2017, 14, e1600411. [Google Scholar] [CrossRef]
- Mao, Z.W.; Zheng, X.; Lin, Y.P.; Hu, C.Y.; Wang, X.L.; Wan, C.P.; Rao, G.X. Design, synthesis and anticancer activity of novel hybrid compounds between benzofuran and N-aryl piperazine. Bioorg. Med. Chem. Lett. 2016, 26, 3421–3424. [Google Scholar] [CrossRef]
- Yang, X.D.; Wan, W.C.; Deng, X.Y.; Li, Y.; Yang, L.J.; Li, L.; Zhang, H.B. Design, synthesis and cytotoxic activities of novel hybrid compounds between 2-phenylbenzofuran and imidazole. Bioorg. Med. Chem. Lett. 2012, 22, 2726–2729. [Google Scholar] [CrossRef]
- Ma, Y.; Zheng, X.; Gao, H.; Wan, C.; Rao, G.; Mao, Z. Design, Synthesis, and Biological Evaluation of Novel Benzofuran Derivatives Bearing N-Aryl Piperazine Moiety. Molecules 2016, 21, 1684. [Google Scholar] [CrossRef]
- Salome, C.; Ribeiro, N.; Chavagnan, T.; Thuaud, F.; Serova, M.; de Gramont, A.; Faivre, S.; Raymond, E.; Desaubry, L. Benzofuran derivatives as anticancer inhibitors of mTOR signaling. Eur. J. Med. Chem. 2014, 81, 181–191. [Google Scholar] [CrossRef]
- Abdelfatah, S.; Berg, A.; Huang, Q.; Yang, L.J.; Hamdoun, S.; Klinger, A.; Greten, H.J.; Fleischer, E.; Berg, T.; Wong, V.K.W.; et al. MCC1019, a selective inhibitor of the Polo-box domain of Polo-like kinase 1 as novel, potent anticancer candidate. Acta Pharm. Sin. B 2019, 9, 1021–1034. [Google Scholar] [CrossRef]
- Amin, K.M.; Syam, Y.M.; Anwar, M.M.; Ali, H.I.; Abdel-Ghani, T.M.; Serry, A.M. Synthesis and molecular docking study of new benzofuran and furo[3,2-g]chromone-based cytotoxic agents against breast cancer and p38alpha MAP kinase inhibitors. Bioorg. Chem. 2018, 76, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Youssif, B.G.M.; Mohamed, A.M.; Osman, E.E.A.; Abou-Ghadir, O.F.; Elnaggar, D.H.; Abdelrahman, M.H.; Treamblu, L.; Gomaa, H.A.M. 5-Chlorobenzofuran-2-carboxamides: From allosteric CB1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, Z.H.; Hu, A.X.; Yan, X.W.; Ding, N.; Ye, J. Design, Synthesis, and Antitumor Activity of (E,Z)-1-(dihydrobenzofuran-5-yl)-3-phenyl-2-(1,2,4-triazol-1-yl)-2-propen-1-ones. Chem. Biol. Drug Des. 2015, 86, 1339–1350. [Google Scholar] [CrossRef]
- Kossakowski, J.; Krawiecka, M.; Kuran, B.; Stefanska, J.; Wolska, I. Synthesis and preliminary evaluation of the antimicrobial activity of selected 3-benzofurancarboxylic acid derivatives. Molecules 2010, 15, 4737–4749. [Google Scholar] [CrossRef] [PubMed]
- Krawiecka, M.; Kuran, B.; Kossakowski, J.; Cieslak, M.; Kazmierczak- Baranska, J.; Krolewska, K.; Nawrot, B. Synthesis and Cytotoxic Properties of Halogen and Aryl-/Heteroarylpiperazinyl Derivatives of Benzofurans. Anti-Cancer Agents Med. Chem. 2015, 15, 115–121. [Google Scholar] [CrossRef]
- Krolewska-Golinska, K.; Cieslak, M.J.; Sobczak, M.; Dolot, R.; Radzikowska-Cieciura, E.; Napiorkowska, M.; Wybranska, I.; Nawrot, B. Novel Benzo[B]Furans with Anti-Microtubule Activity Upregulate Expression of Apoptotic Genes and Arrest Leukemia Cells in G2/M Phase. Anti-Cancer Agents Med. Chem. 2019, 19, 375–388. [Google Scholar] [CrossRef]
- Napiorkowska, M.; Cieslak, M.; Kazmierczak-Baranska, J.; Krolewska-Golinska, K.; Nawrot, B. Synthesis of New Derivatives of Benzofuran as Potential Anticancer Agents. Molecules 2019, 24, 1529. [Google Scholar] [CrossRef] [PubMed]
- Napiorkowska, M.; Otto-Slusarczyk, D.; Kurpios-Piec, D.; Stukan, I.; Gryzik, M.; Wojda, U. BM7, a derivative of benzofuran, effectively fights cancer by promoting cancer cell apoptosis and impacting IL-6 levels. Eur. J. Pharmacol. 2024, 978, 176751. [Google Scholar] [CrossRef]
- Napiorkowska, M.; Kumaravel, P.; Amboo Mahentheran, M.; Kiernozek-Kalinska, E.; Grosicka-Maciag, E. New Derivatives of 1-(3-Methyl-1-Benzofuran-2-yl)Ethan-1-one: Synthesis and Preliminary Studies of Biological Activity. Int. J. Mol. Sci. 2024, 25, 1999. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxidative Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.; Rajeshwari; Hiremathad, A.; Singh, M.; Santos, M.A.; Keri, R.S. A review on antioxidant potential of bioactive heterocycle benzofuran: Natural and synthetic derivatives. Pharmacol. Rep. 2017, 69, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Rindhe, S.S.; Rode, M.A.; Karale, B.K. New benzofuran derivatives as an antioxidant agent. Indian. J. Pharm. Sci. 2010, 72, 231–235. [Google Scholar] [CrossRef]
- Kumar, K.; Mishra, J.P.N.; Singh, R.P. Usnic acid induces apoptosis in human gastric cancer cells through ROS generation and DNA damage and causes up-regulation of DNA-PKcs and γ-H2A.X phosphorylation. Chem.-Biol. Interact. 2020, 315, 108898. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Park, C.; Lee, Y.; Kim, S.; Bose, S.; Choi, M.; Kumar, A.S.; Jung, J.K.; Lee, H. Neuroprotective and antioxidant effects of novel benzofuran-2-carboxamide derivatives. Biomol. Ther. 2015, 23, 275–282. [Google Scholar] [CrossRef]
- Gao, C.; Sun, X.; Wu, Z.; Yuan, H.; Han, H.; Huang, H.; Shu, Y.; Xu, M.; Gao, R.; Li, S.; et al. A Novel Benzofuran Derivative Moracin N Induces Autophagy and Apoptosis Through ROS Generation in Lung Cancer. Front. Pharmacol. 2020, 11, 391. [Google Scholar] [CrossRef]
- Ahmad, J.; Ahamed, M.; Akhtar, M.J.; Alrokayan, S.A.; Siddiqui, M.A.; Musarrat, J.; Al-Khedhairy, A.A. Apoptosis induction by silica nanoparticles mediated through reactive oxygen species in human liver cell line HepG2. Toxicol. Appl. Pharmacol. 2012, 259, 160–168. [Google Scholar] [CrossRef]
- Zhang, Q.; Bao, J.; Yang, J. Genistein-triggered anticancer activity against liver cancer cell line HepG2 involves ROS generation, mitochondrial apoptosis, G2/M cell cycle arrest and inhibition of cell migration. Arch. Med. Sci. 2019, 15, 1001–1009. [Google Scholar] [CrossRef]
- Zou, S.; Wu, Y.; Wen, M.; Liu, J.; Chen, M.; Yuan, J.; Zhou, B. Potential Molecular Mechanism of Illicium simonsii Maxim Petroleum Ether Fraction in the Treatment of Hepatocellular Carcinoma. Pharmaceuticals 2024, 17, 806. [Google Scholar] [CrossRef]
- Gall, J.I.; Goncalves Alves, A.; Carraro Junior, L.R.; da Silva Teixeira Rech, T.; Dos Santos Neto, J.S.; Alves, D.; Pereira Soares, M.S.; Spohr, L.; Spanevello, R.M.; Bruning, C.A.; et al. Insights into serotonergic and antioxidant mechanisms involved in antidepressant-like action of 2-phenyl-3-(phenylselanyl)benzofuran in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 102, 109956. [Google Scholar] [CrossRef]
- Scibetta, S.; Miceli, M.; Iuliano, M.; Stefanuto, L.; Carbone, E.; Piscopo, P.; Petrozza, V.; Romeo, G.; Mangino, G.; Calogero, A.; et al. In Vitro Evaluation of the Antioxidant Capacity of 3,3-Disubstituted-3H-benzofuran-2-one Derivatives in a Cellular Model of Neurodegeneration. Life 2024, 14, 422. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.A.; Dawood, K.M. Anticancer therapeutic potential of benzofuran scaffolds. RSC Adv. 2023, 13, 11096–11120. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Shi, W.; Men, L.; Pi, X.; Jiang, T.; Peng, D.; Huo, S.; Luo, P.; Wang, M.; Guo, J.; Jiang, Y.; et al. Shikonin suppresses colon cancer cell growth and exerts synergistic effects by regulating ADAM17 and the IL-6/STAT3 signaling pathway. Int. J. Oncol. 2021, 59, 99. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Kanneganti, T.D. Caspase-7: A protease involved in apoptosis and inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 21–24. [Google Scholar] [CrossRef]
- Coskun, D.; Erkisa, M.; Ulukaya, E.; Coskun, M.F.; Ari, F. Novel 1-(7-ethoxy-1-benzofuran-2-yl) substituted chalcone derivatives: Synthesis, characterization and anticancer activity. Eur. J. Med. Chem. 2017, 136, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Sithara, T.; Arun, K.B.; Syama, H.P.; Reshmitha, T.R.; Nisha, P. Morin Inhibits Proliferation of SW480 Colorectal Cancer Cells by Inducing Apoptosis Mediated by Reactive Oxygen Species Formation and Uncoupling of Warburg Effect. Front. Pharmacol. 2017, 8, 640. [Google Scholar] [CrossRef]
- Giordano, C.; Rovito, D.; Barone, I.; Mancuso, R.; Bonofiglio, D.; Giordano, F.; Catalano, S.; Gabriele, B.; Ando, S. Benzofuran-2-acetic ester derivatives induce apoptosis in breast cancer cells by upregulating p21(Cip/WAF1) gene expression in p53-independent manner. DNA Repair 2017, 51, 20–30. [Google Scholar] [CrossRef]
- Vaali-Mohammed, M.A.; Abdulla, M.H.; Matou-Nasri, S.; Eldehna, W.M.; Meeramaideen, M.; Elkaeed, E.B.; El-Watidy, M.; Alhassan, N.S.; Alkhaya, K.; Al Obeed, O. The Anticancer Effects of the Pro-Apoptotic Benzofuran-Isatin Conjugate (5a) Are Associated with p53 Upregulation and Enhancement of Conventional Chemotherapeutic Drug Efficiency in Colorectal Cancer Cell Lines. Front. Pharmacol. 2022, 13, 923398. [Google Scholar] [CrossRef]
- Hranjec, M.; Sović, I.; Ratkaj, I.; Pavlović, G.; Ilić, N.; Valjalo, L.; Pavelić, K.; Kraljević Pavelić, S.; Karminski-Zamola, G. Antiproliferative potency of novel benzofuran-2-carboxamides on tumour cell lines: Cell death mechanisms and determination of crystal structure. Eur. J. Med. Chem. 2013, 59, 111–119. [Google Scholar] [CrossRef]
- El-Khouly, O.A.; Henen, M.A.; El-Sayed, M.A.; Shabaan, M.I.; El-Messery, S.M. Synthesis, anticancer and antimicrobial evaluation of new benzofuran based derivatives: PI3K inhibition, quorum sensing and molecular modeling study. Bioorg. Med. Chem. 2021, 31, 115976. [Google Scholar] [CrossRef] [PubMed]
- El-Khouly, O.A.; Henen, M.A.; El-Sayed, M.A.A.; El-Messery, S.M. Design, synthesis and computational study of new benzofuran hybrids as dual PI3K/VEGFR2 inhibitors targeting cancer. Sci. Rep. 2022, 12, 17104. [Google Scholar] [CrossRef]
- Li, Q.; Jian, X.E.; Chen, Z.R.; Chen, L.; Huo, X.S.; Li, Z.H.; You, W.W.; Rao, J.J.; Zhao, P.L. Synthesis and biological evaluation of benzofuran-based 3,4,5-trimethoxybenzamide derivatives as novel tubulin polymerization inhibitors. Bioorg. Chem. 2020, 102, 104076. [Google Scholar] [CrossRef]
- Wang, F.; Feng, K.-R.; Zhao, J.-Y.; Zhang, J.-W.; Shi, X.-W.; Zhou, J.; Gao, D.; Lin, G.-Q.; Tian, P. Identification of novel STAT3 inhibitors bearing 2-acetyl-7-phenylamino benzofuran scaffold for antitumour study. Bioorg. Med. Chem. 2020, 28, 115822. [Google Scholar] [CrossRef]
- Mokenapelli, S.; Thalari, G.; Vadiyaala, N.; Yerrabelli, J.R.; Irlapati, V.K.; Gorityala, N.; Sagurthi, S.R.; Chitneni, P.R. Synthesis, cytotoxicity, and molecular docking of substituted 3-(2-methylbenzofuran-3-yl)-5-(phenoxymethyl)-1,2,4-oxadiazoles. Arch. Pharm. 2020, 353, e2000006. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Tolomeo, M.; Grimaudo, S.; Di Cristina, A.; Pipitone, M.R.; Balzarini, J.; et al. Design, synthesis and structure-activity relationship of 2-(3′,4′,5′-trimethoxybenzoyl)-benzo[b]furan derivatives as a novel class of inhibitors of tubulin polymerization. Bioorg. Med. Chem. 2009, 17, 6862–6871. [Google Scholar] [CrossRef] [PubMed]
- Pieters, L.; Van Dyck, S.; Gao, M.; Bai, R.; Hamel, E.; Vlietinck, A.; Lemiere, G. Synthesis and biological evaluation of dihydrobenzofuran lignans and related compounds as potential antitumor agents that inhibit tubulin polymerization. J. Med. Chem. 1999, 42, 5475–5481. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef]
- Dogan, E.; Kara, H.G.; Kosova, B.; Cetintas, V.B. Targeting Apoptosis to Overcome Chemotherapy Resistance. In Metastasis [Internet]; Sergi, C.M., Ed.; Exon Publications: Brisbane, Australia, 2022; Chapter 12. Available online: https://www.ncbi.nlm.nih.gov/books/NBK580869/ (accessed on 4 June 2025). [CrossRef]
- Zhang, J.; Wang, L.; Zhang, Y. Downregulation of NIMA-related kinase-7 inhibits cell proliferation by inducing cell cycle arrest in human retinoblastoma cells. Exp. Ther. Med. 2018, 15, 1360–1366. [Google Scholar] [CrossRef]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Harper, J.W. Anticancer drug targets: Cell cycle and checkpoint control. J. Clin. Investig. 1999, 104, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anticancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cancer Cells | Normal Cells | ||||||
|---|---|---|---|---|---|---|---|---|
| IC50, µM b | ||||||||
| SW480 c | SW620 d | HCT116 e | HepG2 f | PC3 g | A549 h | MDA i | HUVEC j | |
| 7 | 33.2 ± 1.2 | 26.5 ± 1.8 | 36.9 ± 4.1 | 11 ± 3.2 | 43 ± 2.1 | 6.3 ± 2.5 | 29.5 ± 1.5 | >1000 [15] |
| 8 | 27.6 ± 1.1 | 10.8 ± 0.9 | 58.3 ± 7.9 | 3.8 ± 0.5 | 33.2 ± 1.1 | 3.5 ± 0.6 | 23.2 ± 2.7 | >1000 [15] |
| Dx k | 0.75 ± 0.1 | 0.26 ± 0.1 | 0.6 ± 0.02 | 0.4 ± 0.1 | 0.59 ± 0.1 | 0.63 ± 0.2 | 0.83 ± 0.03 | 1.0 ± 0.03 |
| Cp k | 10.4 ± 0.9 | 6.70 ± 1.1 | 0.6 ± 0.02 | 4.5 ± 1.2 | 13.2 ± 2.1 | 7.10 ± 1.3 | 3.95 ± 1.1 | 12.0 ± 1.8 |
| Cancer Cell Line | Compound | Cell Number × 106 | Cell Number (%) | Viability (%) |
|---|---|---|---|---|
| HepG2 | - | 1.2 ± 0.45 | 100 | 94 ± 1.52 |
| 7 | 0.6 ± 0.02 ** | 56.8 | 92 ± 4.93 | |
| 8 | 0.47 ± 0.01 *** | 39.4 | 55 ± 1.10 | |
| A549 | - | 1.8 ± 0.90 | 100 | 97 ± 2.52 |
| 7 | 0.6 ± 0.10 ** | 33.9 | 91 ± 1.65 | |
| 8 | 0.1 ± 0.02 *** | 6.21 | 85 ± 7.10 |
| Step | Description |
|---|---|
| Cell Lines Used | SW480 (primary colon cancer), SW620 (lymph node colon cancer), HCT116 (colon carcinoma), PC3 (metastatic prostate cancer), HepG2 (liver cancer), A549 (lung cancer), MDA-MB-231 (breast cancer). |
| Cell Source | All cell lines were obtained from ATCC (Manassas, VA, USA). |
| Culture Media |
|
| Media Supplements | 10% FBS (Sigma-Aldrich, St. Louis, MO, USA), 20 mM HEPES (Biowest, Nuaillé, France), 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, Grand Island, NY, USA). |
| Culture Conditions | 37 °C, 5% CO2, humidified incubator; cells used at 80–90% confluence. |
| Seeding for Assay | 1 × 104 cells/well in 96-well plates; adhered for 24 h. |
| Treatment | Cells treated with compounds 7 and 8 at various concentrations (1–100 μM) for 72 h. |
| Cytotoxicity Assay | MTT assay (0.5 mg/mL), incubation for 4 h. |
| Detection | Formazan dissolved in DMSO/isopropanol (1:1, v/v); absorbance measured at 570 nm using a MultiscanGo spectrophotometer (ThermoFisher, Waltham, MA, USA). |
| Data Analysis | % cytotoxicity = [A]/[B] × 100, where [A] = treated absorbance, [B] = control absorbance [54]. IC50 calculated using GraphPad Prism 8.0.1. |
| Step | Description |
|---|---|
| Cell Seeding | 1 × 105 cells per well were seeded in 12-well plates. |
| Incubation | Cells were cultured for 24 h at 37 °C with 5% CO2 to allow for adherence. |
| Treatment | Cells were treated with compounds 7 or 8 at their respective IC50 concentrations. Untreated cells served as controls. |
| Incubation Post Treatment | Cells were incubated for 72 h. |
| Washing | Medium was removed; cells were washed twice with PBS. |
| Cell Harvesting | Cells were trypsinized. |
| Viability and Count Assessment | Cell number and viability were assessed using the trypan blue exclusion assay and an automated cell counter (Countess, Invitrogen). |
| Reproducibility | All experiments were performed in triplicate. |
| Step | Description |
|---|---|
| Cell Seeding | 1 × 104 cells/well in black 96-well plates. |
| Initial Incubation | 24 h at 37 °C with 5% CO2 for cell attachment. |
| Treatment | Cells treated with test compounds at their IC50 concentrations; incubated for an additional 24 h under the same conditions. |
| Probe Incubation | Cells washed with PBS, then incubated with 5 μM DCFH-DA for 30 min at 37 °C in the dark. |
| Controls |
|
| Fluorescence Measurement | Measured using a Microplate Spectrofluorometer (BioTek Synergy, BioTek Instruments, Winooski, VT, USA):
|
| Output | ROS level was quantified in terms of fluorescence intensity (FI). |
| Step | Description |
|---|---|
| Cell Lines Used | HepG2 and A549. |
| Cell Seeding | 5 × 104 cells/well in 12-well plates. |
| Initial Incubation | 24 h at 37 °C, 5% CO2 to allow for cell attachment. |
| Treatment | Cells treated with test compounds at IC50 concentrations for 48 h. Untreated cells served as control. |
| TBARS Measurement | Post treatment, TBARS levels assessed by reading absorbance at 532 nm using a Thermo Scientific MultiscanGo spectrophotometer. |
| Quantification | TBARS expressed as nanomoles of MDA equivalents per milligram of protein, using the molar extinction coefficient of 1.56 × 105 M−1 × cm−1. |
| Protein Determination | Protein content measured by the Bradford assay (separate cultures), with absorbance read at 595 nm (MultiscanGo, ThermoFisher Scientific, Carlsbad, CA, USA). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napiórkowska, M.; Grosicka-Maciąg, E.; Podsadni, P.; Otto-Ślusarczyk, D. Anticancer Potential of Halogen Derivatives of Methyl 6-Acetyl-5-Hydroxy-2-Methyl-1-Benzofuran-3-Carboxylate. Int. J. Mol. Sci. 2025, 26, 5493. https://doi.org/10.3390/ijms26125493
Napiórkowska M, Grosicka-Maciąg E, Podsadni P, Otto-Ślusarczyk D. Anticancer Potential of Halogen Derivatives of Methyl 6-Acetyl-5-Hydroxy-2-Methyl-1-Benzofuran-3-Carboxylate. International Journal of Molecular Sciences. 2025; 26(12):5493. https://doi.org/10.3390/ijms26125493
Chicago/Turabian StyleNapiórkowska, Mariola, Emilia Grosicka-Maciąg, Piotr Podsadni, and Dagmara Otto-Ślusarczyk. 2025. "Anticancer Potential of Halogen Derivatives of Methyl 6-Acetyl-5-Hydroxy-2-Methyl-1-Benzofuran-3-Carboxylate" International Journal of Molecular Sciences 26, no. 12: 5493. https://doi.org/10.3390/ijms26125493
APA StyleNapiórkowska, M., Grosicka-Maciąg, E., Podsadni, P., & Otto-Ślusarczyk, D. (2025). Anticancer Potential of Halogen Derivatives of Methyl 6-Acetyl-5-Hydroxy-2-Methyl-1-Benzofuran-3-Carboxylate. International Journal of Molecular Sciences, 26(12), 5493. https://doi.org/10.3390/ijms26125493