

SARS-CoV-2-Derived RNA Fragment Induces Myocardial Dysfunction via siRNA-like Suppression of Mitochondrial ATP Synthase
 ,
,
Abstract
1. Introduction
2. Results
2.1. Identification of Homologous Sequences Between the SARS-CoV-2 and Human Genomes
2.2. Genomic Localization of the Homologous Sequence
2.3. Danger-Associated Molecular Pattern (DAMP) Activity of ACHF
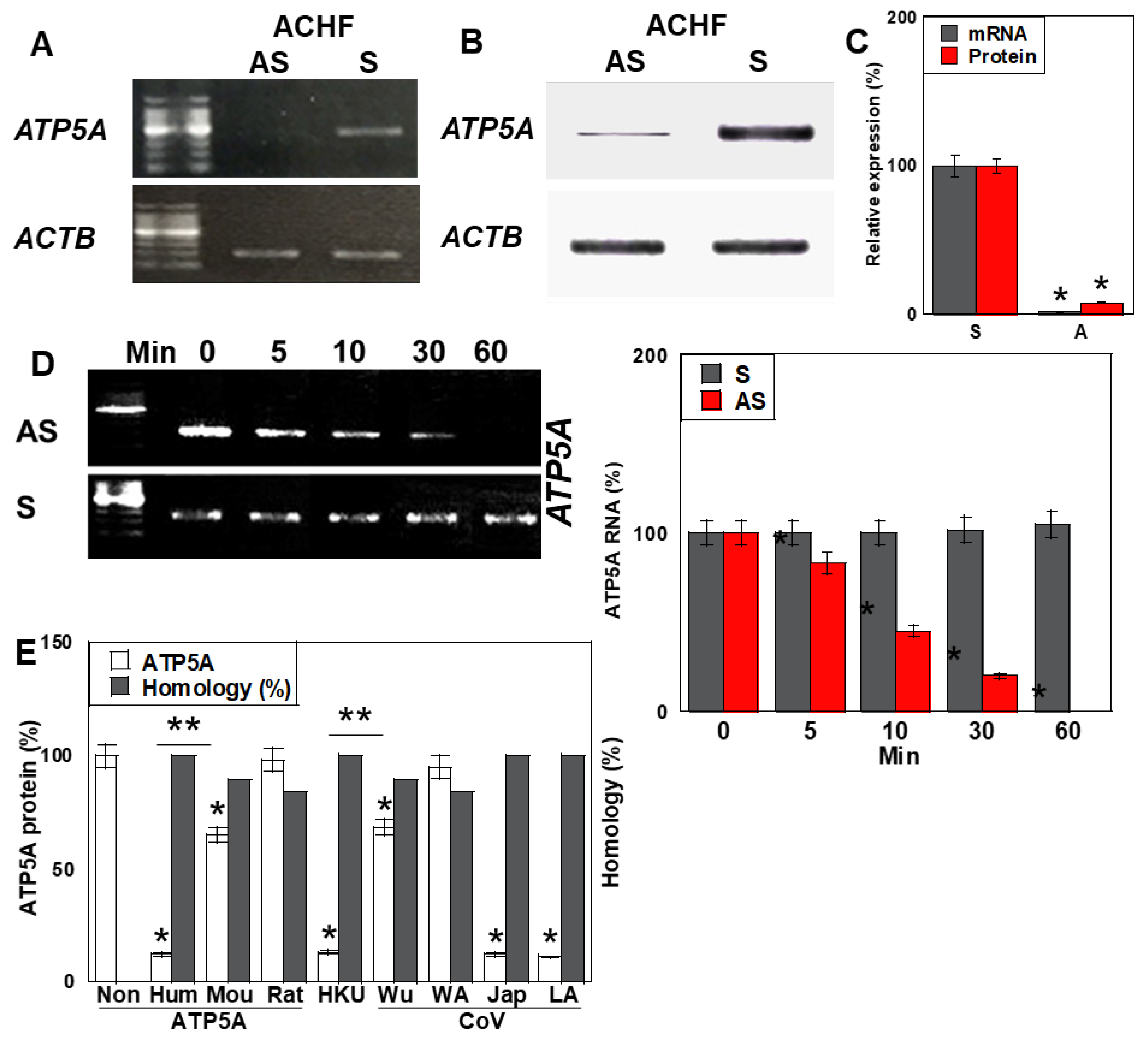
2.4. Antisense Activity of ACHF
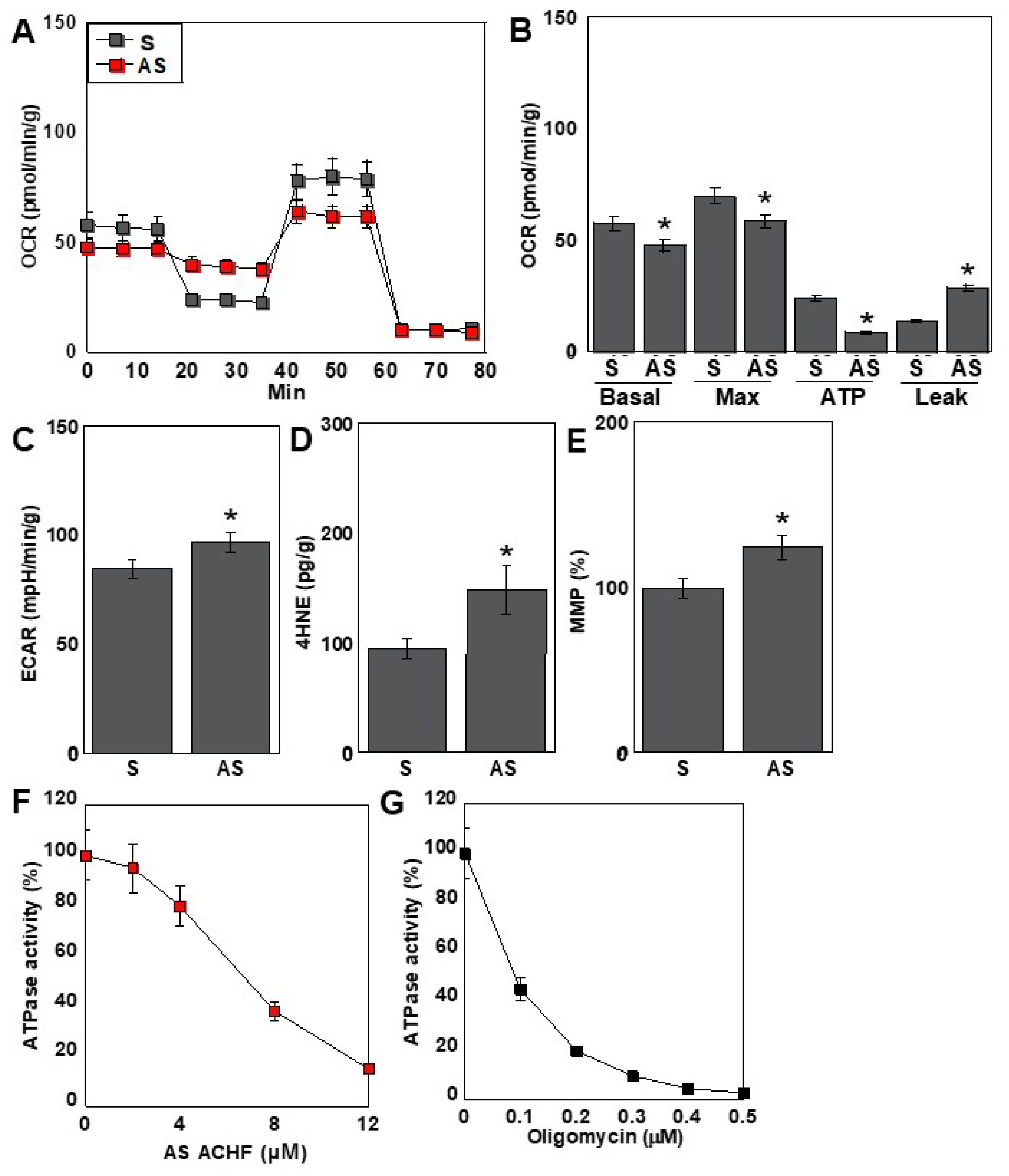
2.5. Effects of the ACHF Sequence on Energy Metabolism
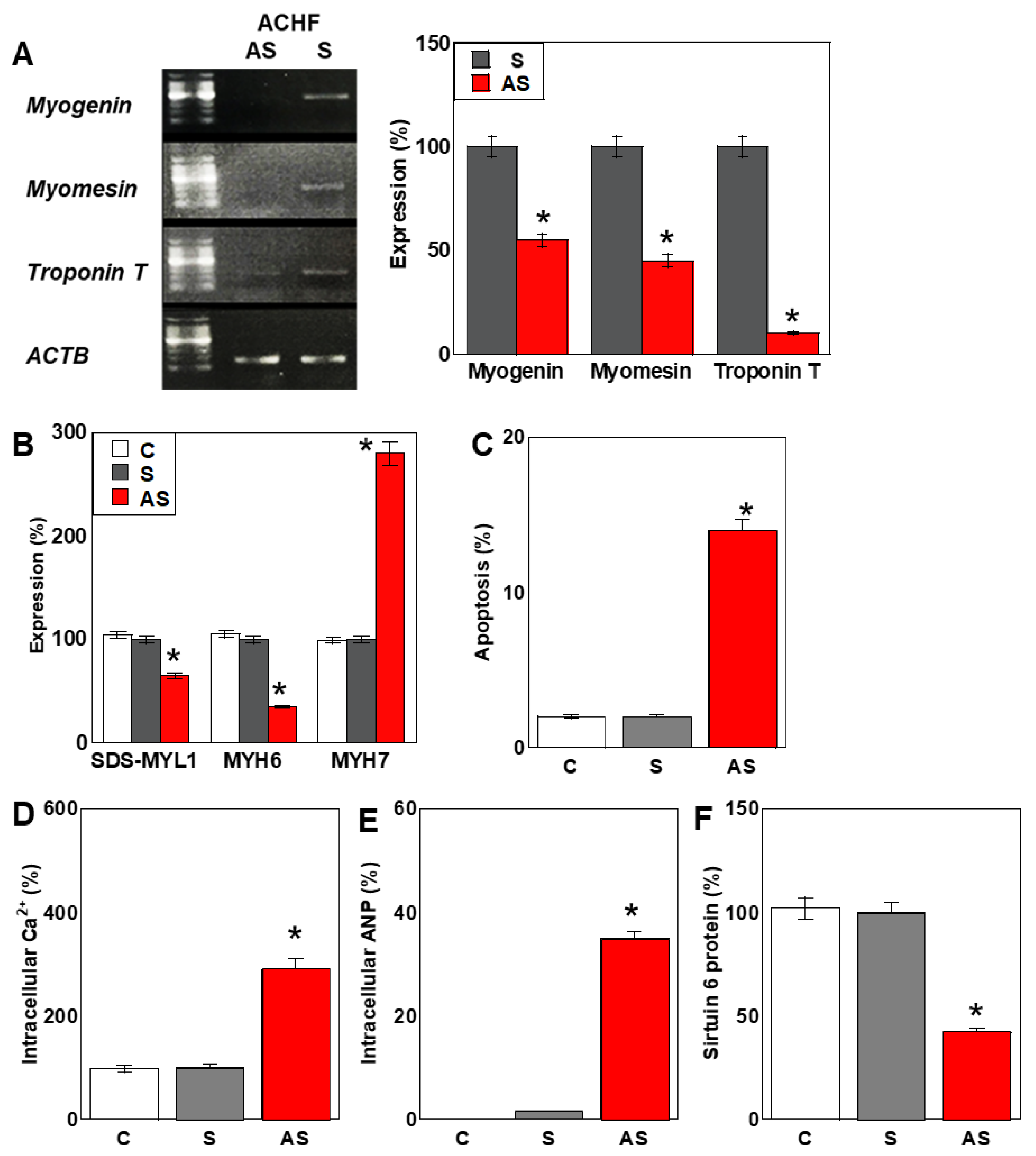
2.6. Effects of the ACHF Sequence on Cardiomyocyte Maturation
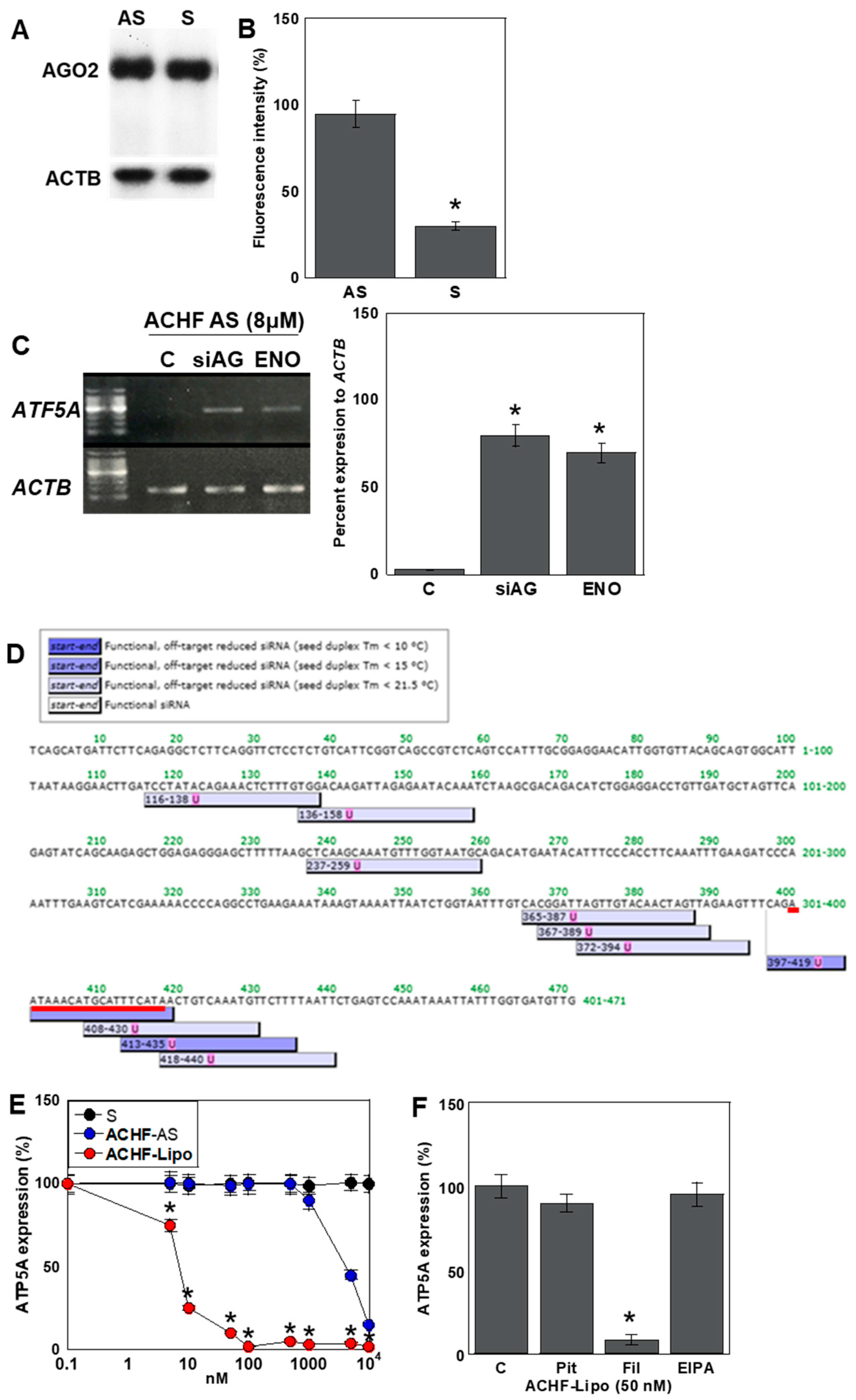
2.7. Mechanism of the Antisense Effect of ACHF-AS RNA
3. Discussion
4. Materials and Methods
4.1. Cell Line
4.2. Assessment of Cell Proliferation and Apoptosis
4.3. Protein Isolation
4.4. Immunoblotting
4.5. Enzyme-Linked Immunosorbent and Fluorometric Assays
4.6. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.7. ATP Synthase Activity Assay
4.8. Assessment of Mitochondrial Membrane Potential (MMP)
4.9. Mitochondrial Stress Analysis (Seahorse Assay)
4.10. Glycolytic Stress Test
4.11. RNA Immunoprecipitation (RIP)
4.12. Sequence Homology and siRNA Prediction
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mallah, S.I.; Ghorab, O.K.; Al-Salmi, S.; Abdellatif, O.S.; Tharmaratnam, T.; Iskandar, M.A.; Sefen, J.A.N.; Sidhu, P.; Atallah, B.; El-Lababidi, R.; et al. COVID-19: Breaking down a global health crisis. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 35. [Google Scholar] [CrossRef] [PubMed]
- Basu-Ray, I.; Almaddah, N.K.; Vaqar, S.; Soos, M.P. Cardiac Manifestations of Coronavirus (COVID-19). In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2025. [Google Scholar]
- Finelli, L.; Gupta, V.; Petigara, T.; Yu, K.; Bauer, K.A.; Puzniak, L.A. Mortality Among US Patients Hospitalized with SARS-CoV-2 Infection in 2020. JAMA Netw. Open 2021, 4, e216556. [Google Scholar] [CrossRef] [PubMed]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. COVID-19 and Cardiovascular Disease. Circulation 2020, 141, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, A.; Wong, F.; Couch, L.S.; Wang, B.X. Cardiac Complications of COVID-19 in Low-Risk Patients. Viruses 2022, 14, 1322. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Qin, M.; Yang, B. Coronavirus Disease 2019 (COVID-19) and Cardiac Injury-Reply. JAMA Cardiol. 2020, 5, 1199–1200. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Tobler, D.L.; Pruzansky, A.J.; Naderi, S.; Ambrosy, A.P.; Slade, J.J. Long-Term Cardiovascular Effects of COVID-19: Emerging Data Relevant to the Cardiovascular Clinician. Curr. Atheroscler. Rep. 2022, 24, 563–570. [Google Scholar] [CrossRef]
- Ammirati, E.; Lupi, L.; Palazzini, M.; Hendren, N.S.; Grodin, J.L.; Cannistraci, C.V.; Schmidt, M.; Hekimian, G.; Peretto, G.; Bochaton, T.; et al. Prevalence, Characteristics, and Outcomes of COVID-19-Associated Acute Myocarditis. Circulation 2022, 145, 1123–1139. [Google Scholar] [CrossRef]
- Giustino, G.; Croft, L.B.; Oates, C.P.; Rahman, K.; Lerakis, S.; Reddy, V.Y.; Goldman, M. Takotsubo Cardiomyopathy in COVID-19. J. Am. Coll. Cardiol. 2020, 76, 628–629. [Google Scholar] [CrossRef]
- Shao, H.H.; Yin, R.X. Pathogenic mechanisms of cardiovascular damage in COVID-19. Mol. Med. 2024, 30, 92. [Google Scholar] [CrossRef] [PubMed]
- Mehra, M.R.; Ruschitzka, F. COVID-19 Illness and Heart Failure: A Missing Link? JACC Heart Fail. 2020, 8, 512–514. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Zapor, M. Persistent Detection and Infectious Potential of SARS-CoV-2 Virus in Clinical Specimens from COVID-19 Patients. Viruses 2020, 12, 1384. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Satterfield, B.A.; Bhatt, D.L.; Gersh, B.J. Cardiac involvement in the long-term implications of COVID-19. Nat. Rev. Cardiol. 2022, 19, 332–341. [Google Scholar] [CrossRef]
- Fox, S.E.; Lameira, F.S.; Rinker, E.B.; Vander Heide, R.S. Cardiac Endotheliitis and Multisystem Inflammatory Syndrome After COVID-19. Ann. Intern. Med. 2020, 173, 1025–1027. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.A.; Cheng, R.K. Cardiovascular disease and COVID-19: Implications for prevention, surveillance and treatment. Heart 2020, 106, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Figliozzi, S.; Masci, P.G.; Ahmadi, N.; Tondi, L.; Koutli, E.; Aimo, A.; Stamatelopoulos, K.; Dimopoulos, M.A.; Caforio, A.L.P.; Georgiopoulos, G. Predictors of adverse prognosis in COVID-19: A systematic review and meta-analysis. Eur. J. Clin. Investig. 2020, 50, e13362. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef]
- Sola, I.; Mateos-Gomez, P.A.; Almazan, F.; Zuñiga, S.; Enjuanes, L. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol. 2011, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Mingaleeva, R.N.; Nigmatulina, N.A.; Sharafetdinova, L.M.; Romozanova, A.M.; Gabdoulkhakova, A.G.; Filina, Y.V.; Shavaliyev, R.F.; Rizvanov, A.A.; Miftakhova, R.R. Biology of the SARS-CoV-2 Coronavirus. Biochemistry 2022, 87, 1662–1678. [Google Scholar] [CrossRef]
- Nunez Lopez, Y.O.; Casu, A.; Pratley, R.E. Investigation of Extracellular Vesicles From SARS-CoV-2 Infected Specimens: A Safety Perspective. Front. Immunol. 2021, 12, 617042. [Google Scholar] [CrossRef]
- Alem, F.; Olanrewaju, A.A.; Omole, S.; Hobbs, H.E.; Ahsan, N.; Matulis, G.; Brantner, C.A.; Zhou, W.; Petricoin, E.F.; Liotta, L.A.; et al. Exosomes originating from infection with the cytoplasmic single-stranded RNA virus Rift Valley fever virus (RVFV) protect recipient cells by inducing RIG-I mediated IFN-B response that leads to activation of autophagy. Cell Biosci. 2021, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Singh, S.; Henderson, J.; Krishnamurthy, P. Mechanisms of COVID-19-induced cardiovascular disease: Is sepsis or exosome the missing link? J. Cell Physiol. 2021, 236, 3366–3382. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Padró, T.; Bollini, S.; Vilahur, G.; Duncker, D.J.; Evans, P.C.; Guzik, T.; Hoefer, I.E.; Waltenberger, J.; Wojta, J.; et al. Progress in cardiac research: From rebooting cardiac regeneration to a complete cell atlas of the heart. Cardiovasc. Res. 2021, 117, 2161–2174. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Liu, M.; Lv, J.; Pan, Z.; Wang, D.; Zhao, L.; Guo, X. Mitochondrial dysfunction in heart failure and its therapeutic implications. Front. Cardiovasc. Med. 2022, 9, 945142. [Google Scholar] [CrossRef]
- Long, Q.; Yang, K.; Yang, Q. Regulation of mitochondrial ATP synthase in cardiac pathophysiology. Am. J. Cardiovasc. Dis. 2015, 5, 19–32. [Google Scholar] [PubMed]
- Wauchop, M.; Rafatian, N.; Zhao, Y.; Chen, W.; Gagliardi, M.; Massé, S.; Cox, B.J.; Lai, P.; Liang, T.; Landau, S.; et al. Maturation of iPSC-derived cardiomyocytes in a heart-on-a-chip device enables modeling of dilated cardiomyopathy caused by R222Q-SCN5A mutation. Biomaterials 2023, 301, 122255. [Google Scholar] [CrossRef]
- Liu, Y.; Song, J.W.; Lin, J.Y.; Miao, R.; Zhong, J.C. Roles of MicroRNA-122 in Cardiovascular Fibrosis and Related Diseases. Cardiovasc. Toxicol. 2020, 20, 463–473. [Google Scholar] [CrossRef]
- Yang, L. Splicing noncoding RNAs from the inside out. Wiley Interdiscip. Rev. RNA 2015, 6, 651–660. [Google Scholar] [CrossRef]
- St Laurent, G.; Shtokalo, D.; Tackett, M.R.; Yang, Z.; Eremina, T.; Wahlestedt, C.; Urcuqui-Inchima, S.; Seilheimer, B.; McCaffrey, T.A.; Kapranov, P. Intronic RNAs constitute the major fraction of the non-coding RNA in mammalian cells. BMC Genom. 2012, 13, 504. [Google Scholar] [CrossRef]
- Azuma-Mukai, A.; Oguri, H.; Mituyama, T.; Qian, Z.R.; Asai, K.; Siomi, H.; Siomi, M.C. Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing. Proc. Natl. Acad. Sci. USA 2008, 105, 7964–7969. [Google Scholar] [CrossRef] [PubMed]
- Orban, T.I.; Izaurralde, E. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA 2005, 11, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, R.L.; Jiang, P.; Gilmore, B.L.; Spengler, R.M.; Tirabassi, R.; Nelson, J.A.; Ross, C.A.; Xing, Y.; Davidson, B.L. Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron 2014, 81, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Lima, W.F.; Wu, H.; Nichols, J.G.; Sun, H.; Murray, H.M.; Crooke, S.T. Binding and cleavage specificities of human Argonaute2. J. Biol. Chem. 2009, 284, 26017–26028. [Google Scholar] [CrossRef]
- Vickers, T.A.; Koo, S.; Bennett, C.F.; Crooke, S.T.; Dean, N.M.; Baker, B.F. Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents. A comparative analysis. J. Biol. Chem. 2003, 278, 7108–7118. [Google Scholar] [CrossRef]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.S.; Khvorova, A. Rational siRNA design for RNA interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar] [CrossRef]
- Ziv, O.; Price, J.; Shalamova, L.; Kamenova, T.; Goodfellow, I.; Weber, F.; Miska, E.A. The Short- and Long-Range RNA-RNA Interactome of SARS-CoV-2. Mol. Cell 2020, 80, 1067–1077.e5. [Google Scholar] [CrossRef]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P.; et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Hackbart, M.; Deng, X.; Baker, S.C. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. USA 2020, 117, 8094–8103. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Ziv, O.; Gabryelska, M.M.; Lun, A.T.L.; Gebert, L.F.R.; Sheu-Gruttadauria, J.; Meredith, L.W.; Liu, Z.Y.; Kwok, C.K.; Qin, C.F.; MacRae, I.J.; et al. COMRADES determines in vivo RNA structures and interactions. Nat. Methods 2018, 15, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.; Kleiman, L. Primer tRNAs for reverse transcription. J. Virol. 1997, 71, 8087–8095. [Google Scholar] [CrossRef] [PubMed]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Abdi, A.; AlOtaiby, S.; Badarin, F.A.; Khraibi, A.; Hamdan, H.; Nader, M. Interaction of SARS-CoV-2 with cardiomyocytes: Insight into the underlying molecular mechanisms of cardiac injury and pharmacotherapy. Biomed. Pharmacother. 2022, 146, 112518. [Google Scholar] [CrossRef]
- Xia, B.; Pan, X.; Luo, R.H.; Shen, X.; Li, S.; Wang, Y.; Zuo, X.; Wu, Y.; Guo, Y.; Xiao, G.; et al. Extracellular vesicles mediate antibody-resistant transmission of SARS-CoV-2. Cell Discov. 2023, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Gonjilashvili, A.; Tatishvili, S. The interplay between SARS-CoV-2 infection related cardiovascular diseases and depression. Common mechanisms, shared symptoms. Am. Heart J. Plus 2024, 38, 100364. [Google Scholar] [CrossRef]
- Jiang, D.; Gao, F.; Zhang, Y.; Wong, D.S.; Li, Q.; Tse, H.F.; Xu, G.; Yu, Z.; Lian, Q. Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. 2016, 7, e2467. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef]
- Sun, W.; Zhao, L.; Song, X.; Zhang, J.; Xing, Y.; Liu, N.; Yan, Y.; Li, Z.; Lu, Y.; Wu, J.; et al. MicroRNA-210 Modulates the Cellular Energy Metabolism Shift During H2O2-Induced Oxidative Stress by Repressing ISCU in H9c2 Cardiomyocytes. Cell Physiol. Biochem. 2017, 43, 383–394. [Google Scholar] [CrossRef]
- Gao, K.; Cheng, M.; Zuo, X.; Lin, J.; Hoogewijs, K.; Murphy, M.P.; Fu, X.D.; Zhang, X. Active RNA interference in mitochondria. Cell Res. 2021, 31, 219–228. [Google Scholar] [CrossRef]
- Blinov, V.M.; Zverev, V.V.; Krasnov, G.S.; Filatov, F.P.; Shargunov, A.V. Viral component of the human genome. Mol. Biol. 2017, 51, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Kazachenka, A.; Kassiotis, G. SARS-CoV-2-Host Chimeric RNA-Sequencing Reads Do Not Necessarily Arise From Virus Integration Into the Host DNA. Front. Microbiol. 2021, 12, 676693. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Liu, X.Z.; He, X.; Zhou, L.Q. Exogenous Coronavirus Interacts With Endogenous Retrotransposon in Human Cells. Front. Cell Infect. Microbiol. 2021, 11, 609160. [Google Scholar] [CrossRef]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. The Interactions of Retroviruses and their Hosts. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Burns, K.H.; Boeke, J.D. Human transposon tectonics. Cell 2012, 149, 740–752. [Google Scholar] [CrossRef]
- Feschotte, C.; Gilbert, C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Koch, B.F. SARS-CoV-2 and human retroelements: A case for molecular mimicry? BMC Genom. Data 2022, 23, 27. [Google Scholar] [CrossRef]
- Shrock, E.; Fujimura, E.; Kula, T.; Timms, R.T.; Lee, I.H.; Leng, Y.; Robinson, M.L.; Sie, B.M.; Li, M.Z.; Chen, Y.; et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 2020, 370, eabd4250. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, C.I.; Galloway, J.; Chu, H.Y.; Shipley, M.M.; Sung, K.; Itell, H.L.; Wolf, C.R.; Logue, J.K.; Magedson, A.; Garrett, M.E.; et al. Epitope profiling reveals binding signatures of SARS-CoV-2 immune response in natural infection and cross-reactivity with endemic human CoVs. Cell Rep. 2021, 35, 109164. [Google Scholar] [CrossRef]
- Kashir, J.; AlKattan, K.; Yaqinuddin, A. COVID-19: Cross-immunity of viral epitopes may influence severity of infection and immune response. Signal Transduct. Target. Ther. 2021, 6, 102. [Google Scholar] [CrossRef]
- Haynes, W.A.; Kamath, K.; Bozekowski, J.; Baum-Jones, E.; Campbell, M.; Casanovas-Massana, A.; Daugherty, P.S.; Dela Cruz, C.S.; Dhal, A.; Farhadian, S.F.; et al. High-resolution epitope mapping and characterization of SARS-CoV-2 antibodies in large cohorts of subjects with COVID-19. Commun. Biol. 2021, 4, 1317. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Reaction of Human Monoclonal Antibodies to SARS-CoV-2 Proteins With Tissue Antigens: Implications for Autoimmune Diseases. Front. Immunol. 2020, 11, 617089. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, S.K.; Dhalla, N.S. Status of Mitochondrial Oxidative Phosphorylation during the Development of Heart Failure. Antioxidants 2023, 12, 1941. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z.; et al. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef]
- Andersson, M.I.; Arancibia-Carcamo, C.V.; Auckland, K.; Baillie, J.K.; Barnes, E.; Beneke, T.; Bibi, S.; Brooks, T.; Carroll, M.; Crook, D.; et al. SARS-CoV-2 RNA detected in blood products from patients with COVID-19 is not associated with infectious virus. Wellcome Open Res. 2020, 5, 181. [Google Scholar] [CrossRef]
- Lawrence Panchali, M.J.; Kim, C.M.; Seo, J.W.; Kim, D.Y.; Yun, N.R.; Kim, D.M. SARS-CoV-2 RNAemia and Disease Severity in COVID-19 Patients. Viruses 2023, 15, 1560. [Google Scholar] [CrossRef]
- Uehata, T.; Takeuchi, O. RNA Recognition and Immunity-Innate Immune Sensing and Its Posttranscriptional Regulation Mechanisms. Cells 2020, 9, 1701. [Google Scholar] [CrossRef] [PubMed]
- Vabret, N.; Bhardwaj, N.; Greenbaum, B.D. Sequence-Specific Sensing of Nucleic Acids. Trends Immunol. 2017, 38, 53–65. [Google Scholar] [CrossRef]
- Forsbach, A.; Nemorin, J.G.; Montino, C.; Müller, C.; Samulowitz, U.; Vicari, A.P.; Jurk, M.; Mutwiri, G.K.; Krieg, A.M.; Lipford, G.B.; et al. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol. 2008, 180, 3729–3738. [Google Scholar] [CrossRef]
- Leonard, J.N.; Ghirlando, R.; Askins, J.; Bell, J.K.; Margulies, D.H.; Davies, D.R.; Segal, D.M. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. USA 2008, 105, 258–263. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Oue, N.; Wakikawa, A.; Shigeishi, H.; Matsutani, N.; Kuraoka, K.; Ito, R.; Yokozaki, H.; Yasui, W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J. Pathol. 2002, 196, 163–170. [Google Scholar] [CrossRef]
- Nukaga, S.; Mori, T.; Miyagawa, Y.; Fujiwara-Tani, R.; Sasaki, T.; Fujii, K.; Mori, S.; Goto, K.; Kishi, S.; Nakashima, C.; et al. Combined administration of lauric acid and glucose improved cancer-derived cardiac atrophy in a mouse cachexia model. Cancer Sci. 2020, 111, 4605–4615. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara-Tani, R.; Fujii, K.; Mori, S.; Kishi, S.; Sasaki, T.; Ohmori, H.; Nakashima, C.; Kawahara, I.; Nishiguchi, Y.; Mori, T.; et al. Role of Clostridium perfringens Enterotoxin on YAP Activation in Colonic Sessile Serrated Adenoma/Polyps with Dysplasia. Int. J. Mol. Sci. 2020, 21, 3840. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Accession Number | 1st Base | Sequence |
|---|---|---|---|
| Human ATP5A | M37104.1 | 400 | AAT AAA CAT GCA TTT CAT A |
| Mouse ATP5j | NM_001358500.1 | 626 | AAT AAA CAT TCA TTT CAC A |
| Rat ATP5PF | NM_053602.2 | 552 | AAT AAA CAT TCA CTT CAC A |
| HKU1B.9 strain SC2521 | ON461763.1 | 19314 | AAT AAA CAT GCA TTT CAT A |
| SARS-CoV-2/Wuhan | NC_045512.2 | 19303 | AAT AAA CAT GCA TTC CAC A |
| SARS-CoV-2/USA/WA -PHL-001844 | OV278871.2 | 19303 | AAT AAA CAT GCA TTC CAA C |
| SARS-CoV-2/Japan/sb_ncgms _cov_2_02761/2022 | BS012486.1 | 19273 | AAT AAA CAT GCA TTT CAT A |
| SARS-CoV-2/USA/LA-CDC -STM-94SR8FMQV/2022 | OP399780.1 | 19283 | AAT AAA CAT GCA TTT CAT A |
| RT-PCR Primers | |||
|---|---|---|---|
| Gene | ID | Upper | Lower |
| ATP5A | NM_001101.3 | GGACTTCGAGCAAGAGATGG | AGCACTGTGTTGGCGTACAG |
| Myomesin | AJ621424.1 | GATACAGCTCAGTACCGGGC | CTTTGTTGGCCTCCAAGCAC |
| Myogenin | NM_002479.6 | TACCAGGAACCCCGCTTCTA | GTGATGCTGTCCACGATGGA |
| Troponin T | S69208.1 | TTCGATGACATCCACCGCAA | TTTCAGCTTCGCCATCAGGT |
| Antibodies | |||
| Target | Cat No. | Company | |
| IRF3 | ab68481 | Abcam, Waltham, MA, USA | |
| IRF7 | 22392-1-AP | Proteintech, Rosemont, IL, USA | |
| ATP5A | ab110273 | Abcam, Waltham, MA, USA | |
| β-actin | sc-47778 | Santa Cruz Biotechnologies, Santa Cruz, CA, USA | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nukaga, S.; Fujiwara-Tani, R.; Mori, T.; Kawahara, I.; Nishida, R.; Miyagawa, Y.; Goto, K.; Ohmori, H.; Fujii, K.; Sasaki, T.; et al. SARS-CoV-2-Derived RNA Fragment Induces Myocardial Dysfunction via siRNA-like Suppression of Mitochondrial ATP Synthase. Int. J. Mol. Sci. 2025, 26, 5392. https://doi.org/10.3390/ijms26115392
Nukaga S, Fujiwara-Tani R, Mori T, Kawahara I, Nishida R, Miyagawa Y, Goto K, Ohmori H, Fujii K, Sasaki T, et al. SARS-CoV-2-Derived RNA Fragment Induces Myocardial Dysfunction via siRNA-like Suppression of Mitochondrial ATP Synthase. International Journal of Molecular Sciences. 2025; 26(11):5392. https://doi.org/10.3390/ijms26115392
Chicago/Turabian StyleNukaga, Shota, Rina Fujiwara-Tani, Takuya Mori, Isao Kawahara, Ryoichi Nishida, Yoshihiro Miyagawa, Kei Goto, Hitoshi Ohmori, Kiyomu Fujii, Takamitsu Sasaki, and et al. 2025. "SARS-CoV-2-Derived RNA Fragment Induces Myocardial Dysfunction via siRNA-like Suppression of Mitochondrial ATP Synthase" International Journal of Molecular Sciences 26, no. 11: 5392. https://doi.org/10.3390/ijms26115392
APA StyleNukaga, S., Fujiwara-Tani, R., Mori, T., Kawahara, I., Nishida, R., Miyagawa, Y., Goto, K., Ohmori, H., Fujii, K., Sasaki, T., Nakashima, C., Luo, Y., Mori, S., Kishi, S., Ogata, R., & Kuniyasu, H. (2025). SARS-CoV-2-Derived RNA Fragment Induces Myocardial Dysfunction via siRNA-like Suppression of Mitochondrial ATP Synthase. International Journal of Molecular Sciences, 26(11), 5392. https://doi.org/10.3390/ijms26115392