
Molecular Screening Reveals De Novo Loss-of-Function NR4A2 Variants in Saudi Children with Autism Spectrum Disorders: A Single-Center Study

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. General Demographic and Clinical Phenotypes of the Study Cohort
2.2. Genetic Analysis: Frequency of De Novo NR4A2 Variants Among Children with ASD
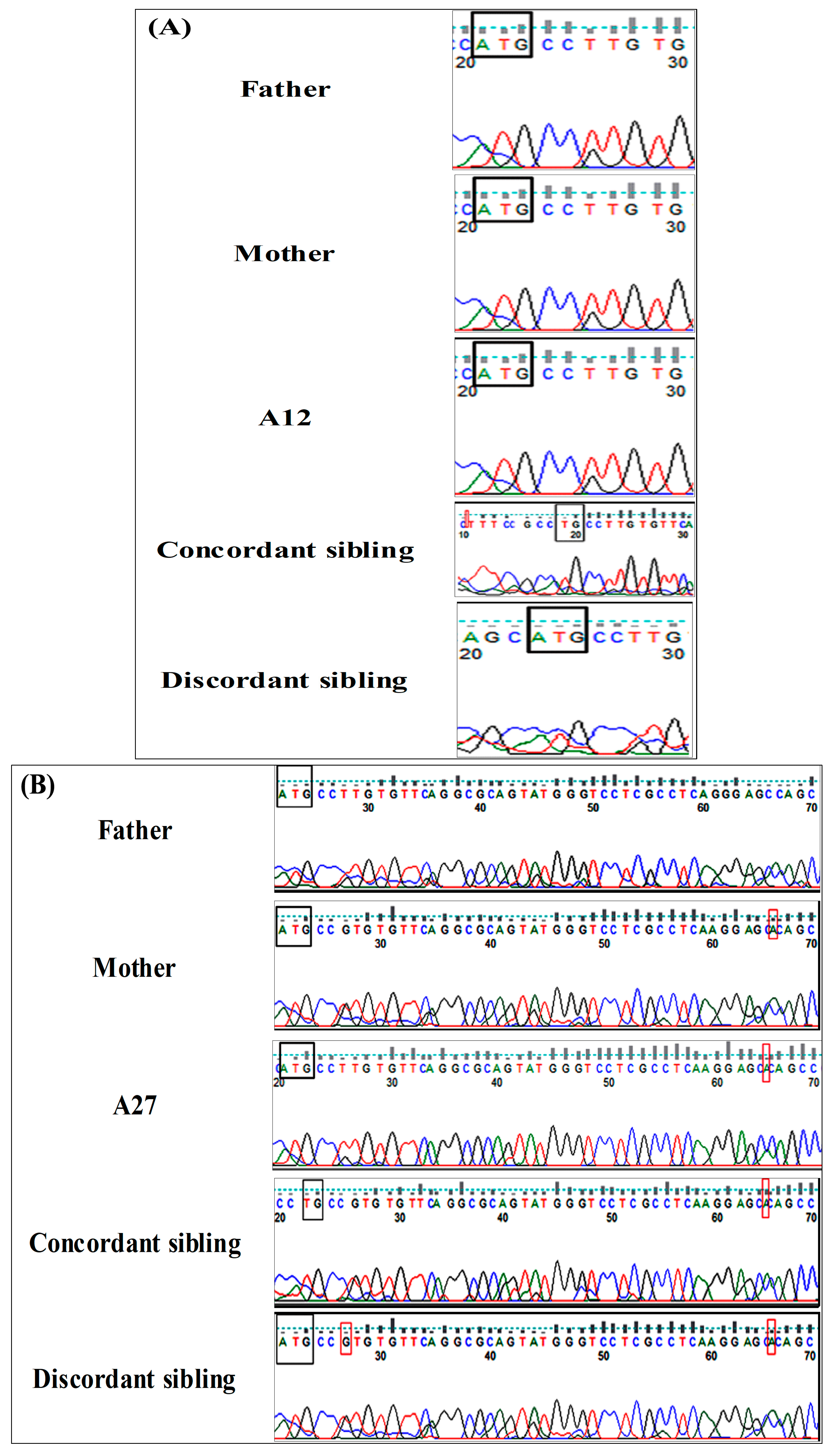
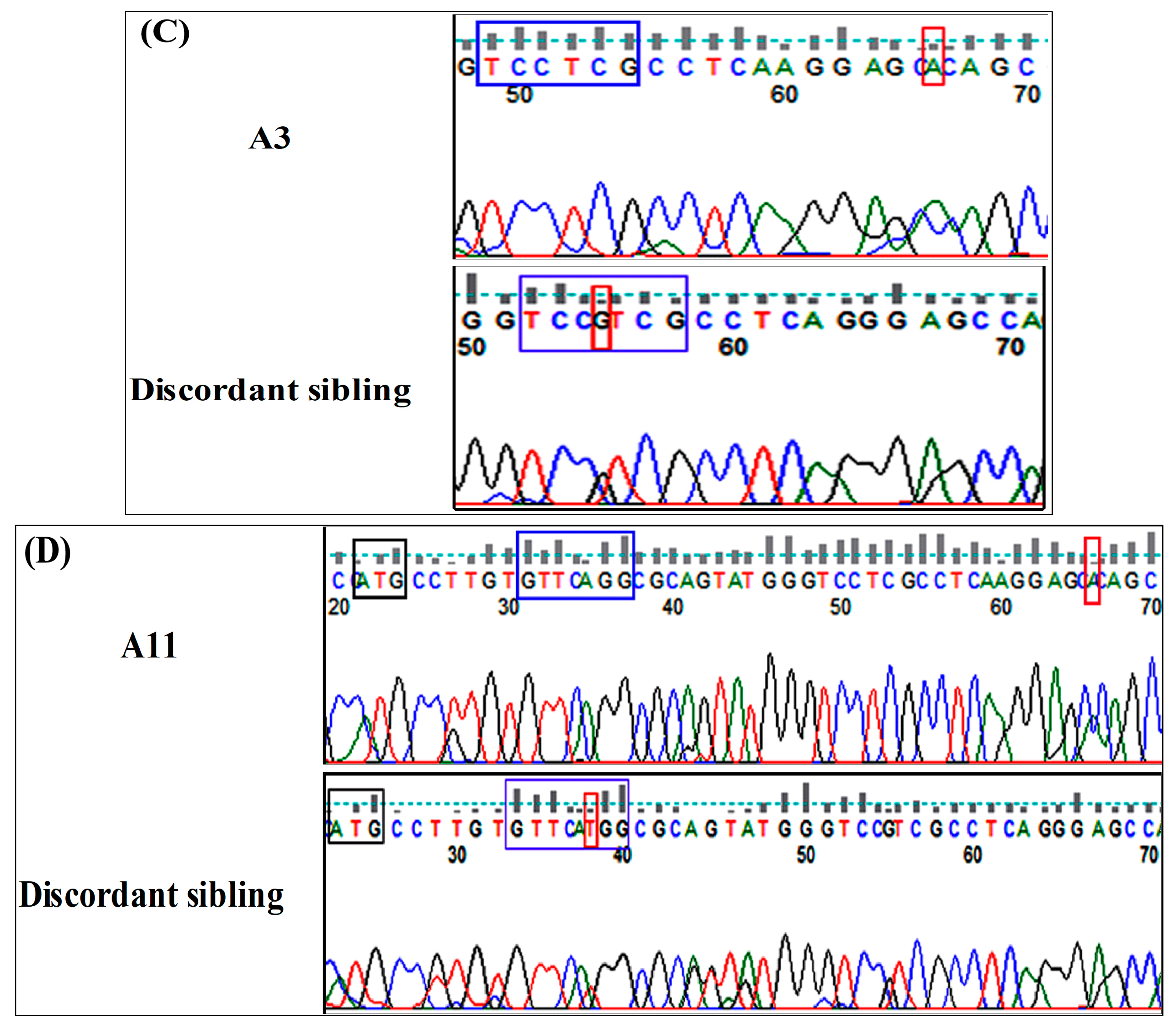
2.3. Molecular Screening of Concordant and Discordant Siblings Reveals Deleterious NR4A2 Variants
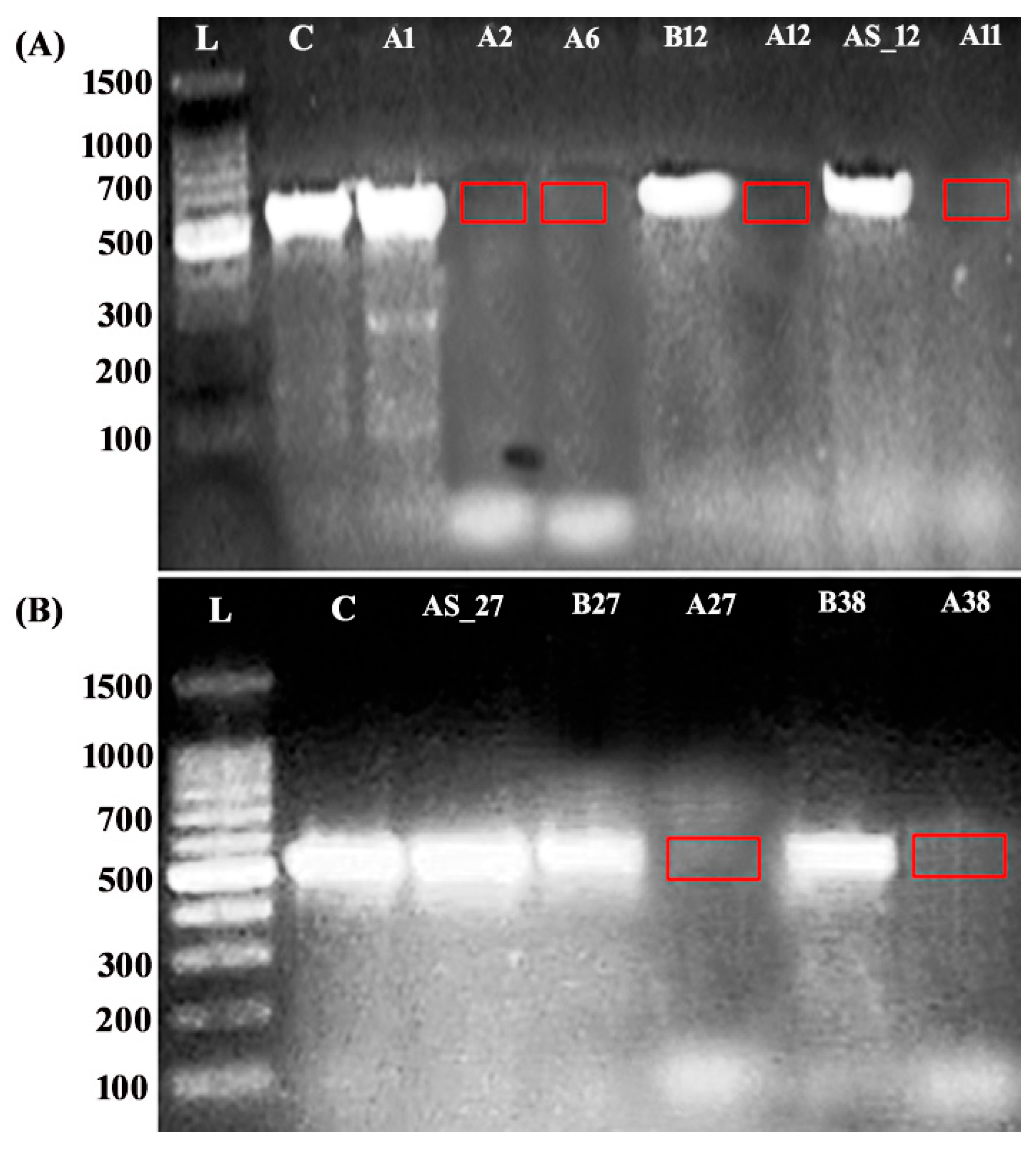
2.4. Functional Consequences of the Identified NR4A2 Variants
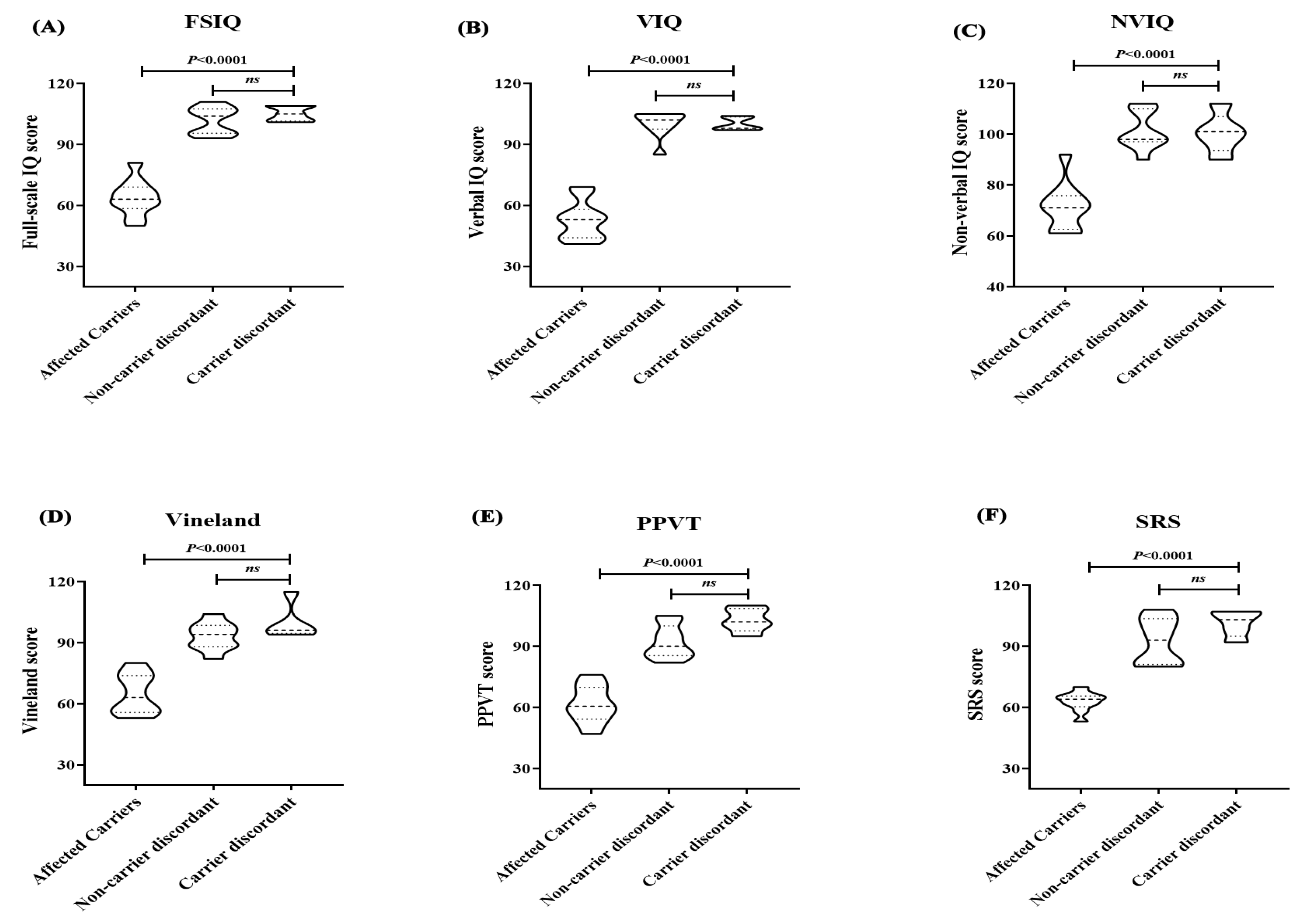
2.5. Clinical Phenotypes and Intellectual Profiles of NR4A2 Subjects
3. Discussion
4. Materials and Methods
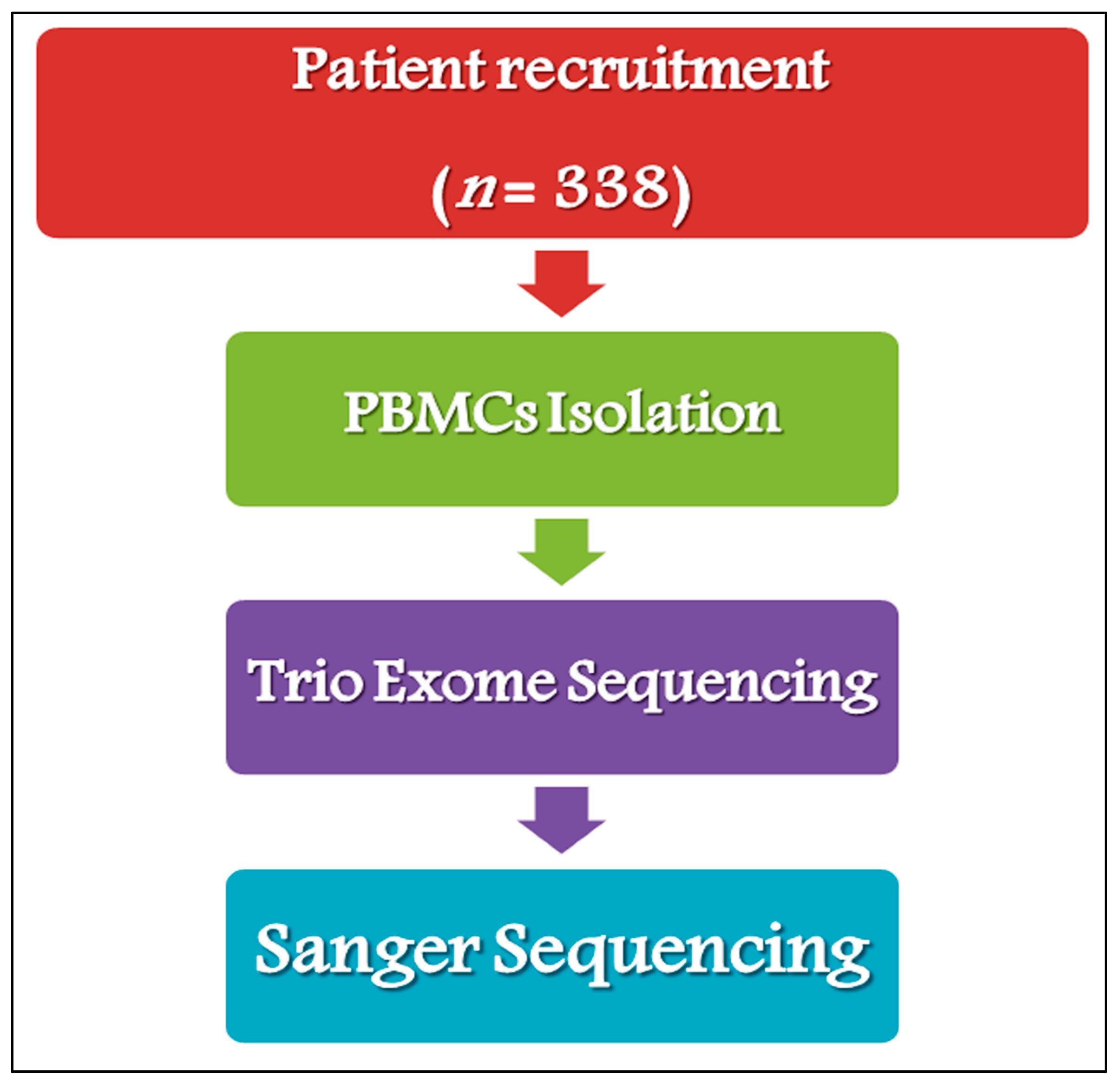
4.1. Study Cohort
4.2. Study Design
4.3. Genetic Analysis and Variant Validation
4.4. Functional Analysis of Splice Region Variants in ASD Patients with Chromosomal Deletions
4.5. Variant Annotation and Classification
4.6. Statistical Analysis
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACMG/AMP | American College of Medical Genetics and Genomics and Association for Molecular Pathology |
| ADHD | Attention Deficit and Hyperactivity Disorder |
| ASD | Autism Spectrum Disorder |
| Bp | Base pair |
| DBD | DNA-binding domain |
| DSM-5 | Diagnostic and Statistical Manual of Mental Disorders (5th edition) |
| EDS | Ehlers–Danlos syndrome |
| ES | Exome sequencing |
| FSIQ | Full-Scale Intelligence Quotient |
| IBD | Inflammatory Bowel Disease |
| ID | Intellectual disability |
| LBD | Ligand binding domain |
| LOF | Loss of function |
| NDD | Neurodevelopmental delay |
| NMD | Nonsense-mediated mRNA decay |
| NR4A2 | Nuclear receptor superfamily 4 group A member 2 |
| NTD | N-terminal domain |
| Nurr1 | Nuclear receptor-related 1 |
| NVIQ | Non-Verbal Intelligence Quotient |
| OCD | Obsessive–Compulsive Disorder |
| PBMCs | Peripheral Blood Mononuclear Cells |
| PCR | Polymerase chain reaction |
| PPVT | Peabody Picture Vocabulary Test |
| RT–PCR | Reverse transcription polymerase chain reaction |
| SB-5 | Stanford–Binet Intelligence Scales (5th edition) |
| SRS | Social Responsiveness Scale |
| VIQ | Verbal Intelligence Quotient |
| WGS | Whole-genome sequencing |
| WHO | World Health Organization |
References
- World Health Organization. WHO Fact Sheet: Autism, November 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/autism-spectrum-disorders (accessed on 16 April 2025).
- Iles, A. Autism Spectrum Disorders. Prim. Care 2021, 48, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Khachadourian, V.; Mahjani, B.; Sandin, S.; Kolevzon, A.; Buxbaum, J.D.; Reichenberg, A.; Janecka, M. Comorbidities in Autism Spectrum Disorder and Their Etiologies. Transl. Psychiatry 2023, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Togher, K.; Jay, S. Disclosing an autism diagnosis: A social identity approach. Autism Res. 2023, 16, 1934–1945. [Google Scholar] [CrossRef]
- Chiurazzi, P.; Kiani, A.K.; Miertus, J.; Paolacci, S.; Barati, S.; Manara, E.; Stuppia, L.; Gurrieri, F.; Bertelli, M. Genetic Analysis of Intellectual Disability and Autism. Acta Biomed. 2020, 91, e2020003. [Google Scholar] [CrossRef]
- Martini, M.I.; Butwicka, A.; Du Rietz, E.; Kanina, A.; Rosenqvist, M.A.; Larsson, H.; Lichtenstein, P.; Taylor, M.J. Age Effects on Autism Heritability and Etiological Stability of Autistic Traits. J. Child Psychol. Psychiatry 2024, 65, 1135–1144. [Google Scholar] [CrossRef]
- Genovese, A.; Butler, M.G. Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD). Int. J. Mol. Sci. 2020, 2, 4726. [Google Scholar] [CrossRef]
- Iannuccelli, M.; Vitriolo, A.; Licata, L.; Lo Surdo, P.; Contino, S.; Cheroni, C.; Capocefalo, D.; Castagnoli, L.; Testa, G.; Cesareni, G.; et al. Curation of Causal Interactions Mediated by Genes Associated With Autism Accelerates the Understanding of Gene-Phenotype Relationships Underlying Neurodevelopmental Disorders. Mol. Psychiatry 2024, 29, 197. [Google Scholar] [CrossRef]
- Verhelst, H.; Verloo, P.; Dheedene, A.; Callewaert, B.; Menten, B. NR4A2 Causes an Autism Spectrum Disorder. Eur. J. Paediatr. Neurol. 2017, 21, e49. [Google Scholar] [CrossRef]
- Ramos, L.L.P.; Monteiro, F.P.; Sampaio, L.P.B.; Costa, L.A.; Ribeiro, M.D.O.; Freitas, E.L.; Kitajima, J.P.; Kok, F. Heterozygous Loss of Function of NR4A2 is Associated With Intellectual Deficiency, Rolandic Epilepsy, and Language Impairment. Clin. Case Rep. 2019, 7, 1582–1584. [Google Scholar] [CrossRef]
- Wirth, T.; Mariani, L.L.; Bergant, G.; Baulac, M.; Habert, M.O.; Drouot, N.; Ollivier, E.; Hodžić, A.; Rudolf, G.; Nitschke, P.; et al. Loss-of-Function Mutations in NR4A2 Cause Dopa-Responsive Dystonia Parkinsonism. Mov. Disord. 2020, 35, 880–885. [Google Scholar] [CrossRef]
- Singh, S.; Gupta, A.; Zech, M.; Sigafoos, A.N.; Clark, K.J.; Dincer, Y.; Wagner, M.; Humberson, J.B.; Green, S.; van Gassen, K.; et al. De Novo Variants of NR4A2 are Associated with Neurodevelopmental Disorder and Epilepsy. Genet. Med. 2020, 22, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xu, W.; Xiao, M.; Lu, Y.; Lan, X.; Tang, X.; Xu, N.; Yu, G.; Zhang, H.; Wu, S. Two Novel Heterozygous Truncating Variants in NR4A2 Identified in Patients with Neurodevelopmental Disorder and Brief Literature Review. Front. Neurosci. 2022, 16, 956429. [Google Scholar] [CrossRef]
- Yu, X.; Shang, J.; Kojetin, D.J. Molecular Basis of Ligand-Dependent Nurr1-RXRα activation. eLife 2023, 12, e85039. [Google Scholar] [CrossRef]
- Català-Solsona, J.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J. Nr4a2 Transcription Factor in Hippocampal Synaptic Plasticity, Memory and Cognitive Dysfunction: A Perspective Review. Front. Mol. Neurosci. 2021, 14, 786226. [Google Scholar] [CrossRef] [PubMed]
- García-Yagüe, Á.J.; Cuadrado, A. Mechanisms of NURR1 Regulation: Consequences for Its Biological Activity and Involvement in Pathology. Int. J. Mol. Sci. 2023, 24, 12280. [Google Scholar] [CrossRef]
- Delobel-Ayoub, M.; Saemundsen, E.; Gissler, M.; Ego, A.; Moilanen, I.; Ebeling, H.; Rafnsson, V.; Klapouszczak, D.; Thorsteinsson, E.; Arnaldsdóttir, K.M.; et al. Prevalence of Autism Spectrum Disorder in 7-9-Year-Old Children in Denmark, Finland, France and Iceland: A Population-Based Registries Approach Within the ASDEU Project. J. Autism Dev. Disord. 2020, 50, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Vaez, M.; Montalbano, S.; Calle Sánchez, X.; Georgii Hellberg, K.L.; Dehkordi, S.R.; Krebs, M.D.; Meijsen, J.; Shorter, J.; Byberg-Grauholm, J.; Mortensen, P.B.; et al. Population-Based Risk of Psychiatric Disorders Associated With Recurrent Copy Number Variants. JAMA Psychiatry 2024, 81, e241453. [Google Scholar] [CrossRef]
- Al-Sarraj, Y.; Taha, R.Z.; Al-Dous, E.; Ahram, D.; Abbasi, S.; Abuazab, E.; Shaath, H.; Habbab, W.; Errafii, K.; Bejaoui, Y.; et al. The Genetic Landscape of Autism Spectrum Disorder in the Middle Eastern Population. Front. Genet. 2024, 15, 1363849. [Google Scholar] [CrossRef]
- AlBatti, T.H.; Alsaghan, L.B.; Alsharif, M.F.; Alharbi, J.S.; BinOmair, A.I.; Alghurair, H.A.; Aleissa, G.A.; Bashiri, F.A. Prevalence of Autism Spectrum Disorder Among Saudi Children Between 2 and 4 Years Old in Riyadh. Asian J. Psychiatr. 2022, 71, 103054. [Google Scholar] [CrossRef]
- Lévy, J.; Grotto, S.; Mignot, C.; Maruani, A.; Delahaye-Duriez, A.; Benzacken, B.; Keren, B.; Haye, D.; Xavier, J.; Heulin, M.; et al. NR4A2 Haploinsufficiency is Associated with Intellectual Disability and Autism Spectrum Disorder. Clin. Genet. 2018, 94, 264–268. [Google Scholar] [CrossRef]
- Gabaldon-Albero, A.; Mayo, S.; Martinez, F. NR4A2 as a Novel Target Gene for Developmental and Epileptic Encephalopathy: A Systematic Review of Related Disorders and Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 5198. [Google Scholar] [CrossRef] [PubMed]
- Elsabbagh, M.; Divan, G.; Koh, Y.J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.D.; Marrus, N.; Maloney, S.E.; Yip, B.; Sandin, S.; Turner, T.N.; Selmanovic, D.; Kroll, K.L.; Gutmann, D.H.; Constantino, J.N.; et al. Can the “female protective effect” liability threshold model explain sex differences in autism spectrum disorder? Neuron 2022, 110, 3243–3262. [Google Scholar] [CrossRef] [PubMed]
- Grondhuis, S.N.; Lecavalier, L.; Arnold, L.E.; Handen, B.L.; Scahill, L.; McDougle, C.J.; Aman, M.G. Differences in verbal and nonverbal IQ test scores in children with autism spectrum disorder. Res. Autism Spectr. Disord. 2018, 49, 47–55. [Google Scholar] [CrossRef]
- Al-Mamari, W.; Idris, A.B.; Gabr, A.; Jalees, S.; Al-Jabri, M.; Abdulrahim, R.; Al-Mujaini, A.; Islam, M.M.; Al-Alawi, M.; Al-Adawi, S. Intellectual profile of children with autism spectrum disorder: Identification of verbal and nonverbal subscales predicting intelligence quotient. Sultan Qaboos Univ. Med. J. 2021, 21, 386–393. [Google Scholar] [CrossRef]
- Zyoud, S.H.; Shakhshir, M.; Abushanab, A.S.; Koni, A.; Shahwan, M.; Jairoun, A.A.; Abu Taha, A.; Al-Jabi, S.W. Gut microbiota and autism spectrum disorders: Where do we stand? Gut Pathog. 2023, 15, 50. [Google Scholar] [CrossRef]
- Sotelo-Orozco, J.; Hertz-Picciotto, I. The Association between gastrointestinal issues and psychometric scores in children with autism spectrum disorder, developmental delays, Down syndrome, and typical development. J. Autism Dev. Disord. 2024. [Google Scholar] [CrossRef]
- Yousefi, Y.; Baines, K.J.; Maleki Vareki, S. Microbiome bacterial influencers of host immunity and response to immunotherapy. Cell Rep. Med. 2024, 5, 101487. [Google Scholar] [CrossRef]
- Ullrich, K.A.; Schulze, L.L.; Paap, E.M.; Müller, T.M.; Neurath, M.F.; Zundler, S. Immunology of IL-12: An update on functional activities and implications for disease. EXCLI J. 2020, 19, 1563–1589. [Google Scholar] [CrossRef]
- Loureiro, L.O.; Howe, J.L.; Reuter, M.S.; Iaboni, A.; Calli, K.; Roshandel, D.; Pritišanac, I.; Moses, A.; Forman-Kay, J.D.; Trost, B.; et al. A Recurrent SHANK3 Frameshift Variant in Autism Spectrum Disorder. NPJ Genom. Med. 2021, 6, 91. [Google Scholar] [CrossRef]
- Kissel, L.T.; Pochareddy, S.; An, J.Y.; Sestan, N.; Sanders, S.J.; Wang, X.; Werling, D.M. Sex-Differential Gene Expression in Developing Human Cortex and Its Intersection With Autism Risk Pathways. Biol. Psychiatry Glob. Open Sci. 2024, 4, 100321. [Google Scholar] [CrossRef]
- Lu, X.; Song, Y.; Wang, J.; Cai, Y.; Peng, S.; Lin, J.; Lai, B.; Sun, J.; Liu, T.; Chen, G.; et al. Developmental Dopaminergic Signaling Modulates Neural Circuit Formation and Contributes to Autism Spectrum Disorder-Related Phenotypes. Am. J. Pathol. 2024, 194, 1062–1077. [Google Scholar] [CrossRef] [PubMed]
- Leppa, V.M.; Kravitz, S.N.; Martin, C.L.; Andrieux, J.; Le Caignec, C.; Martin-Coignard, D.; DyBuncio, C.; Sanders, S.J.; Lowe, J.K.; Cantor, R.M.; et al. Rare Inherited and De Novo CNVs Reveal Complex Contributions to ASD Risk in Multiplex Families. Am. J. Hum. Genet. 2016, 99, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, E.K.; Pérez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866. [Google Scholar] [CrossRef] [PubMed]
- Ozonoff, S.; Young, G.S.; Bradshaw, J.; Charman, T.; Chawarska, K.; Iverson, J.M.; Klaiman, C.; Landa, R.J.; McDonald, N.; Messinger, D.; et al. Familial Recurrence of Autism: Updates from the Baby Siblings Research Consortium. Pediatrics 2024, 154, e2023065297. [Google Scholar] [CrossRef] [PubMed]
- D’Gama, A.M. Somatic Mosaicism and Autism Spectrum Disorder. Genes 2021, 12, 1699. [Google Scholar] [CrossRef]
- Ye, K.; Iossifov, I.; Levy, D.; Yamrom, B.; Buja, A.; Krieger, A.M.; Wigler, M. Measuring Shared Variants in Cohorts of Discordant Siblings with Applications to Autism. Proc. Natl. Acad. Sci. USA 2017, 114, 7073–7076. [Google Scholar] [CrossRef]
- Antaki, D.; Guevara, J.; Maihofer, A.X.; Klein, M.; Gujral, M.; Grove, J.; Carey, C.E.; Hong, O.; Arranz, M.J.; Hervas, A.; et al. A Phenotypic Spectrum of Autism is Attributable to the Combined Effects of Rare Variants, Polygenic Risk and Sex. Nat. Genet. 2022, 54, 1284–1292. [Google Scholar] [CrossRef]
- Al-Mamari, W.; Idris, A.B.; Al-Thihli, K.; Abdulrahim, R.; Jalees, S.; Al-Jabri, M.; Gabr, A.; Al Murshedi, F.; Al Kindy, A.; Al-Hadabi, I.; et al. Applying Whole Exome Sequencing in A Consanguineous Population with Autism Spectrum Disorder. Int. J. Dev. Disabil. 2021, 69, 190–200. [Google Scholar] [CrossRef]
- Bordoni, L.; Petracci, I.; Calleja-Agius, J.; Lalor, J.G.; Gabbianelli, R. NURR1 Alterations in Perinatal Stress: A First Step Towards Late-Onset Diseases? A Narrative Review. Biomedicines 2020, 8, 584. [Google Scholar] [CrossRef]
- Di Gesù, C.M.; Buffington, S.A. The Early Life Exposome and Autism Risk: A Role for the Maternal Microbiome? Gut Microbes 2024, 16, 2385117. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Gilman, S.R.; Chiang, A.H.; Sanders, S.J.; Vitkup, D. Genotype to Phenotype Relationships in Autism Spectrum Disorders. Nat. Neurosci. 2015, 18, 191–198. [Google Scholar] [CrossRef]
- Mordaunt, C.E.; Jianu, J.M.; Laufer, B.I.; Zhu, Y.; Hwang, H.; Dunaway, K.W.; Bakulski, K.M.; Feinberg, J.I.; Volk, H.E.; Lyall, K.; et al. Cord Blood DNA Methylome in Newborns Later Diagnosed with Autism Spectrum Disorder Reflects Early Dysregulation of Neurodevelopmental and X-Linked Genes. Genome Med. 2020, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Kausel, L.; Michon, M.; Soto-Icaza, P.; Aboitiz, F. A Multimodal Interface for Speech Perception: The Role of the Left Superior Temporal Sulcus in Social Cognition and Autism. Cereb. Cortex 2024, 34, 84–93. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Sun, Q.; Tan, Y.Y.; Hu, Y.Y.; Zhan, W.W.; Li, D.H.; Wang, Y.; Xiao, Q.; Liu, J.; Chen, S.D. Substantia Nigra Echogenicity Correlated with Clinical Features of Parkinson’s Disease. Park. Relat. Disord. 2016, 24, 28–33. [Google Scholar] [CrossRef]
- Han, Y.F.; Cao, G.W. Role of Nuclear Receptor NR4A2 in Gastrointestinal Inflammation and Cancers. World J. Gastroenterol. 2012, 18, 6865–6873. [Google Scholar] [CrossRef]
- Li, R.; Chen, S.; Gu, X.; An, S.; Wang, Z. Role of the Nuclear Receptor Subfamily 4A in Mast Cells in the Development of Irritable Bowel Syndrome. Comput. Struct. Biotechnol. J. 2022, 20, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Montarolo, F.; Martire, S.; Chiara, F.; Allegra, S.; De Francia, S.; Hoxha, E.; Tempia, F.; Capobianco, M.A.; Bertolotto, A. NURR1-Deficient Mice Have Age- and Sex-Specific Behavioral Phenotypes. J. Neurosci. Res. 2022, 100, 1747–1754. [Google Scholar] [CrossRef]
- Hodges, H.; Fealko, C.; Soares, N. Autism Spectrum Disorder: Definition, Epidemiology, Causes, and Clinical Evaluation. Transl. Pediatr. 2020, 9, S55–S65. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5*), 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013; Available online: https://psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596 (accessed on 3 June 2025).
- Grondhuis, S.N.; Mulick, J.A. Comparison of the Leiter International Performance Scale-Revised and the Stanford-Binet Intelligence Scales, 5th Edition, in Children with Autism Spectrum Disorders. Am. J. Intellect. Dev. Disabil. 2013, 118, 44–54. [Google Scholar] [CrossRef]
- Constantino, J.N. Social Responsiveness Scale. In Encyclopedia of Autism Spectrum Disorders; Springer International Publishing: Cham, Switzerland, 2021; pp. 4457–4467. [Google Scholar]
- Kover, S.T.; McDuffie, A.S.; Hagerman, R.J.; Abbeduto, L. Receptive Vocabulary in Boys with Autism Spectrum Disorder: Cross-Sectional Developmental Trajectories. J. Autism Dev. Disord. 2013, 43, 2696–2709. [Google Scholar] [CrossRef] [PubMed]
- Farmer, C.; Adedipe, D.; Bal, V.H.; Chlebowski, C.; Thurm, A. Concordance of the Vineland Adaptive Behavior Scales, Second and Third Editions. J. Intellect. Disabil. Res. 2020, 64, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Xiaozhen, S.; Fan, Y.; Fang, Y.; Xiaoping, L.; Jia, J.; Wuhen, X.; Xiaojun, T.; Jun, S.; Yucai, C.; Hong, Z.; et al. Novel Truncating and Missense Variants in SEMA6B in Patients with Early-Onset Epilepsy. Front. Cell Dev. Biol. 2021, 9, 633819. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, Y.; He, H.; Shen, J. Clinical Interpretation of Sequence Variants. Curr. Protoc. Hum. Genet. 2020, 106, e98. [Google Scholar] [CrossRef]
- Eapen, V.; Mabrouk, A.A.; Zoubeidi, T.; Yunis, F. Prevalence of Pervasive Developmental Disorders in Preschool Children in the UAE. J. Trop. Pediatr. 2007, 53, 202–205. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | |
| Gender [n, %] | |
| Male | 244 (72.19%) |
| Female | 94 (27.81%) |
| Age (Years) [n, %] | |
| 3–5 | 74 (21.89%) |
| 6–9 | 125 (36.98%) |
| 10–14 | 139 (41.13%) |
| Family history | |
| Simplex | 293 (93.01%) |
| Quad | 21 (6.66%) |
| Quintet | 1 (0.317%) |
| Total | 315 (100%) |
| FSIQ classification [n, %] | |
| Average and above IQ (90–129) | 33 (9.76%) |
| Low average IQ (80–89) | 59 (17.46%) |
| Borderline IQ (70–79) | 74 (21.89%) |
| Mildly impaired IQ (55–69) | 84 (24.85%) |
| Moderately impaired IQ (40–54) | 72 (21.3%) |
| Severely impaired IQ (>35) | 16 (4.74%) |
| IQ subscales scoring | |
| FSIQ score | 84 (68.5–100.5) * |
| VIQ score | 81.5 (63–101.5) |
| NVIQ score | 91 (75–104) |
| PPVT score | 77 (67.5–88.5) |
| SRS score | 77 (67–90) |
| Vineland score (AE) | |
| <4.5 Years | 102 (97–112) |
| >4.5 Years | 69 (57–83) |
| Clinical Phenotypes (current) [n, %] | |
| Gastrointestinal disorders | 182 (76.47%) |
| Sleeping disorders | 165 (48.82%) |
| Language/speech impairment | 160 (47.34%) |
| Motor difficulties | 148 (43.79%) |
| Feeding difficulties | 136 (40.23%) |
| Short stature | 77 (22.78%) |
| Seizures/epilepsy | 35 (10.35%) |
| Autoimmune diseases | 33 (9.76%) |
| Facial dimorphism | 17 (5.03%) |
| Psychiatric/behavioral problems (current) [n, %] | |
| Hyperactivity/ADHD | 185 (54.74%) |
| Anxiety | 143 (42.3%) |
| Sensory processing difficulties | 123 (36.39%) |
| Learning difficulties | 115 (34.02%) |
| Attachment disorder | 97 (28.7%) |
| OCD | 33 (13.87%) |
| Aggressive behavior | 15 (4.43%) |
|
Proband
(Biosample Accession) | Age (Y)/Sex | Variant (NM_006186.4) | Clingen Identifier/ClinVar Accession | Protein Alteration |
SNV/Indel/
CNV | Protein Domain/Region | ACMG Classification/Evidences | Clinical Phenotypes/Behavioral Problems | Seizures? |
|---|---|---|---|---|---|---|---|---|---|
| A1 (SAMN43045798) | 6/M | c.-2-2del | VCV003338047.1 | Splicing change | Splice region variant | Intron 2 | Pathogenic (12 ACMG Points: 12P and 0B. PVS, PM2, PP3 (moderate)) | Mild ID, absent speech, mild ataxia, stereotypical body rocking, irregular sleeping, social phobia, attachment disorder, hypersensitivity to external stimuli | No |
| c:1del | CA2837995541/VCV003338025.1 | p.(Met1del) | Indel | NTD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | ||||
| c.548del | CA645529278, VCV003338477.1 | p.(Pro183Leufs*20) | |||||||
| A2 (SAMN43045230) | 11/M | c.44_45insA | CA2837995542, VCV003338013.1 | p.(Ser16Glnfs*28) | Indel | NTD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | Moderate ID, significant language impairment, Rolandic epilepsy, feeding problems, sleeping problems, choreoathetotic movements, incontinence, hyperactivity, learning difficulties | Yes |
| c.1159-81_1540+67del | CA2838010100, VCV003338024.1 | p.(Phe387Argfs*19)/Splicing change | CNV | LBD | |||||
| A3 (SAMN43045749) | 11/M | c.44_45insA | CA2837995542, VCV003338013.1 | p.(Ser16Glnfs*28) | Indel | NTD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | Mild ID, delayed speech and motor development, mild ataxia, dystonia, chronic dyspepsia and constipation, hyperactivity, anxiety | No |
| c.536del | CA2837995544, VCV003338030.1 | p.(Lys179Serfs*24) | |||||||
| A6 (SAMN43046049) | 4/F | c.44_45insA | CA2837995542, VCV003338013.1 | p.(Ser16Glnfs*28) | Indel | NTD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | Mild ID, speech dyslexia, delayed walking, mild hypotonia, chronic diarrhea, hyposensitivity to temperature and pain, attachment disorders | No |
| c.1159-81_1540+67del | CA2838010100, VCV003338024.1 | p.(Phe387Argfs*19)/Splicing change | CNV | LBD | |||||
| A11 (SAMN43045260) | 7/M | c.44_45insA | CA2837995542, VCV003338013.1 | p.(Ser16Glnfs*28) | indel | NTD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | Mild to moderate ID, speech and motor delay, learning difficulties, generalized hypotonia, frequent diarrhea, hypermobile EDS, skin hyperextensibility, mild scoliosis, sleeping problems | Infantile spasms |
| c.1159-81_1540+67del | CA2838010100, VCV003338024.1 | p.(Phe387Argfs*19)/Splicing change | CNV | LBD | |||||
| A12 (SAMN43046050) | 10/F | c.5_6delCTinsTG | CA2837995543, VCV003338026.1 | p.(Pro2Leu) | SNV | NTD | VUS (3 ACMG points: 3P and 0B. PM2, PP3). SIFT score = 0 (deleterious), PolyPhen score = 0.929 (probably damaging) | Moderate ID, prominent speech impairment, supported walking, overall poor growth, progressive hypotonia, short stature, facial dimorphism, IBD, sleeping disorders | Lennox-Gastaut syndrome |
| c.534del | CA2837995545, VCV003338016.1 | p.(Phe178Leufs*25) | Indel | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | |||||
| c.1159-81_1540+67del | CA2838010100, VCV003338024.1 | p.(Phe387Argfs*19)/Splicing change | CNV | LBD | |||||
| AS_12 (SAMN43045274) | 9/M | c.-2-8del | VCV003338474.1 | Non coding | Splice polypyrimidine tract variant | Intron 2 | VUS/Likely Benign (-1 ACMG points: 1P and 2B. BP4, PM2) | Mild ID, speech apraxia, delayed echolalia, choreoathetotic movements, supported walking, IBD, incontinence, anxiety, irregular sleeping | No |
| c:1del | CA2837995541/VCV003338025.1 | p.(Met1del) | Indel | NTD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | ||||
| A27 (SAMN43045261) | 8/M | c.44_45insA | CA2837995542, VCV003338013.1 | p.(Ser16Glnfs*28) | Indel | NTD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | Mild ID, moderate speech impairment, disproportionate motor development, generalized hypotonia, ataxic gait, anxiety, attachment disorders | No |
| c.537G>C | CA348682205, VCV003338475.1 | p.(Lys179Asn) | SNV | VUS (2 ACMG points: 2P and 0B. PM2, PP2). TraP score = 0.338 | |||||
| c.1159-81_1540+67del | CA2838010100, VCV003338024.1 | p.(Phe387Argfs*19)/Splicing change | CNV | LBD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | ||||
| AS_27 (SAMN43046045) | 5/M | c:1del | CA2837995541/VCV003338025.1 | p.(Met1del) | Indel | NTD | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | Mild ID, receptive-expressive language delay, motor delay, mild infantile hypotonia, motor tics, IBD, anxiety, hyperactivity | No |
| c.44_45insA | CA2837995542, VCV003338013.1 | p.(Ser16Glnfs*28) | |||||||
| A38 (SAMN43045262) | 10/M | c.39A>G | CA429727752 | p.(Gln13=) | Synonymous | NTD | Likely benign (3 ACMG points: 2P and 5B. PM2, PB4, BP7) | Mild to moderate ID, significant language impairment, ataxic gait, dystonia, sleeping disorders, self injury, aggressive behavior | Febrile |
| c.44_45insA | CA2837995542, VCV003338013.1 | p.(Ser16Glnfs*28) | Indel | Pathogenic (10 ACMG Points: 10P and 0B. PVS1, PM2) | |||||
| c.1159-81_1540+67del | CA2838010100, VCV003338024.1 | p.(Phe387Argfs*19)/Splicing change | CNV | LBD |
| Patient (Biosample Accession) | Age (Y)/Sex | Variant (NM_006186.4) | Clingen Identifier/ClinVar Accession | Protein Alteration | SNV/Indel | Protein Domain/Region | ACMG Classification | ACMG Evidences |
|---|---|---|---|---|---|---|---|---|
| B3_2 (SAMN43942449) | 6/F | c.30_31insG | CA2838140773/ VCV003338478.1 | p.(Ser11Valfs*33) | Indel | NTD | Pathogenic | 10 ACMG Points: 10P and 0B. PVS1, PM2 (moderate) |
| B11_2 (SAMN43942450) | 3/F | c.14_15insT | CA2838140772/ VCV003338476.1 | p.(Gln5Hisfs*39) | Indel | Pathogenic | 10 ACMG Points: 10P and 0B. PVS1, PM2 (moderate) | |
| B12 (SAMN43942448) | 7/M | c.536_537delCTinsGC | PA2838140774/VCV003338031.1 | p.(Lys179Ser) | SNV | VUS | 2 ACMG points: 2P and 0B. PM2 (moderate) | |
| B27 (SAMN43370295) | 4/F | c.5_6delCTinsTG | CA2837995543, VCV003338026.1 | p.(Pro2Leu) | SNV | VUS | 3 ACMG points: 3P and 0B. PM2, PP3). SIFT score = 0 (deleterious), PolyPhen score = 0.929 (probably damaging) | |
| c.44_45insA | CA2837995542/VCV003338013.1 | p.(Ser16Glnfs*28) | Indel | Pathogenic | 10 ACMG Points: 10P and 0B. PVS1, PM2 (moderate) | |||
| B38 (SAMN43370296) | 6/F | c.39A>G | CA429727752 | p.(Gln13=) | Synonymous | Likely benign | 3 ACMG points: 2P and 5B. PM2 (Supporting), PB4 (strong), BP7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alharbi, N.M.; Baaboud, W.F.; Shawky, H.; Alrofaidi, A.A.; Farsi, R.M.; Algothmi, K.M.; Hassoubah, S.A.; Basingab, F.S.; Azhari, S.A.; Alharbi, M.G.; et al. Molecular Screening Reveals De Novo Loss-of-Function NR4A2 Variants in Saudi Children with Autism Spectrum Disorders: A Single-Center Study. Int. J. Mol. Sci. 2025, 26, 5468. https://doi.org/10.3390/ijms26125468
Alharbi NM, Baaboud WF, Shawky H, Alrofaidi AA, Farsi RM, Algothmi KM, Hassoubah SA, Basingab FS, Azhari SA, Alharbi MG, et al. Molecular Screening Reveals De Novo Loss-of-Function NR4A2 Variants in Saudi Children with Autism Spectrum Disorders: A Single-Center Study. International Journal of Molecular Sciences. 2025; 26(12):5468. https://doi.org/10.3390/ijms26125468
Chicago/Turabian StyleAlharbi, Najwa M., Wejdan F. Baaboud, Heba Shawky, Aisha A. Alrofaidi, Reem M. Farsi, Khloud M. Algothmi, Shahira A. Hassoubah, Fatemah S. Basingab, Sheren A. Azhari, Mona G. Alharbi, and et al. 2025. "Molecular Screening Reveals De Novo Loss-of-Function NR4A2 Variants in Saudi Children with Autism Spectrum Disorders: A Single-Center Study" International Journal of Molecular Sciences 26, no. 12: 5468. https://doi.org/10.3390/ijms26125468
APA StyleAlharbi, N. M., Baaboud, W. F., Shawky, H., Alrofaidi, A. A., Farsi, R. M., Algothmi, K. M., Hassoubah, S. A., Basingab, F. S., Azhari, S. A., Alharbi, M. G., Yahya, R., & Alhazmi, S. (2025). Molecular Screening Reveals De Novo Loss-of-Function NR4A2 Variants in Saudi Children with Autism Spectrum Disorders: A Single-Center Study. International Journal of Molecular Sciences, 26(12), 5468. https://doi.org/10.3390/ijms26125468