
Proteomic Analysis of ARID1A-Deficient Ovarian Clear Cell Carcinoma Cells Reveals Differential Mitochondria ETC Subunit Abundances and Targetable Mitochondrial Pathways
 , , , ,
, , , , 

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Generation of ARID1A Knockout (KO) Clones
2.3. Cell Doubling Time
2.4. Mitochondrial Labeling and Quantification of Mitochondrial Membrane Potential
2.5. Cell Viability Assay
2.6. Western Blot
2.7. Protein Extraction and Enzymatic Digestion for Proteomic Analysis
2.8. Basic Reversed-Phase Liquid Chromatography
2.9. TMT Labeling
2.10. LC-MS/MS Data Acquisition
2.11. Mass Spectrometry Data Analysis
2.12. Bioinformatic Analysis of Proteomic Data
3. Results
3.1. Generation and Characterization of ARID1A Knockout (KO) Clones for Proteomic Analysis
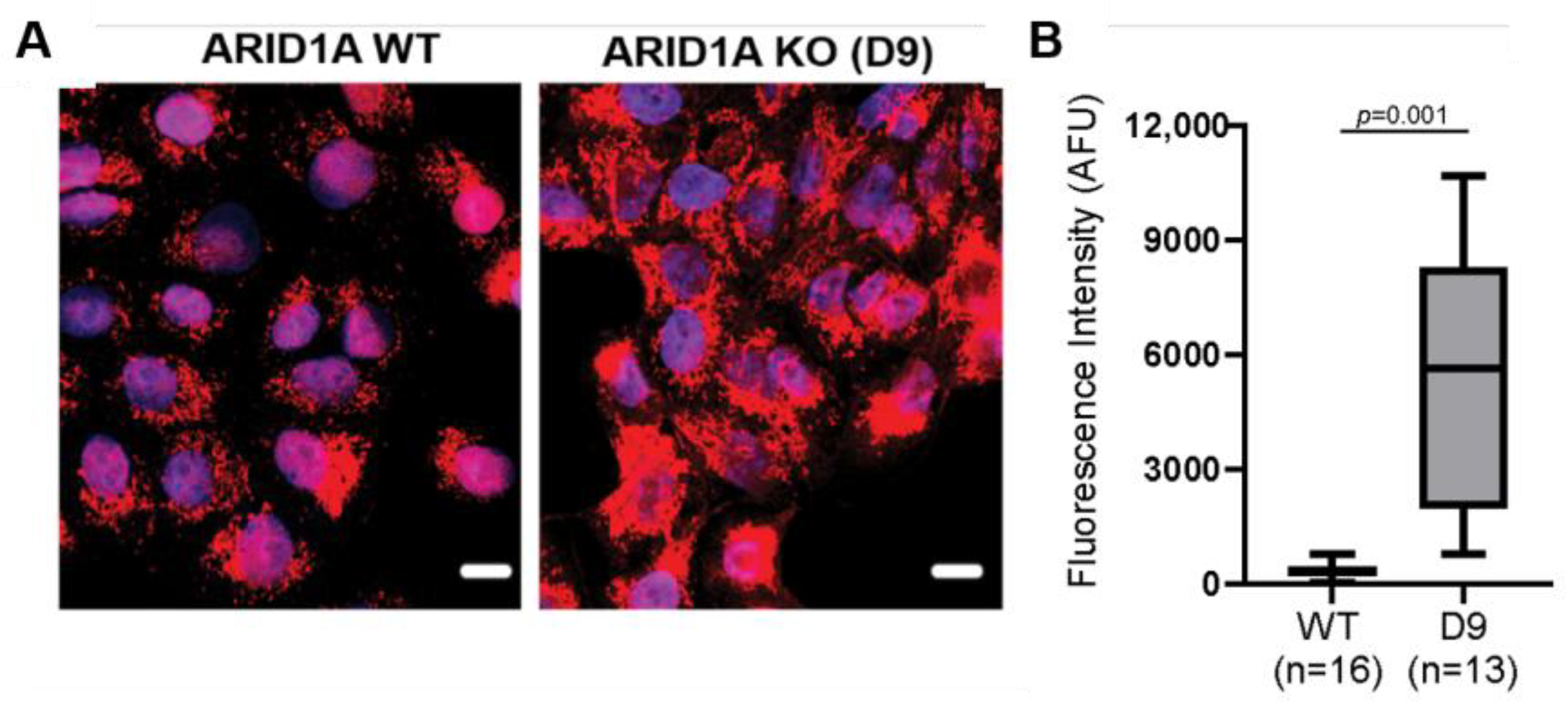
3.2. Global OCCC Proteome Profile of Loss of ARID1A Expression Reveals an Increased Relative Abundance of Mitochondrial Proteins
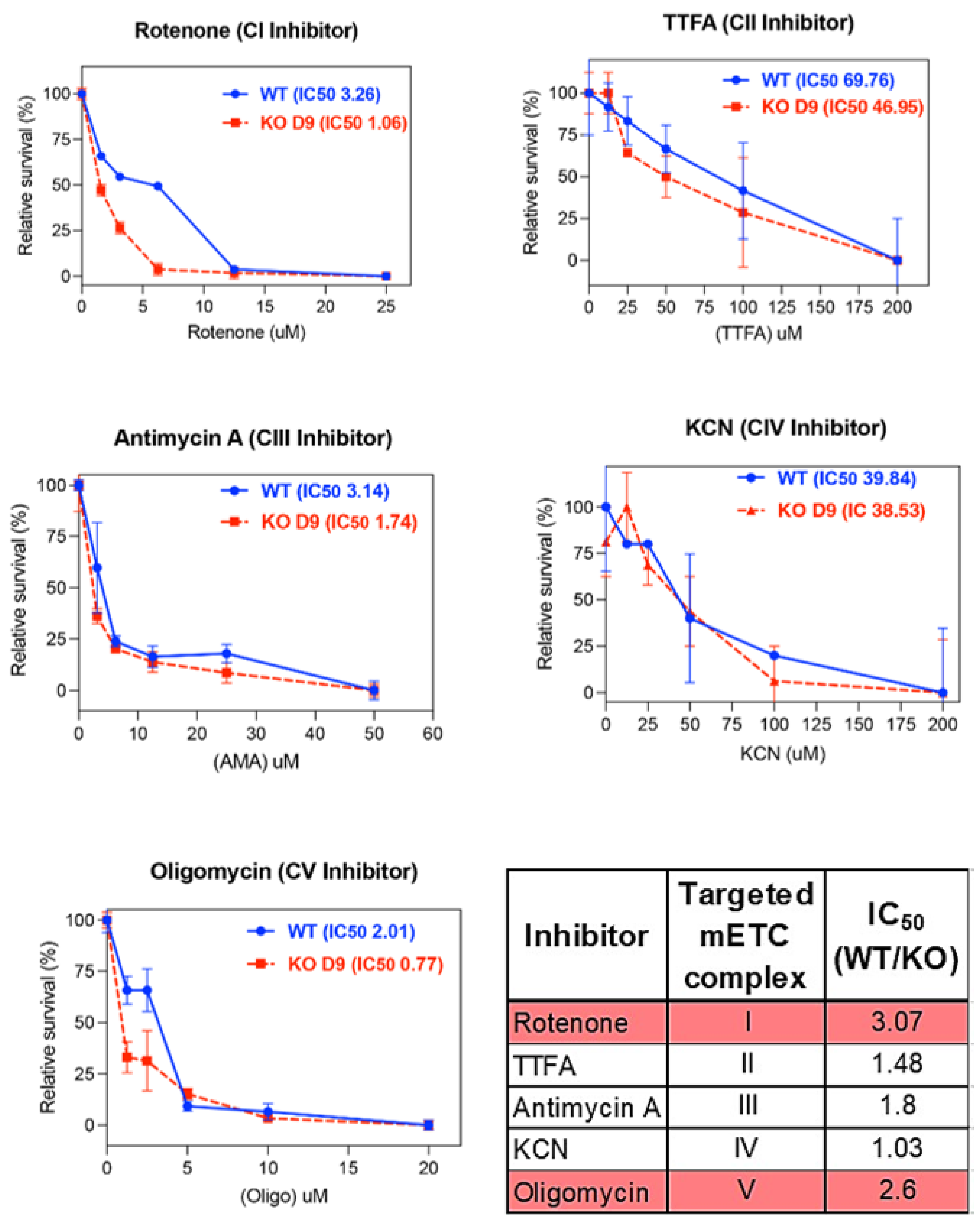
3.3. ARID1A KO Results in Increased Sensitivity to Complex I and Complex V Mitochondrial Inhibitors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Berns, K.; Caumanns, J.J.; Hijmans, E.M.; Gennissen, A.M.C.; Severson, T.M.; Evers, B.; Wisman, G.B.A.; Jan Meersma, G.; Lieftink, C.; Beijersbergen, R.L.; et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene 2018, 37, 4611–4625. [Google Scholar] [CrossRef] [PubMed]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.J.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, A.; Nakayama, K.; Rahman, M.T.; Rahman, M.; Katagiri, H.; Nakayama, N.; Ishikawa, M.; Ishibashi, T.; Iida, K.; Kobayashi, H.; et al. Loss of ARID1A expression is related to shorter progression-free survival and chemoresistance in ovarian clear cell carcinoma. Mod. Pathol. 2012, 25, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef]
- Zhang, X.; Shetty, M.; Clemente, V.; Linder, S.; Bazzaro, M. Targeting Mitochondrial Metabolism in Clear Cell Carcinoma of the Ovaries. Int. J. Mol. Sci. 2021, 22, 4750. [Google Scholar] [CrossRef]
- Emmings, E.; Mullany, S.; Chang, Z.; Landen, C.N., Jr.; Linder, S.; Bazzaro, M. Targeting Mitochondria for Treatment of Chemoresistant Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 229. [Google Scholar] [CrossRef]
- Lissanu Deribe, Y.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.A.; Khor, T.; Feng, N.; et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef]
- Dier, U.; Shin, D.H.; Hemachandra, L.P.; Uusitalo, L.M.; Hempel, N. Bioenergetic analysis of ovarian cancer cell lines: Profiling of histological subtypes and identification of a mitochondria-defective cell line. PLoS ONE 2014, 9, e98479. [Google Scholar] [CrossRef]
- Das, S.; Zea, M.P.; Russon, M.P.; Xing, Z.; Torregrosa-Allen, S.; Cervantes, H.E.; Harper, H.A.; Elzey, B.D.; Tran, E.J. Supinoxin blocks small cell lung cancer progression by inhibiting mitochondrial respiration through DDX5. iScience 2025, 28, 112219. [Google Scholar] [CrossRef]
- Chen, X.; Kadier, M.; Shi, M.; Li, K.; Chen, H.; Xia, Y.; Wang, Q.; Li, R.; Long, Y.; Qin, J.; et al. Targeting Melatonin to Mitochondria Mitigates Castration-Resistant Prostate Cancer by Inducing Pyroptosis. Small 2025, e2408996. [Google Scholar] [CrossRef]
- Jia, M.; Fu, Z.; Ye, C.; Xu, W.; Liu, J.; Wu, C.; Yan, H. Targeting MTHFD2 alters metabolic homeostasis and synergizes with bortezomib to inhibit multiple myeloma. Cell Death Discov. 2025, 11, 201. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhu, Y.; Guan, G.; Liu, M. Napabucasin targets resistant triple negative breast cancer through suppressing STAT3 and mitochondrial function. Cancer Chemother. Pharmacol. 2025, 95, 51. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Daver, N.; Mahendra, M.; Zhang, J.; Kamiya-Matsuoka, C.; Meric-Bernstam, F.; Kantarjian, H.M.; Ravandi, F.; Collins, M.E.; Francesco, M.E.D.; et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: Phase I trials. Nat. Med. 2023, 29, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, C.; Moreira-Barbosa, C.; Brunelli, L.; Ostano, P.; Panini, N.; Lupi, M.; Anastasia, A.; Fiordaliso, F.; Salio, M.; Formenti, L.; et al. PGC1α/β Expression Predicts Therapeutic Response to Oxidative Phosphorylation Inhibition in Ovarian Cancer. Cancer Res. 2022, 82, 1423–1434. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Shi, B.; Hu, X.; He, H.; Fang, W. Metformin suppresses breast cancer growth via inhibition of cyclooxygenase-2. Oncol. Lett. 2021, 22, 615. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Xiao, B.; Deng, X.; Zhou, W.; Tan, E.K. Flow Cytometry-Based Assessment of Mitophagy Using MitoTracker. Front. Cell Neurosci. 2016, 10, 76. [Google Scholar] [CrossRef]
- Feng, X.; Tang, M.; Dede, M.; Su, D.; Pei, G.; Jiang, D.; Wang, C.; Chen, Z.; Li, M.; Nie, L.; et al. Genome-wide CRISPR screens using isogenic cells reveal vulnerabilities conferred by loss of tumor suppressors. Sci. Adv. 2022, 8, eabm6638. [Google Scholar] [CrossRef]
- Fournier, L.A.; Kalantari, F.; Wells, J.P.; Lee, J.S.; Trigo-Gonzalez, G.; Moksa, M.M.; Smith, T.; White, J.; Shanks, A.; Wang, S.L.; et al. Genome-Wide CRISPR Screen Identifies KEAP1 Perturbation as a Vulnerability of ARID1A-Deficient Cells. Cancers 2024, 16, 2949. [Google Scholar] [CrossRef]
- Morgenstern, M.; Peikert, C.D.; Lübbert, P.; Suppanz, I.; Klemm, C.; Alka, O.; Steiert, C.; Naumenko, N.; Schendzielorz, A.; Melchionda, L.; et al. Quantitative high-confidence human mitochondrial proteome and its dynamics in cellular context. Cell Metab. 2021, 33, 2464–2483.e18. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [PubMed]
- Erdem, A.; Marin, S.; Pereira-Martins, D.A.; Geugien, M.; Cunningham, A.; Pruis, M.G.; Weinhauser, I.; Gerding, A.; Bakker, B.M.; Wierenga, A.T.J.; et al. Inhibition of the succinyl dehydrogenase complex in acute myeloid leukemia leads to a lactate-fuelled respiratory metabolic vulnerability. Nat. Commun. 2022, 13, 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, T.; Ding, X.; Chang, Y.; Liu, C.; Zhang, Y.; Hao, S.; Yin, Q.; Jiang, B. Inhibition of mitochondrial complex III induces differentiation in acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2021, 547, 162–168. [Google Scholar] [CrossRef]
- Gandhirajan, R.K.; Rodder, K.; Bodnar, Y.; Pasqual-Melo, G.; Emmert, S.; Griguer, C.E.; Weltmann, K.D.; Bekeschus, S. Cytochrome C oxidase Inhibition and Cold Plasma-derived Oxidants Synergize in Melanoma Cell Death Induction. Sci. Rep. 2018, 8, 12734. [Google Scholar] [CrossRef]
- Wang, T.; Ma, F.; Qian, H.L. Defueling the cancer: ATP synthase as an emerging target in cancer therapy. Mol. Ther. Oncolytics 2021, 23, 82–95. [Google Scholar] [CrossRef]
- Chowdhury, S.; Kennedy, J.J.; Ivey, R.G.; Murillo, O.D.; Hosseini, N.; Song, X.; Petralia, F.; Calinawan, A.; Savage, S.R.; Berry, A.B.; et al. Proteogenomic analysis of chemo-refractory high-grade serous ovarian cancer. Cell 2023, 186, 3476–3498.e35. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, J.M.; Ryu, J.; Khan, H.; Shetty, M.; Parker, E.; D’Arcy, P.; Zhu, S.; Bazzaro, M.; Thomas, S.N. Proteomic Analysis of ARID1A-Deficient Ovarian Clear Cell Carcinoma Cells Reveals Differential Mitochondria ETC Subunit Abundances and Targetable Mitochondrial Pathways. Int. J. Mol. Sci. 2025, 26, 5466. https://doi.org/10.3390/ijms26125466
Perez JM, Ryu J, Khan H, Shetty M, Parker E, D’Arcy P, Zhu S, Bazzaro M, Thomas SN. Proteomic Analysis of ARID1A-Deficient Ovarian Clear Cell Carcinoma Cells Reveals Differential Mitochondria ETC Subunit Abundances and Targetable Mitochondrial Pathways. International Journal of Molecular Sciences. 2025; 26(12):5466. https://doi.org/10.3390/ijms26125466
Chicago/Turabian StylePerez, Jesenia M., Joohyun Ryu, Hannah Khan, Mihir Shetty, Emma Parker, Padraig D’Arcy, Shijia Zhu, Martina Bazzaro, and Stefani N. Thomas. 2025. "Proteomic Analysis of ARID1A-Deficient Ovarian Clear Cell Carcinoma Cells Reveals Differential Mitochondria ETC Subunit Abundances and Targetable Mitochondrial Pathways" International Journal of Molecular Sciences 26, no. 12: 5466. https://doi.org/10.3390/ijms26125466
APA StylePerez, J. M., Ryu, J., Khan, H., Shetty, M., Parker, E., D’Arcy, P., Zhu, S., Bazzaro, M., & Thomas, S. N. (2025). Proteomic Analysis of ARID1A-Deficient Ovarian Clear Cell Carcinoma Cells Reveals Differential Mitochondria ETC Subunit Abundances and Targetable Mitochondrial Pathways. International Journal of Molecular Sciences, 26(12), 5466. https://doi.org/10.3390/ijms26125466