
Molecular Mechanisms of Biochanin A in AML Cells: Apoptosis Induction and Pathway-Specific Regulation in U937 and THP-1

Abstract
1. Introduction
2. Results
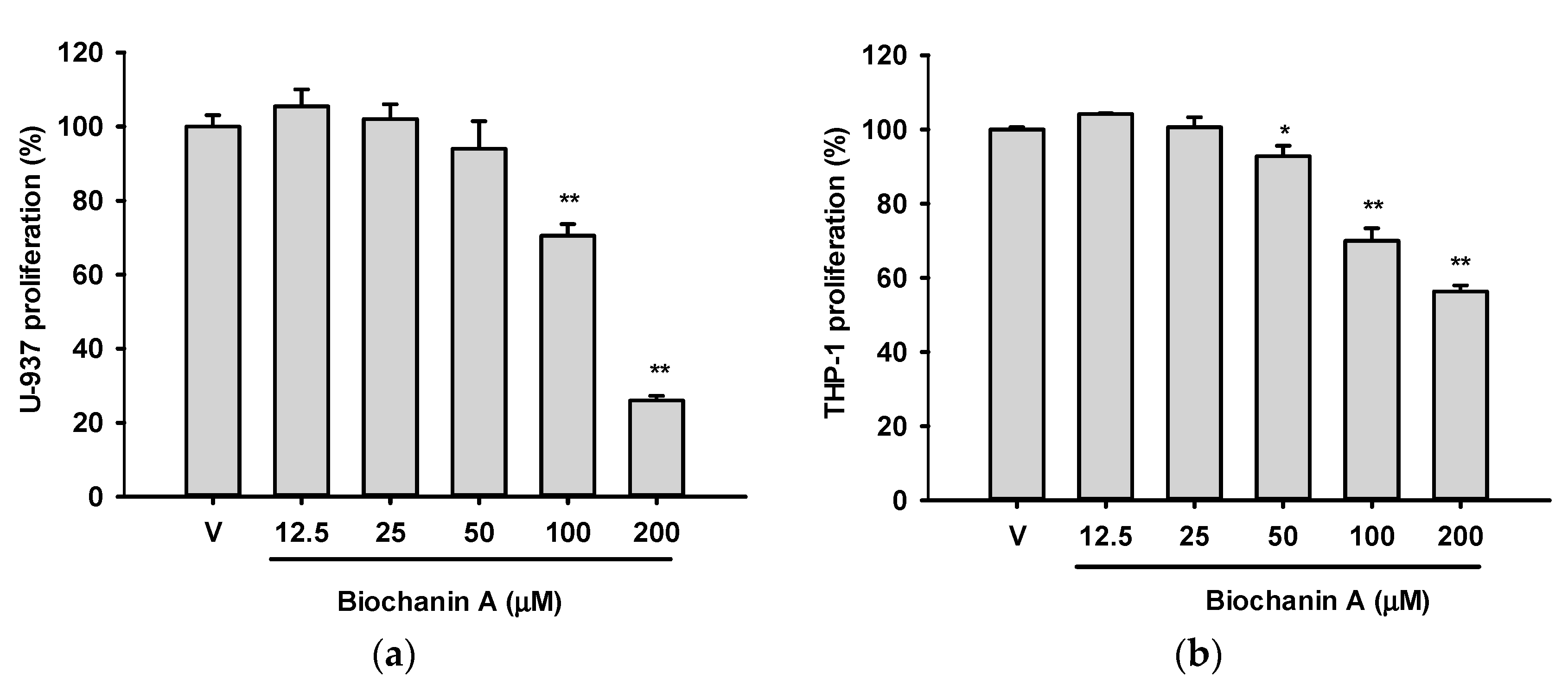
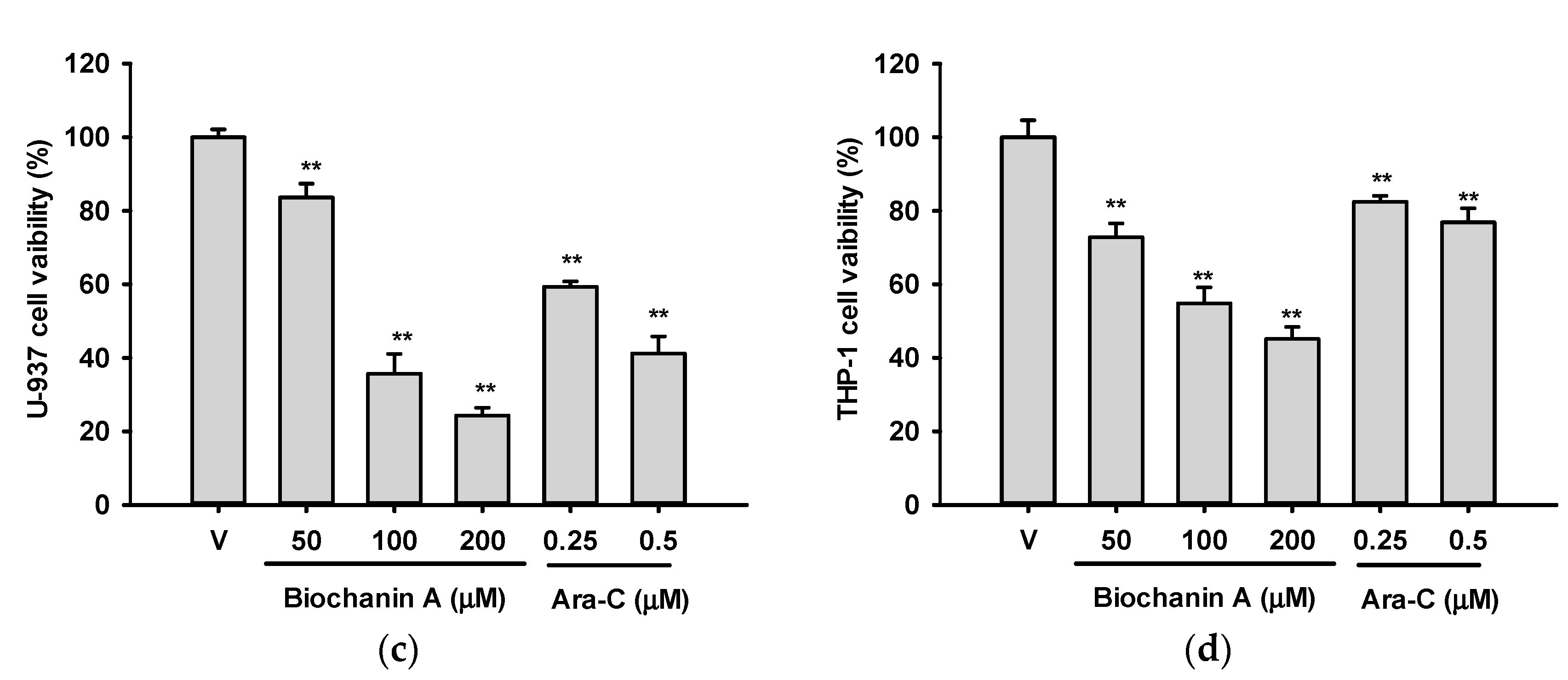
2.1. Effects of Biochanin A on the Viabilities of U937 and THP-1 Cells
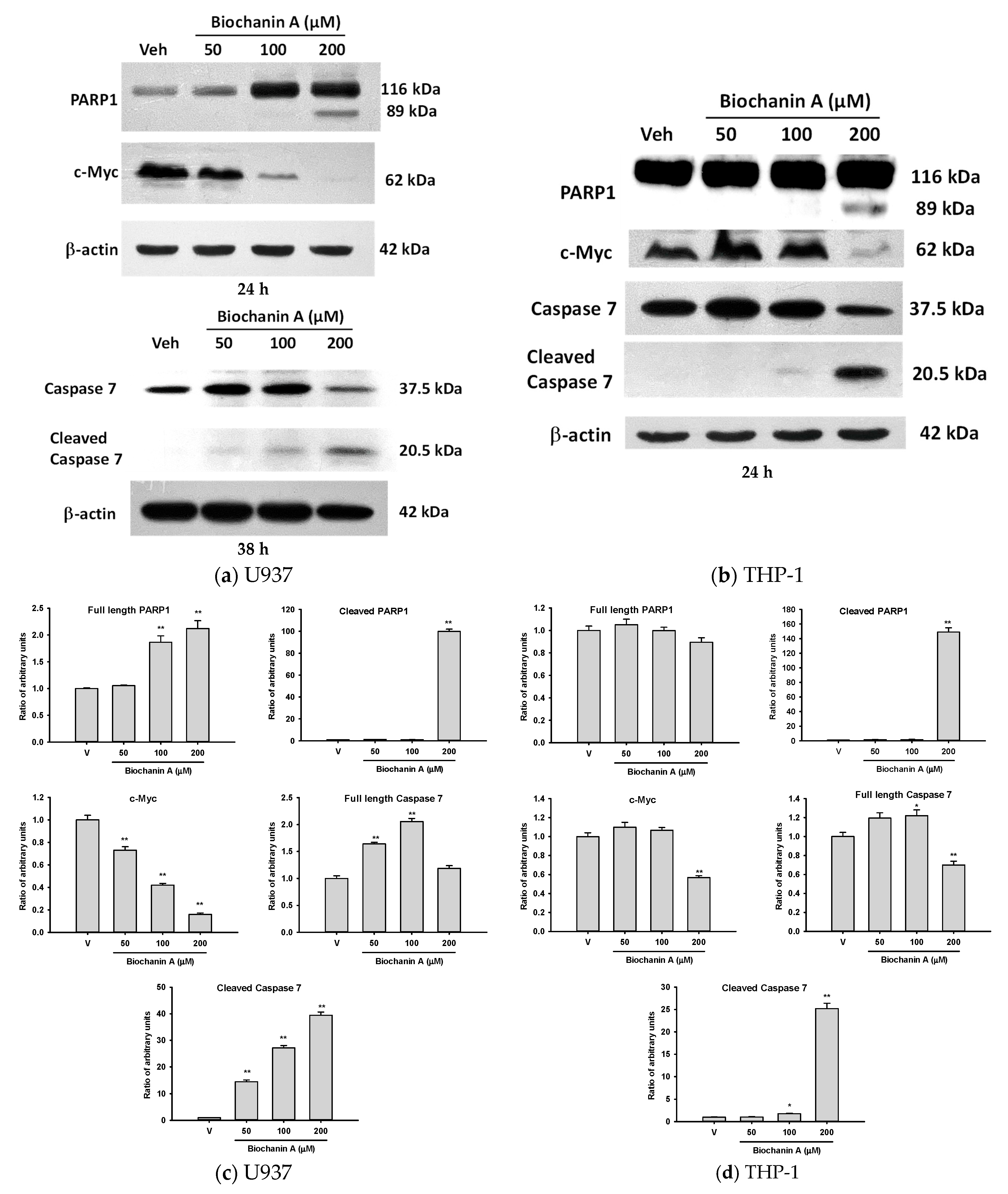
2.2. Effects of Biochanin A on Cell Proliferation- and Apoptosis-Related Protein Expression in U937 and THP-1 Cells
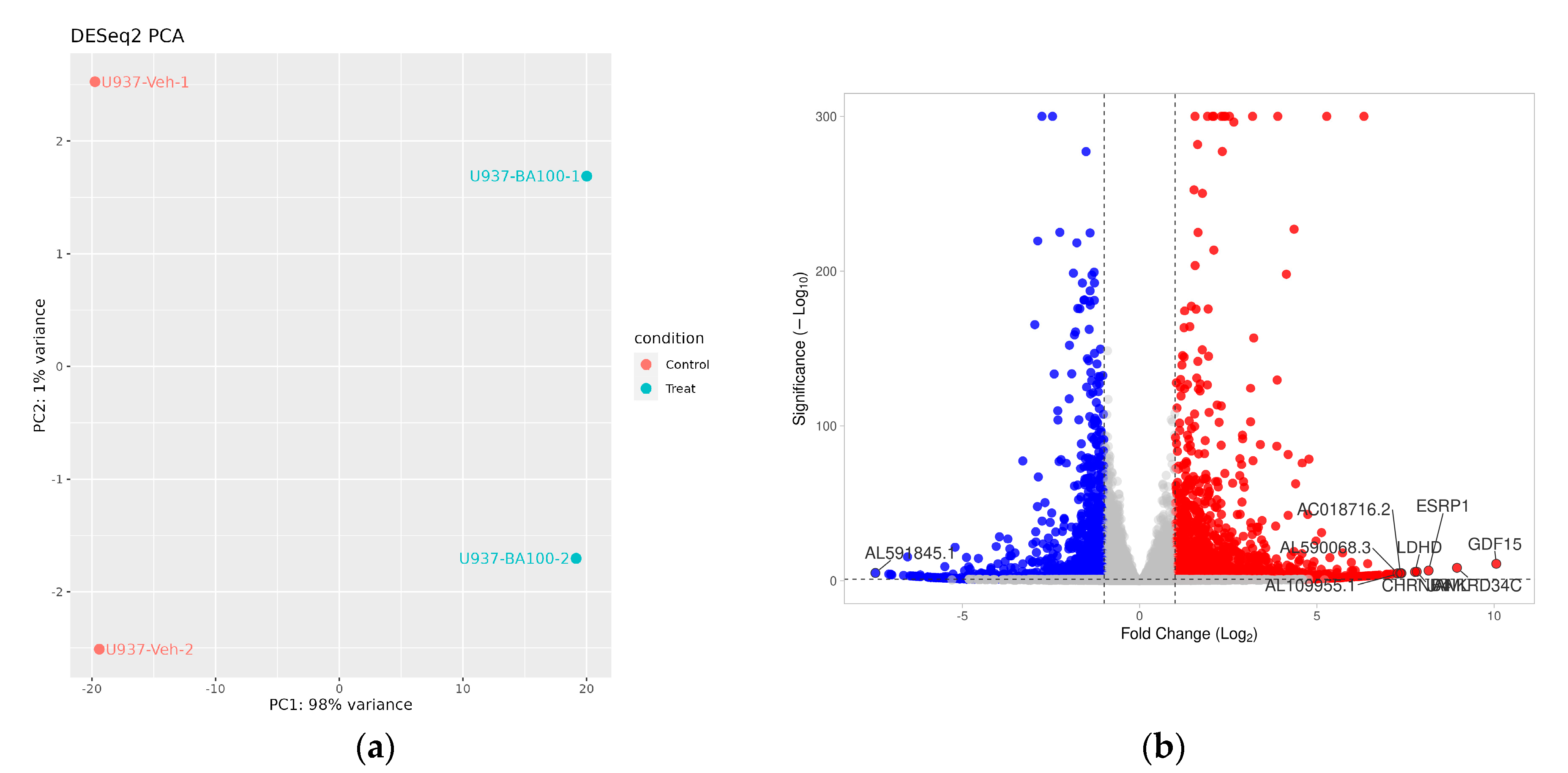
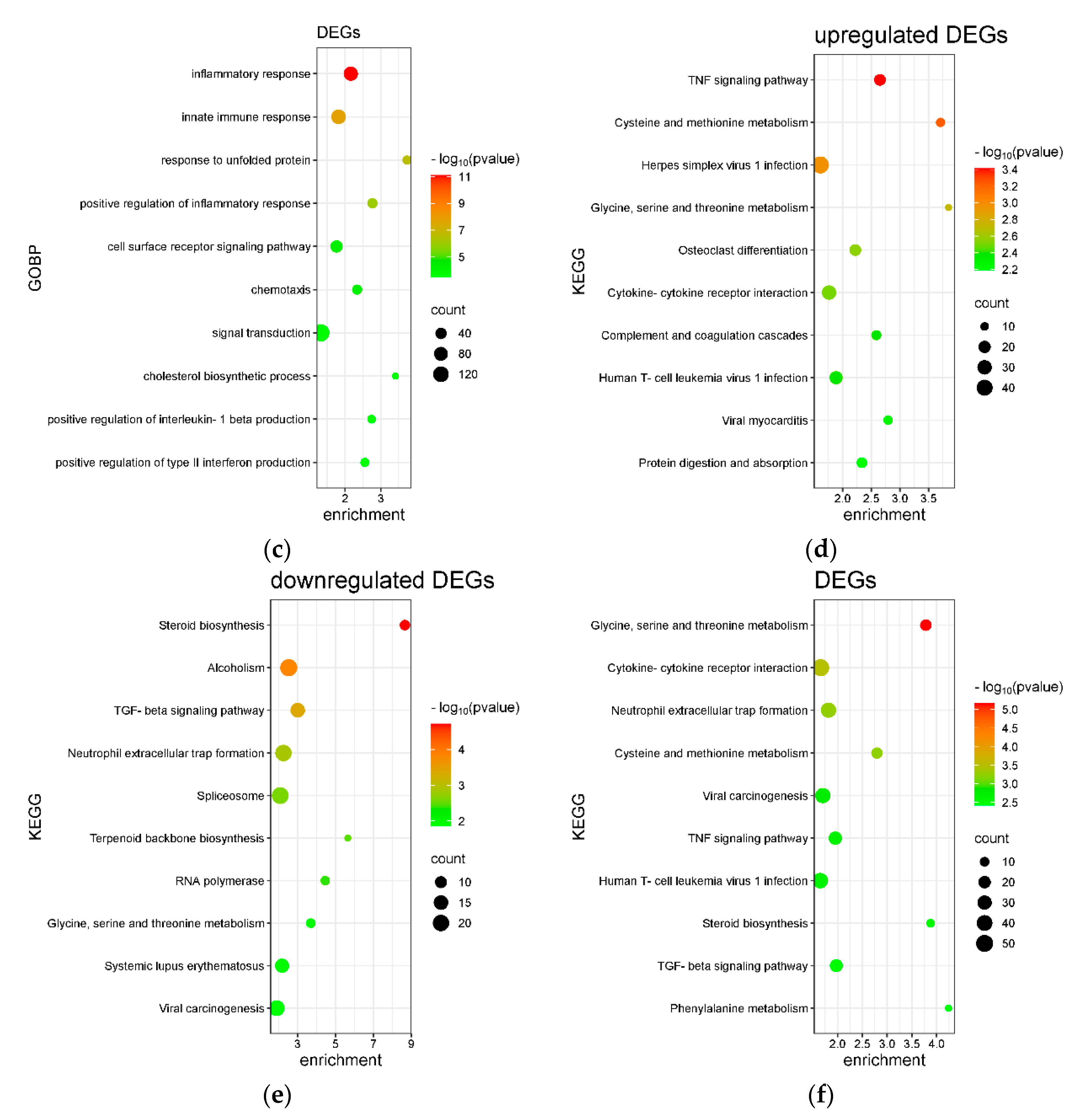
2.3. Transcriptome Analysis for Biochanin A-Treated U937 Cells: Over-Representation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA)
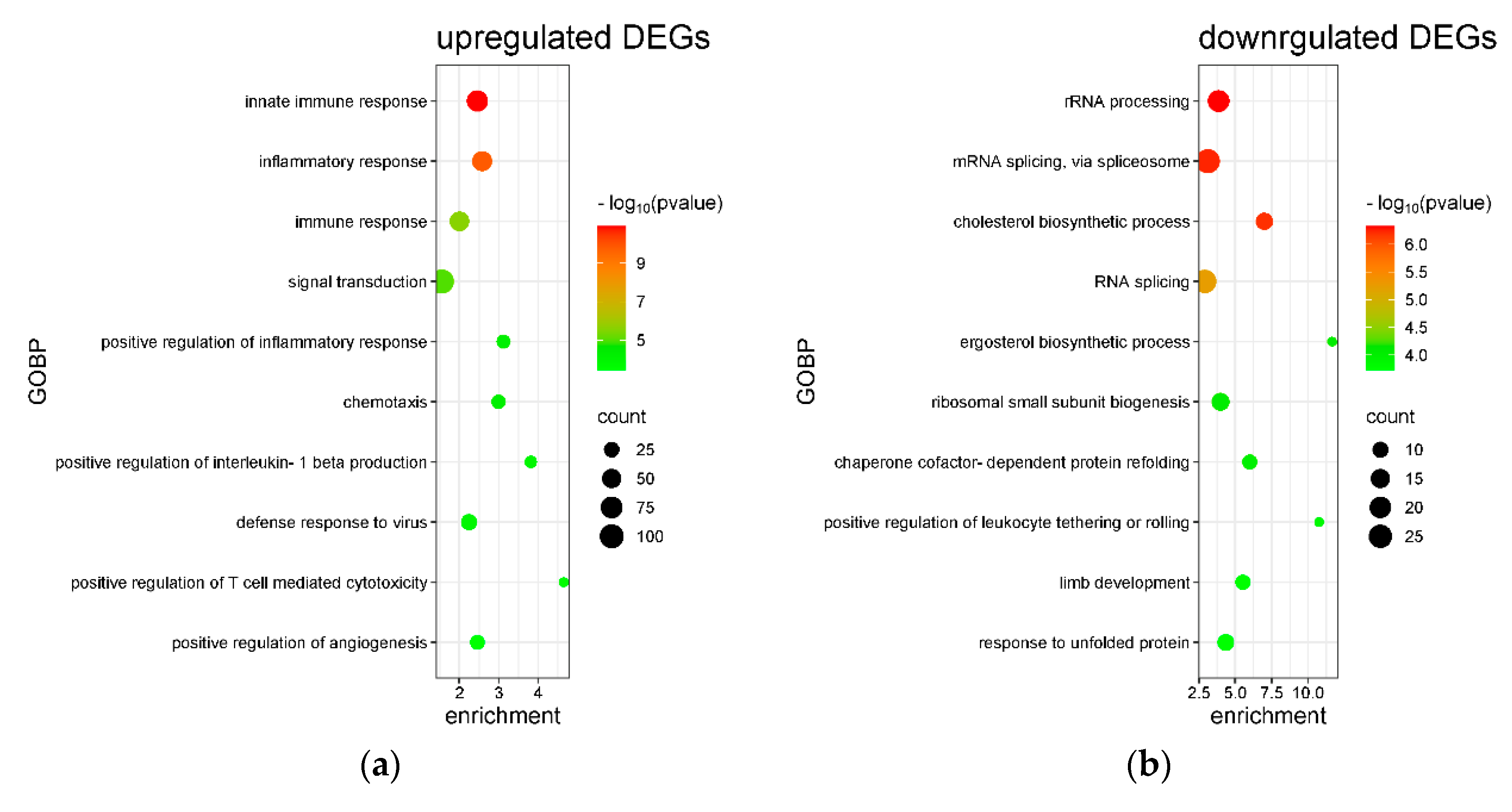
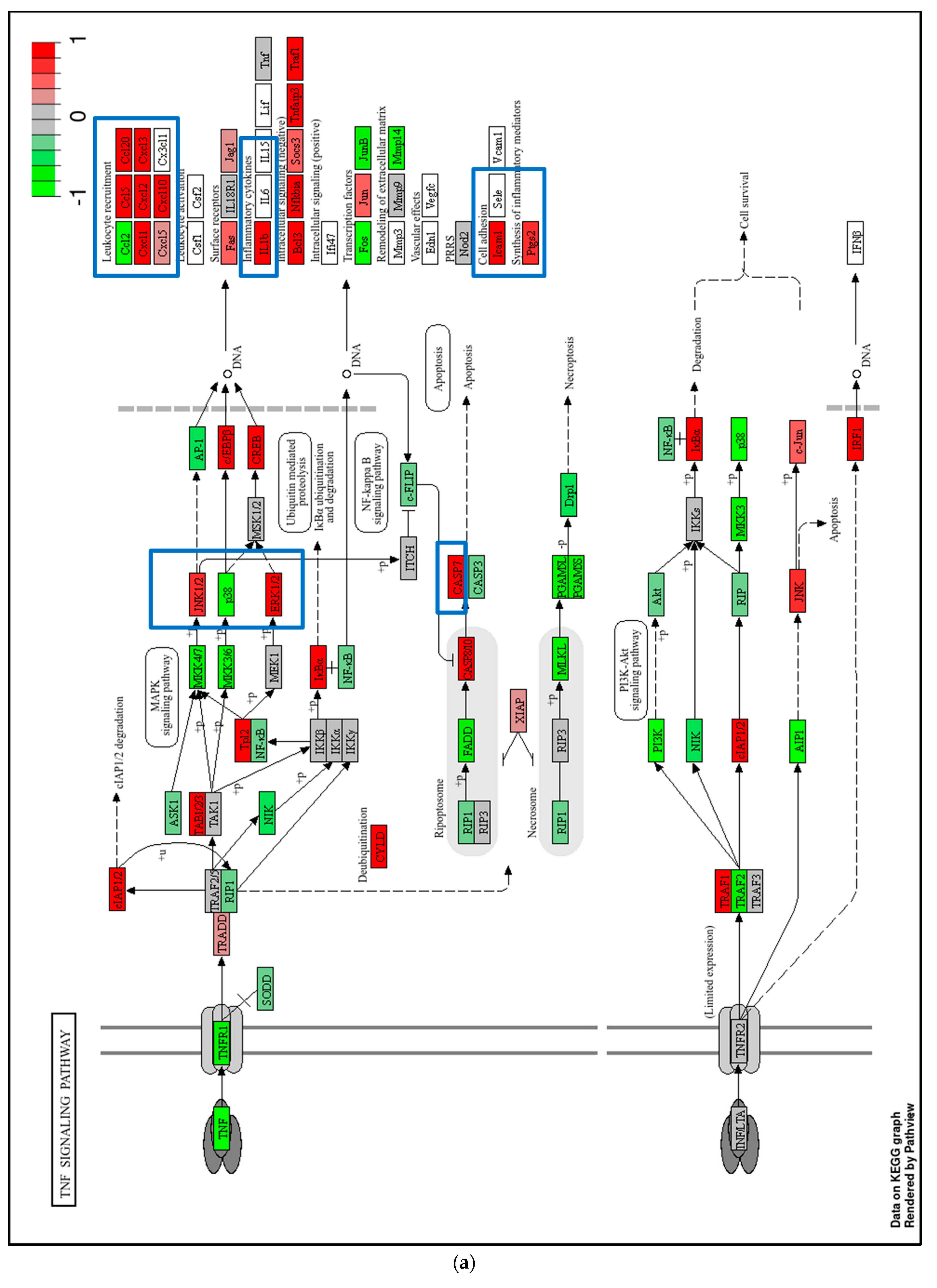
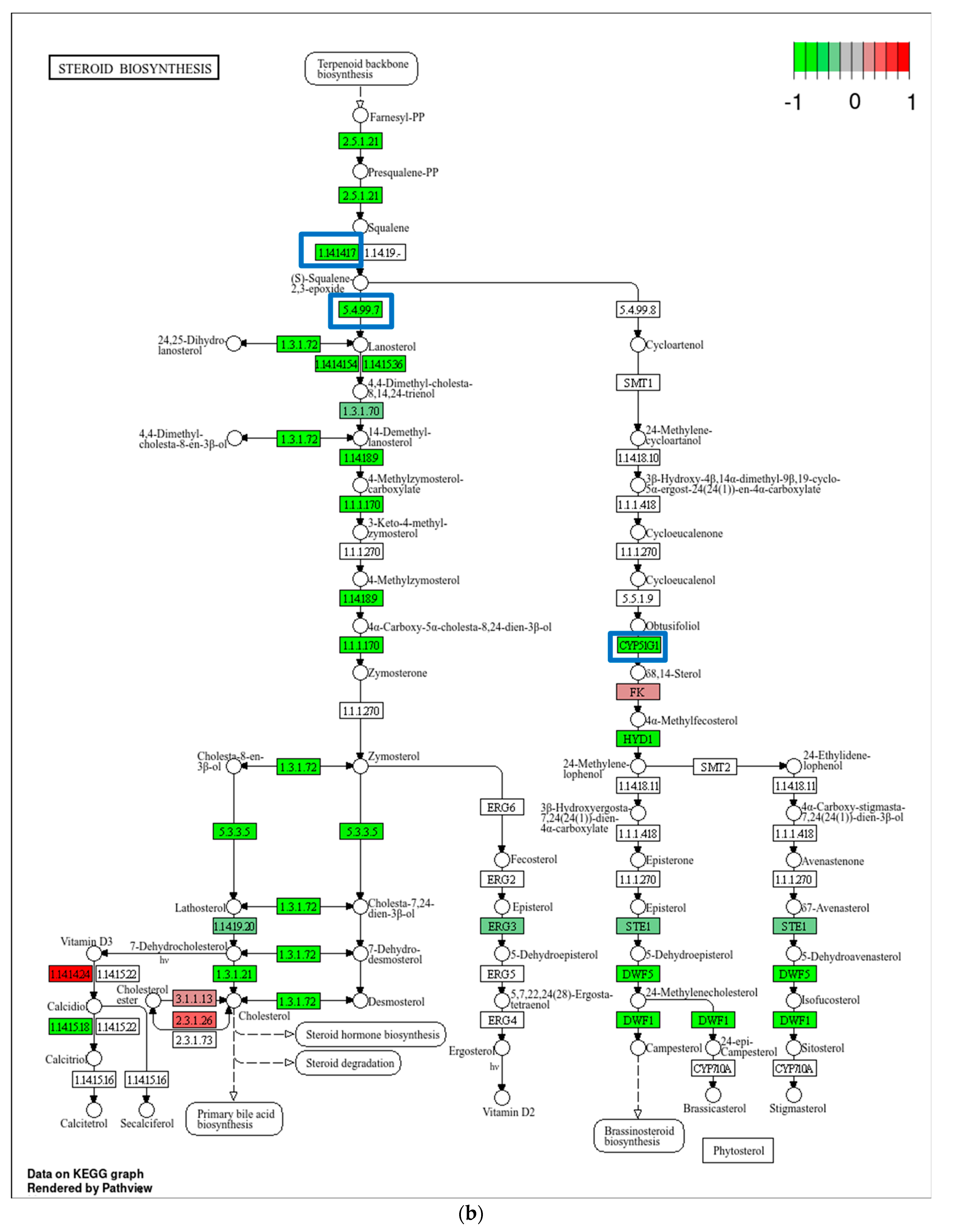
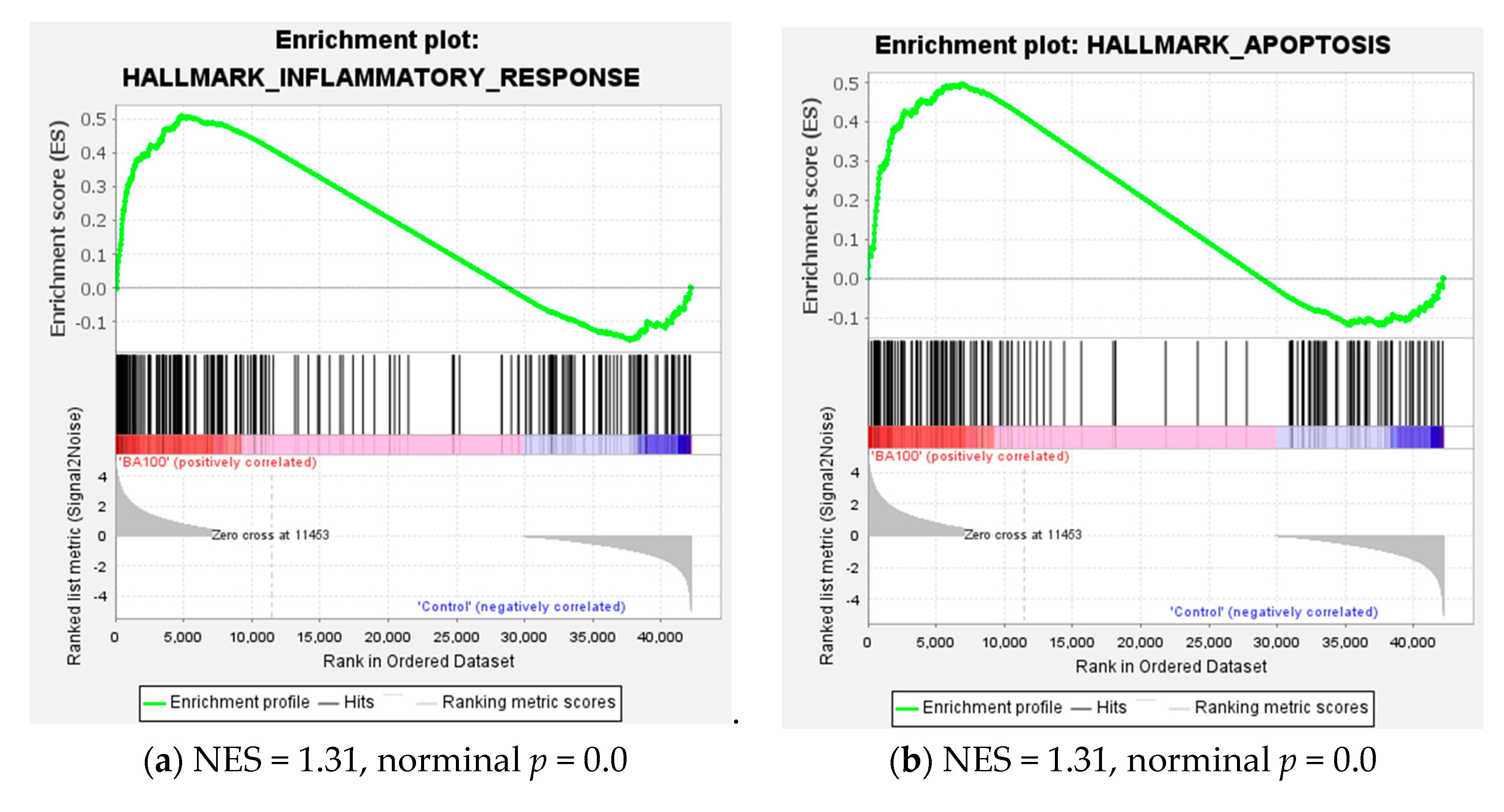
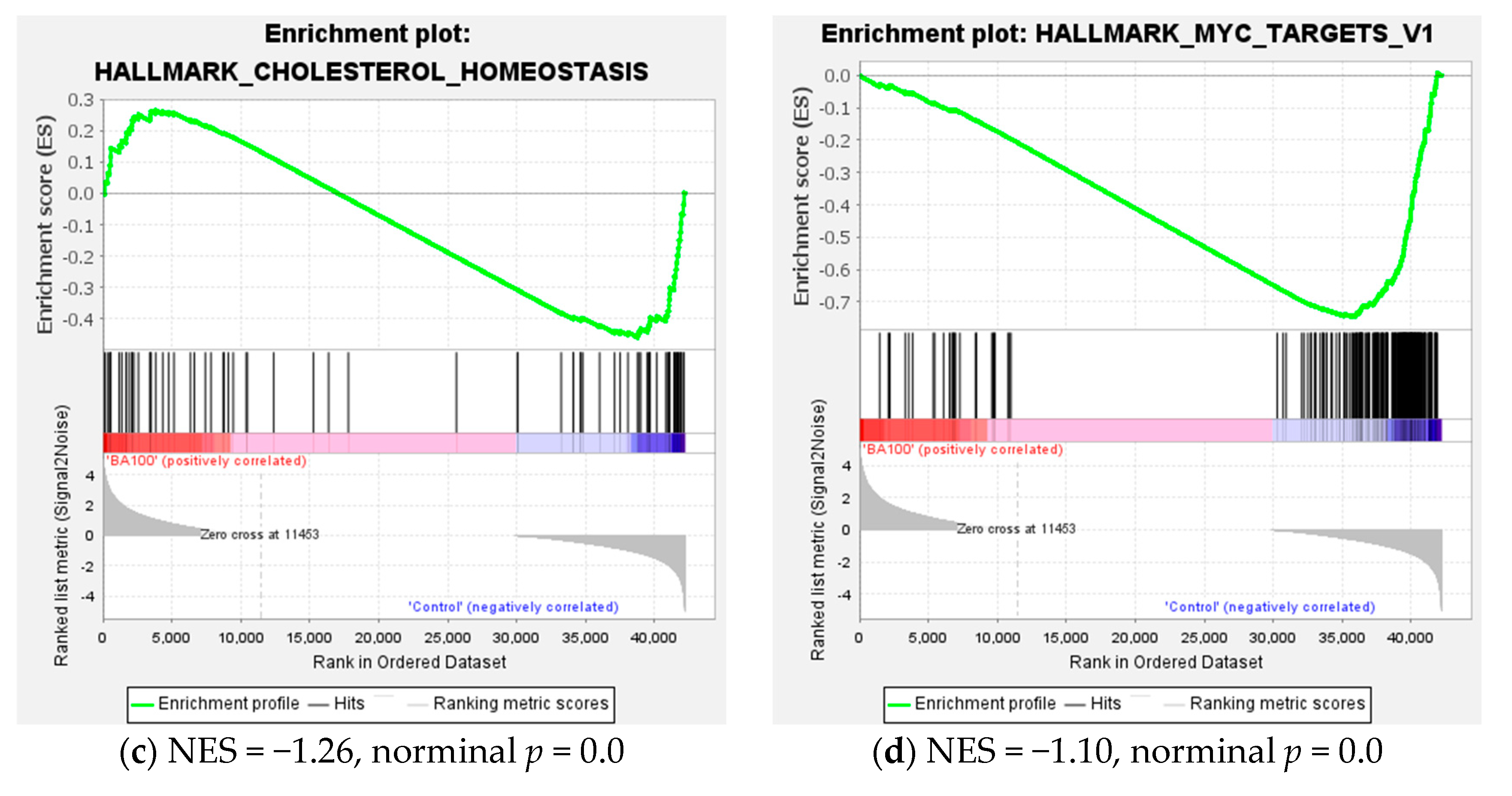
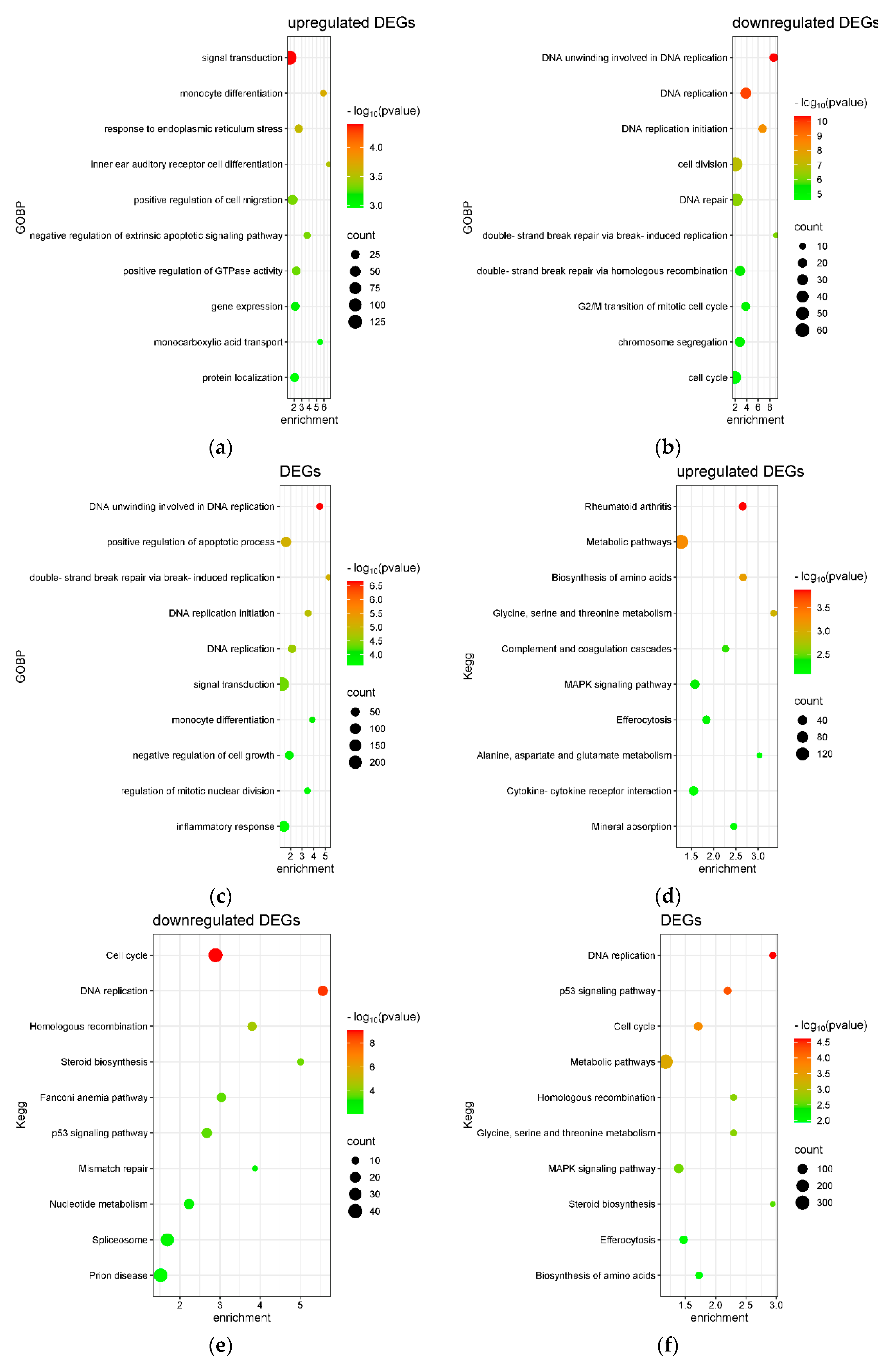
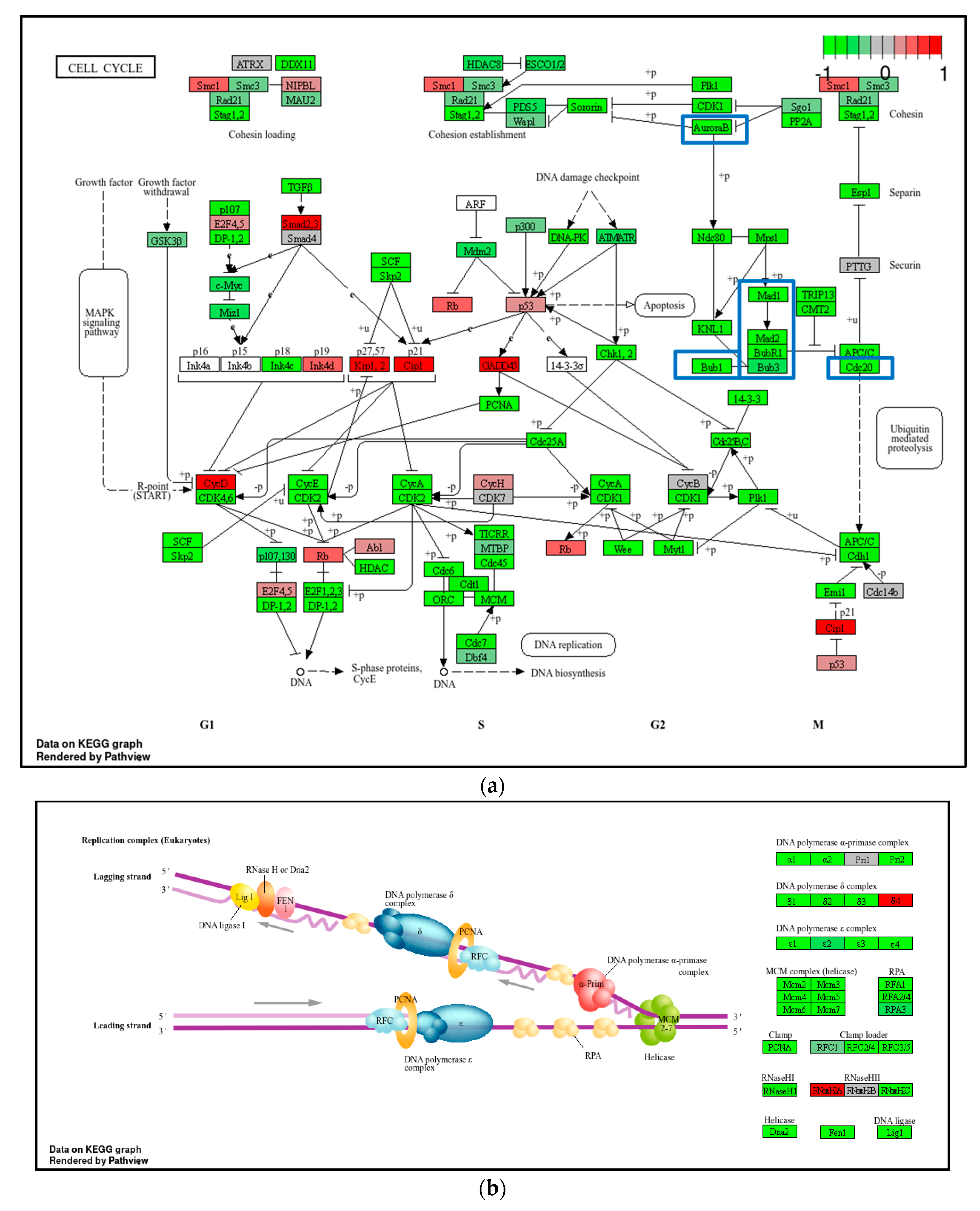
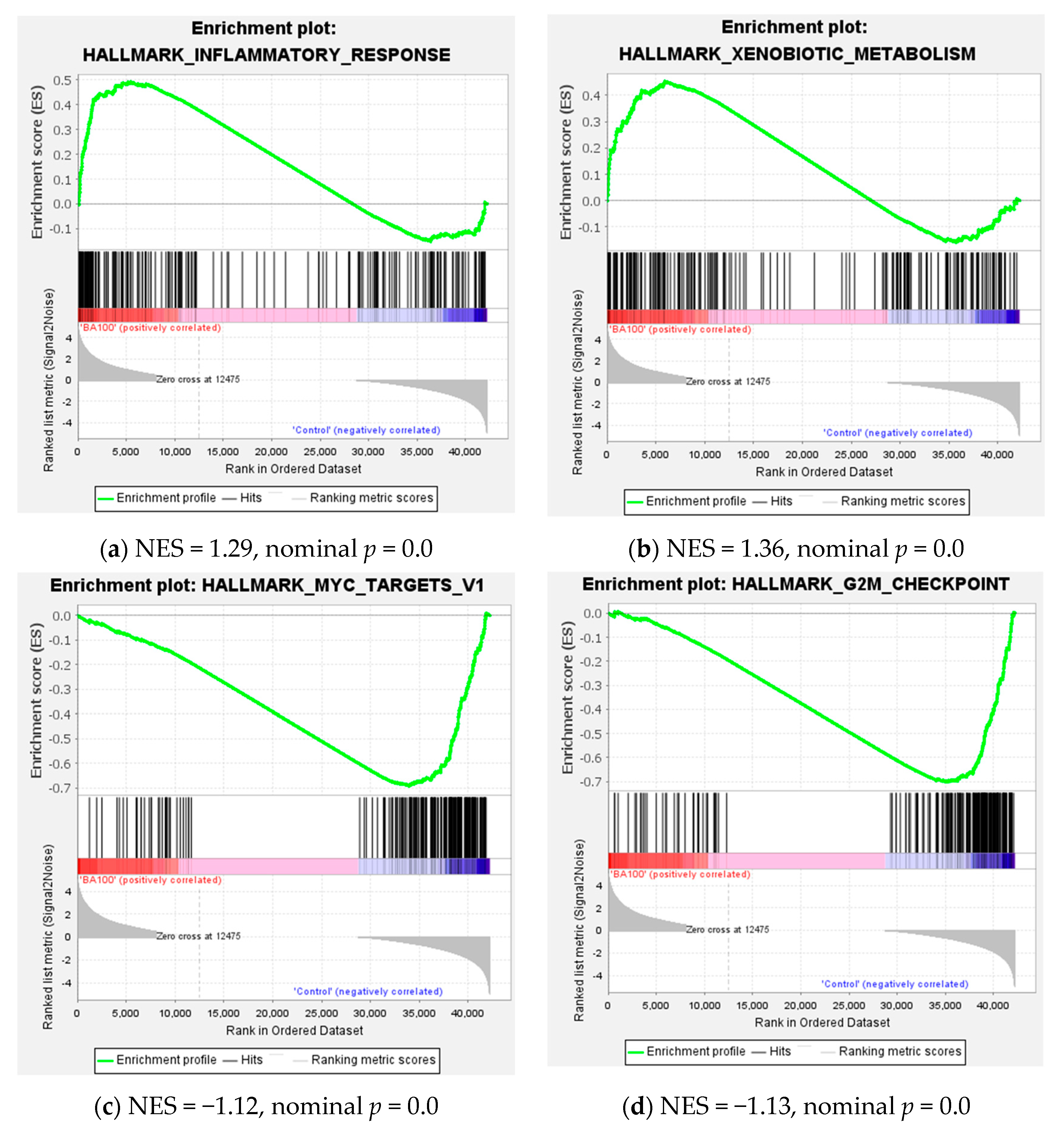
2.4. Transcriptome Analysis for Biochanin A-Treated THP-1 Cells: ORA and GSEA
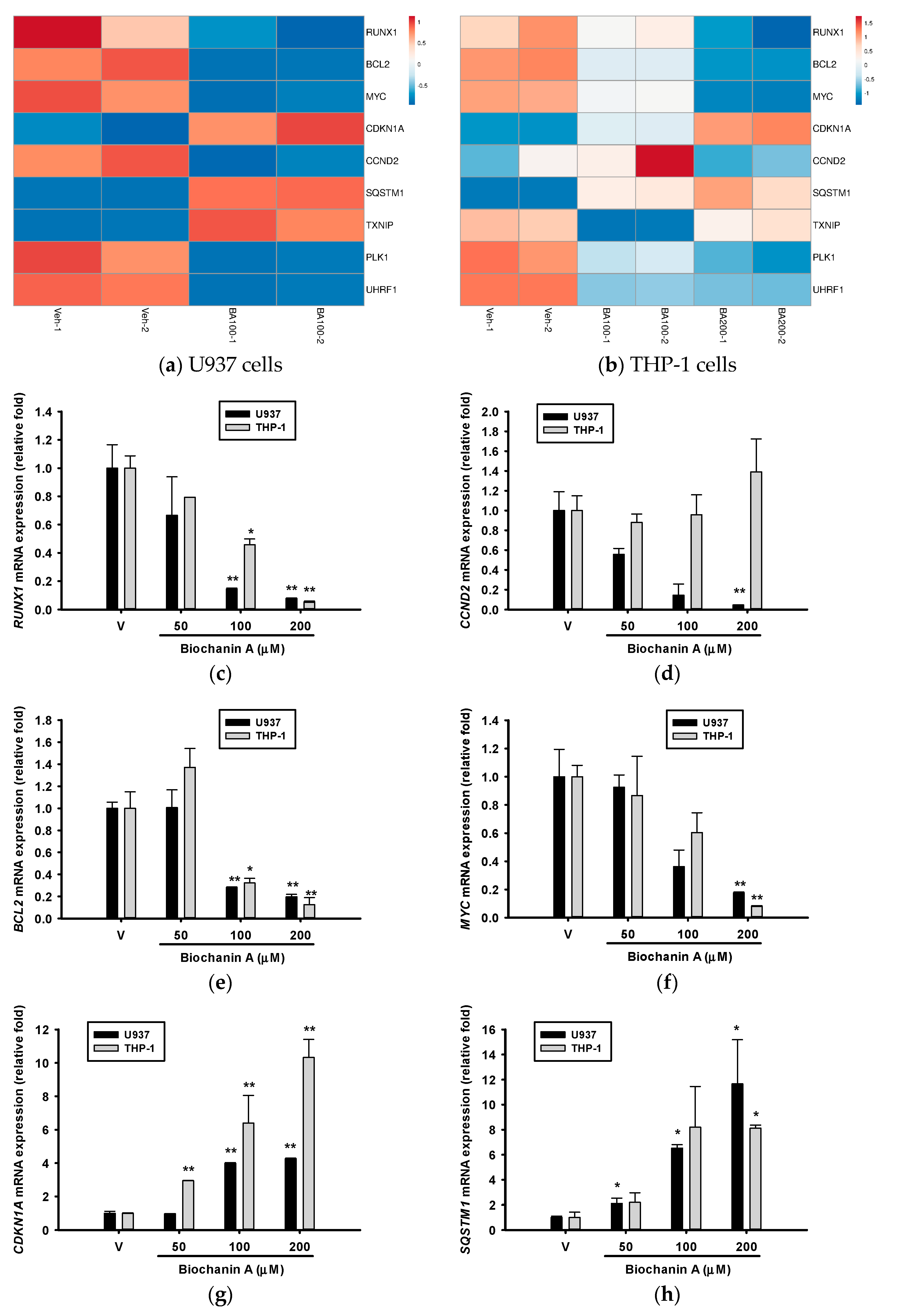
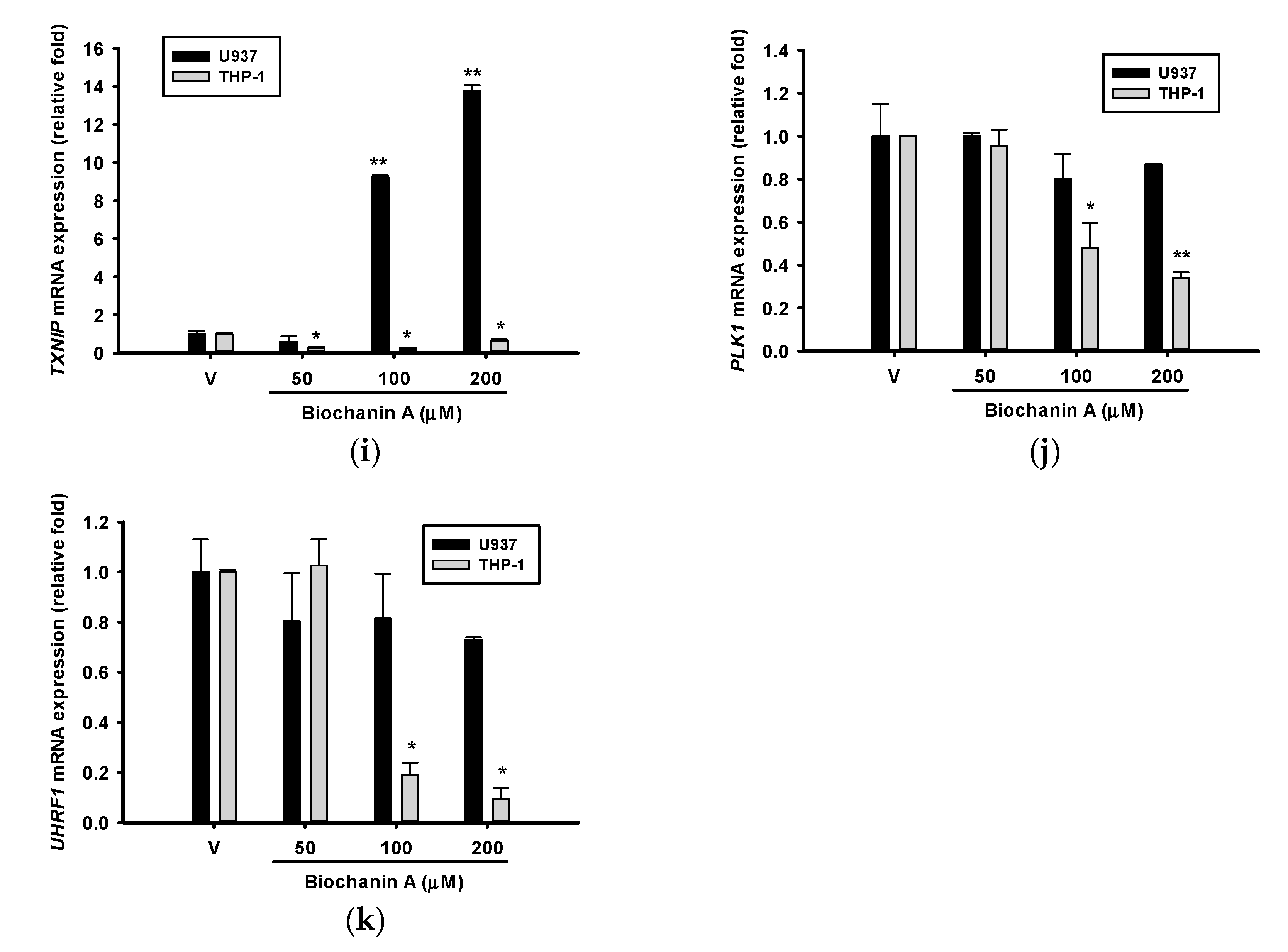
2.5. Effects of Biochanin A on AML-Related Gene Expression
3. Discussion
3.1. Transcriptomic Effects of Biochanin A in U937 and THP-1 Cells
3.2. Possible Cell-Type-Specific Mechanisms of Action of Biochanin A
3.3. Potential Off-Target Effects of Biochanin A Based on Transcriptomic Changes
3.4. Potential Usage of Biochanin A in TP53-Mutant AML Cells
3.5. Limitations of the Currrent Study
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Proliferation and Viability Analysis
4.4. Western Blot Analysis
4.5. Total RNA Isolation and RNA Sequencing
4.6. Overrepresentation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA)
4.7. Reverse Transcription Real-Time PCR (RT-qPCR)
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, C.; Zhang, P.; Lou, L.; Wang, Y. Perspectives Regarding the Role of Biochanin A in Humans. Front. Pharmacol. 2019, 10, 793. [Google Scholar] [CrossRef] [PubMed]
- Sohel, M. Comprehensive exploration of Biochanin A as an oncotherapeutics potential in the treatment of multivarious cancers with molecular insights. Phytother. Res. 2024, 38, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Sarfraz, A.; Javeed, M.; Shah, M.A.; Hussain, G.; Shafiq, N.; Sarfraz, I.; Riaz, A.; Sadiqa, A.; Zara, R.; Zafar, S.; et al. Biochanin A: A novel bioactive multifunctional compound from nature. Sci. Total Environ. 2020, 722, 137907. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, 52, W422–W431. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Sundaresan, A.; Radhiga, T.; Deivasigamani, B. Biological activity of biochanin A: A review. Asian J. Pharm. Pharmacol. 2018, 4, 1–5. [Google Scholar] [CrossRef]
- Gürbüz, P.; Şöhretoğlu, D.; Biochanin, A. Advances on Resources, Biosynthetic Pathway, Bioavailability, Bioactivity, and Pharmacology. In Handbook of Dietary Flavonoids; Xiao, J., Ed.; Springer International Publishing: Cham, Switzerland, 2023; pp. 1–27. [Google Scholar]
- Torrens-Mas, M.; Roca, P. Phytoestrogens for Cancer Prevention and Treatment. Biology 2020, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Liu, B.; Edgerton, S.; Yang, X.; Kim, A.; Ordonez-Ercan, D.; Mason, T.; Alvarez, K.; McKimmey, C.; Liu, N.; Thor, A. Low-Dose Dietary Phytoestrogen Abrogates Tamoxifen-Associated Mammary Tumor Prevention. Cancer Res. 2005, 65, 879–886. [Google Scholar] [CrossRef]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DiNardo, C.D.; Kadia, T.M.; Daver, N.G.; Altman, J.K.; Stein, E.M.; Jabbour, E.; Schiffer, C.A.; Lang, A.; Ravandi, F. Acute myeloid leukemia management and research in 2025. CA. Cancer J. Clin. 2025, 75, 46–67. [Google Scholar] [CrossRef] [PubMed]
- Sundström, C.; Nilsson, K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 1976, 17, 565–577. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef]
- Chiou, J.T.; Hsu, C.C.; Hong, Y.C.; Lee, Y.C.; Chang, L.S. Cytarabine-induced destabilization of MCL1 mRNA and protein triggers apoptosis in leukemia cells. Biochem. Pharmacol. 2023, 211, 115494. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, C.R.; Rodrigues Fernandes, N.A.; Gonzalez Maldonado, L.A.; Rossa Junior, C. Comparison of monocytic cell lines U937 and THP-1 as macrophage models for in vitro studies. Biochem. Biophys. Rep. 2022, 32, 101383. [Google Scholar] [CrossRef]
- van der Meer, D.; Barthorpe, S.; Yang, W.; Lightfoot, H.; Hall, C.; Gilbert, J.; Francies, H.E.; Garnett, M.J. Cell Model Passports-a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res. 2019, 47, D923–D929. [Google Scholar] [CrossRef]
- The Gene Ontology, C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Zambon, A.C.; Gaj, S.; Ho, I.; Hanspers, K.; Vranizan, K.; Evelo, C.T.; Conklin, B.R.; Pico, A.R.; Salomonis, N. GO-Elite: A flexible solution for pathway and ontology over-representation. Bioinformatics 2012, 28, 2209–2210. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Bruggisser, R.; von Daeniken, K.; Jundt, G.; Schaffner, W.; Tullberg-Reinert, H. Interference of plant extracts, phytoestrogens and antioxidants with the MTT tetrazolium assay. Planta Med. 2002, 68, 445–448. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Yamada, S.; Negi, S.; Bu, X.; Kato, J.; et al. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Grewal, S.S.; Li, L.; Orian, A.; Eisenman, R.N.; Edgar, B.A. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat. Cell Biol. 2005, 7, 295–302. [Google Scholar] [CrossRef]
- Verbist, B.; Klambauer, G.; Vervoort, L.; Talloen, W.; Consortium, Q.; Shkedy, Z.; Thas, O.; Bender, A.; Gohlmann, H.W.; Hochreiter, S. Using transcriptomics to guide lead optimization in drug discovery projects: Lessons learned from the QSTAR project. Drug Discov. Today 2015, 20, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.S.; Wang, C.Y.; Hsu, H.J.; Yen, J.H.; Wu, M.J. 8-Hydroxydaidzein Induces Apoptosis and Inhibits AML-Associated Gene Expression in U-937 Cells: Potential Phytochemical for AML Treatment. Biomolecules 2023, 13, 1575. [Google Scholar] [CrossRef]
- Wu, P.S.; Yen, J.H.; Wang, C.Y.; Chen, P.Y.; Hung, J.H.; Wu, M.J. 8-Hydroxydaidzein, an Isoflavone from Fermented Soybean, Induces Autophagy, Apoptosis, Differentiation, and Degradation of Oncoprotein BCR-ABL in K562 Cells. Biomedicines 2020, 8, 506. [Google Scholar] [CrossRef]
- Wu, P.S.; Wang, C.Y.; Chen, P.S.; Hung, J.H.; Yen, J.H.; Wu, M.J. 8-Hydroxydaidzein Downregulates JAK/STAT, MMP, Oxidative Phosphorylation, and PI3K/AKT Pathways in K562 Cells. Biomedicines 2021, 9, 1907. [Google Scholar] [CrossRef]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Ciciro, Y.; Ragusa, D.; Sala, A. Expression of the checkpoint kinase BUB1 is a predictor of response to cancer therapies. Sci. Rep. 2024, 14, 4461. [Google Scholar] [CrossRef]
- Maytin, E.V.; Ubeda, M.; Lin, J.C.; Habener, J.F. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp. Cell Res. 2001, 267, 193–204. [Google Scholar] [CrossRef]
- Feral, K.; Jaud, M.; Philippe, C.; Di Bella, D.; Pyronnet, S.; Rouault-Pierre, K.; Mazzolini, L.; Touriol, C. ER Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules 2021, 11, 199. [Google Scholar] [CrossRef]
- Kishtagari, A.; Levine, R.L.; Viny, A.D. Driver mutations in acute myeloid leukemia. Curr. Opin. Hematol. 2020, 27, 49–57. [Google Scholar] [CrossRef]
- Martinez-Soria, N.; McKenzie, L.; Draper, J.; Ptasinska, A.; Issa, H.; Potluri, S.; Blair, H.J.; Pickin, A.; Isa, A.; Chin, P.S.; et al. The Oncogenic Transcription Factor RUNX1/ETO Corrupts Cell Cycle Regulation to Drive Leukemic Transformation. Cancer Cell 2018, 34, 626–642.e628. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.; Spinella, J.-F.; Yao, C.-Y.; Lavallée, V.-P.; Boivin, I.; Boucher, G.; Audemard, E.; Bordeleau, M.-E.; Lemieux, S.; Hébert, J.; et al. High frequency of germline RUNX1 mutations in patients with RUNX1-mutated AML. Blood 2020, 135, 1882–1886. [Google Scholar] [CrossRef]
- Mill, C.P.; Fiskus, W.; DiNardo, C.D.; Qian, Y.; Raina, K.; Rajapakshe, K.; Perera, D.; Coarfa, C.; Kadia, T.M.; Khoury, J.D.; et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood 2019, 134, 59–73. [Google Scholar] [CrossRef]
- Gerritsen, M.; Yi, G.; Tijchon, E.; Kuster, J.; Schuringa, J.J.; Martens, J.H.A.; Vellenga, E. RUNX1 mutations enhance self-renewal and block granulocytic differentiation in human in vitro models and primary AMLs. Blood Adv. 2019, 3, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, F.; Barthélémy, A.; Peyrouze, P.; Fenwarth, L.; Preudhomme, C.; Duployez, N.; Cheok, M.H. Targeting RUNX1 in acute myeloid leukemia: Preclinical innovations and therapeutic implications. Expert Opin. Ther. Targets 2021, 25, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Tsuchiya, A.; Tamura, M.; Yamamoto, K.; Kawata, S.; Ishihara-Sugano, M.; Kato, M.; Kitamura, T.; Goyama, S. RUNX1 Inhibition Using Lipid Nanoparticle-Mediated Silencing RNA Delivery as an Effective Treatment for Acute Leukemias. Exp. Hematol. 2022, 112, 1–8. [Google Scholar] [CrossRef]
- Eisfeld, A.K.; Kohlschmidt, J.; Schwind, S.; Nicolet, D.; Blachly, J.S.; Orwick, S.; Shah, C.; Bainazar, M.; Kroll, K.W.; Walker, C.J.; et al. Mutations in the CCND1 and CCND2 genes are frequent events in adult patients with t(8;21)(q22;q22) acute myeloid leukemia. Leukemia 2017, 31, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Destefanis, F.; Manara, V.; Bellosta, P. Myc as a Regulator of Ribosome Biogenesis and Cell Competition: A Link to Cancer. Int. J. Mol. Sci. 2020, 21, 4037. [Google Scholar] [CrossRef]
- Jung, P.; Menssen, A.; Mayr, D.; Hermeking, H. AP4 encodes a c-MYC-inducible repressor of p21. Proc. Natl. Acad. Sci. USA 2008, 105, 15046–15051. [Google Scholar] [CrossRef]
- El-Deiry, W.S. p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res. 2016, 76, 5189–5191. [Google Scholar] [CrossRef]
- Local, A.; Zhang, H.; Benbatoul, K.D.; Folger, P.; Sheng, X.; Tsai, C.Y.; Howell, S.B.; Rice, W.G. APTO-253 Stabilizes G-quadruplex DNA, Inhibits MYC Expression, and Induces DNA Damage in Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2018, 17, 1177–1186. [Google Scholar] [CrossRef]
- Trocoli, A.; Bensadoun, P.; Richard, E.; Labrunie, G.; Merhi, F.; Schlafli, A.M.; Brigger, D.; Souquere, S.; Pierron, G.; Pasquet, J.M.; et al. p62/SQSTM1 upregulation constitutes a survival mechanism that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ. 2014, 21, 1852–1861. [Google Scholar] [CrossRef]
- Wang, X.; Nachliely, M.; Harrison, J.S.; Danilenko, M.; Studzinski, G.P. Participation of vitamin D-upregulated protein 1 (TXNIP)-ASK1-JNK1 signalosome in the enhancement of AML cell death by a post-cytotoxic differentiation regimen. J. Steroid Biochem. Mol. Biol. 2019, 187, 166–173. [Google Scholar] [CrossRef]
- Zhou, J.; Bi, C.; Cheong, L.L.; Mahara, S.; Liu, S.C.; Tay, K.G.; Koh, T.L.; Yu, Q.; Chng, W.J. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 2011, 118, 2830–2839. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef]
- Renner, A.G.; Dos Santos, C.; Recher, C.; Bailly, C.; Créancier, L.; Kruczynski, A.; Payrastre, B.; Manenti, S. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 2009, 114, 659–662. [Google Scholar] [CrossRef]
- Liu, Q.; Subedi, A.; Yao, S.; Xu, C.; Voisin, V.; Bader, G.D.; Chan, S.M.; Wang, J.C.Y. A Stemness-Based Screen Identifies PLK1 Inhibitors for Targeting Leukemia Stem Cells in AML. Blood 2020, 136, 34–35. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Ridinger, M.; Croucher, P.J.P.; Samuëlsz, E.; Erlander, M.; Ruffner, K.L.; Wang, E.S. Predictive Biomarkers of Response to the Polo-like Kinase 1 (PLK1) Inhibitor, Onvansertib, in Combination with Decitabine in Relapsed or Refractory Acute Myeloid Leukemia (R/R AML). Blood 2021, 138, 3431. [Google Scholar] [CrossRef]
- Kolosenko, I.; Goroshchuk, O.; Vidarsdottir, L.; Bjorklund, A.C.; Dowdy, S.F.; Palm-Apergi, C. RNAi prodrugs decrease elevated mRNA levels of Polo-like kinase 1 in ex vivo cultured primary cells from pediatric acute myeloid leukemia patients. FASEB J. 2021, 35, e21476. [Google Scholar] [CrossRef]
- Hu, C.L.; Chen, B.Y.; Li, Z.; Yang, T.; Xu, C.H.; Yang, R.; Yu, P.C.; Zhao, J.; Liu, T.; Liu, N.; et al. Targeting UHRF1-SAP30-MXD4 axis for leukemia initiating cell eradication in myeloid leukemia. Cell Res. 2022, 32, 1105–1123. [Google Scholar] [CrossRef]
- Park, J.; Luo, Y.; Park, J.W.; Kim, S.H.; Hong, Y.J.; Lim, Y.; Seo, Y.J.; Bae, J.; Seo, S.B. Downregulation of DNA methylation enhances differentiation of THP-1 cells and induces M1 polarization of differentiated macrophages. Sci. Rep. 2023, 13, 13132. [Google Scholar] [CrossRef]
- Houliston, R.S.; Lemak, A.; Iqbal, A.; Ivanochko, D.; Duan, S.; Kaustov, L.; Ong, M.S.; Fan, L.; Senisterra, G.; Brown, P.J.; et al. Conformational dynamics of the TTD-PHD histone reader module of the UHRF1 epigenetic regulator reveals multiple histone-binding states, allosteric regulation, and druggability. J. Biol. Chem. 2017, 292, 20947–20959. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Cartron, P.F.; Liutkevičiūtė, Z.; Klimašauskas, S.; Matulis, D.; Bronner, C.; Martinet, N.; Mikros, E. Tandem virtual screening targeting the SRA domain of UHRF1 identifies a novel chemical tool modulating DNA methylation. Eur. J. Med. Chem. 2016, 114, 390–396. [Google Scholar] [CrossRef]
- Seo, J.S.; Choi, Y.H.; Moon, J.W.; Kim, H.S.; Park, S.H. Hinokitiol induces DNA demethylation via DNMT1 and UHRF1 inhibition in colon cancer cells. BMC Cell Biol. 2017, 18, 14. [Google Scholar] [CrossRef]
- Gazova, I.; Lefevre, L.; Bush, S.J.; Clohisey, S.; Arner, E.; de Hoon, M.; Severin, J.; van Duin, L.; Andersson, R.; Lengeling, A.; et al. The Transcriptional Network That Controls Growth Arrest and Macrophage Differentiation in the Human Myeloid Leukemia Cell Line THP-1. Front. Cell Dev. Biol. 2020, 8, 498. [Google Scholar] [CrossRef]
- Wolff, L.; Schmidt, M.; Koller, R.; Haviernik, P.; Watson, R.; Bies, J.; Maciag, K. Three Genes with Different Functions in Transformation are Regulated by c-Myb in Myeloid Cells. Blood Cells. Mol. Dis. 2001, 27, 483–488. [Google Scholar] [CrossRef]
- Ciciro, Y.; Sala, A. MYB oncoproteins: Emerging players and potential therapeutic targets in human cancer. Oncogenesis 2021, 10, 19. [Google Scholar] [CrossRef]
- Choi, A.; Illendula, A.; Pulikkan, J.A.; Roderick, J.E.; Tesell, J.; Yu, J.; Hermance, N.; Zhu, L.J.; Castilla, L.H.; Bushweller, J.H.; et al. RUNX1 is required for oncogenic Myb and Myc enhancer activity in T-cell acute lymphoblastic leukemia. Blood 2017, 130, 1722–1733. [Google Scholar] [CrossRef]
- Fuster, O.; Llop, M.; Dolz, S.; García, P.; Such, E.; Ibáñez, M.; Luna, I.; Gómez, I.; López, M.; Cervera, J.; et al. Adverse prognostic value of MYBL2 overexpression and association with microRNA-30 family in acute myeloid leukemia patients. Leuk. Res. 2013, 37, 1690–1696. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Choi, E.H.; Park, S.J. TXNIP: A key protein in the cellular stress response pathway and a potential therapeutic target. Exp. Mol. Med. 2023, 55, 1348–1356. [Google Scholar] [CrossRef]
- Takaki, T.; Trenz, K.; Costanzo, V.; Petronczki, M. Polo-like kinase 1 reaches beyond mitosis--cytokinesis, DNA damage response, and development. Curr. Opin. Cell Biol. 2008, 20, 650–660. [Google Scholar] [CrossRef]
- Bronner, C.; Achour, M.; Arima, Y.; Chataigneau, T.; Saya, H.; Schini-Kerth, V.B. The UHRF family: Oncogenes that are drugable targets for cancer therapy in the near future? Pharmacol. Ther. 2007, 115, 419–434. [Google Scholar] [CrossRef]
- Hu, A.; Sun, L.; Lin, H.; Liao, Y.; Yang, H.; Mao, Y. Harnessing innate immune pathways for therapeutic advancement in cancer. Signal Transduct. Target. Ther. 2024, 9, 68. [Google Scholar] [CrossRef]
- Dvinge, H.; Bradley, R.K. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015, 7, 45. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Ren, F.; Ma, Y.; Zhang, K.; Luo, Y.; Pan, R.; Zhang, J.; Kan, C.; Hou, N.; Han, F.; Sun, X. Exploring the multi-targeting phytoestrogen potential of Calycosin for cancer treatment: A review. Medicine 2024, 103, e38023. [Google Scholar] [CrossRef]
- Cimmino, A.; Fasciglione, G.F.; Gioia, M.; Marini, S.; Ciaccio, C. Multi-Anticancer Activities of Phytoestrogens in Human Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 13344. [Google Scholar] [CrossRef]
- Banerjee, S.; Li, Y.; Wang, Z.; Sarkar, F.H. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008, 269, 226–242. [Google Scholar] [CrossRef]
- DiGennaro, J.; Sallman, D.A. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Current Guidelines, Therapies, and Future Considerations. Acta Haematol. 2024, 147, 175–185. [Google Scholar] [CrossRef]
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604. [Google Scholar] [CrossRef]
- Santini, V.; Stahl, M.; Sallman, D.A. TP53 Mutations in Acute Leukemias and Myelodysplastic Syndromes: Insights and Treatment Updates. Am. Soc. Clin. Oncol. Educ. Book. 2024, 44, e432650. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Rahimian, E.; Rahimi, S.; Zarandi, B.; Bahraini, M.; Soleymani, M.; Safdari, S.M.; Shabannezhad, A.; Jaafari, N.; Safa, M. From regulation to deregulation of p53 in hematologic malignancies: Implications for diagnosis, prognosis and therapy. Biomark. Res. 2024, 12, 137. [Google Scholar] [CrossRef]
- Mihailidou, C.; Papazian, I.; Papavassiliou, A.G.; Kiaris, H. CHOP-dependent regulation of p21/waf1 during ER stress. Cell. Physiol. Biochem. 2010, 25, 761–766. [Google Scholar] [CrossRef]
- Han, H.K.; Lee, B.J.; Lee, H.K. Enhanced dissolution and bioavailability of biochanin A via the preparation of solid dispersion: In vitro and in vivo evaluation. Int. J. Pharm. 2011, 415, 89–94. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, H.; Zhou, K.; Wang, L.; Dong, S.; Wang, D.; Chen, W. Nanostructured lipid carriers as a delivery system of biochanin A. Drug Deliv. 2013, 20, 331–337. [Google Scholar] [CrossRef]
- Wang, L.; Luo, Q.; Lin, T.; Li, R.; Zhu, T.; Zhou, K.; Ji, Z.; Song, J.; Jia, B.; Zhang, C.; et al. PEGylated nanostructured lipid carriers (PEG-NLC) as a novel drug delivery system for biochanin A. Drug Dev. Ind. Pharm. 2015, 41, 1204–1212. [Google Scholar] [CrossRef]
- Wu, X.; Ge, W.; Shao, T.; Wu, W.; Hou, J.; Cui, L.; Wang, J.; Zhang, Z. Enhancing the oral bioavailability of biochanin A by encapsulation in mixed micelles containing Pluronic F127 and Plasdone S630. Int. J. Nanomed. 2017, 12, 1475–1483. [Google Scholar] [CrossRef]
- Singh, S.K.; Rashid, M.; Bhalala, K.; Malik, Y.; Chaturvedi, S.; Raju, K.S.R.; Sultana, N.; Mitra, K.; Gayen, J.R.; Wahajuddin, M. A novel nanosized phospholipid complex of Biochanin A for improving oral bioavailability: Preparation and in-vitro/in-vivo characterizations. J. Drug Deliv. Sci. Technol. 2021, 61, 102254. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3-B. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Catalog Number |
|---|---|---|
| β-Actin | Proteintech | 20536-1-AP |
| Caspase 7 | Cell Signaling | 9492 |
| Cleaved Caspase 7 | Cell Signaling | 9491 |
| BCL-2 | Genetex | GTX100064 |
| CHOP/GADD153 | Genetex | GTX112827 |
| c-Myc | Genetex | GTX103436 |
| PARP-1 | Santa cruz | sc-7150 |
| Gene | Primer Sequence (5′-3′) | Size (bp) | |
|---|---|---|---|
| GAPDH | F | CATGAGAAGTATGACAACAGCCT | 113 |
| R | AGTCCTTCCACGATACCAAAGT | ||
| CCND2 | F | TTTGCCATGTACCCACCGTC | 104 |
| R | AGGGCATCACAAGTGAGCG | ||
| BCL2 | F | GGTGGGGTCATGTGTGTGG | 89 |
| R | GTCGTTTCTTGCCACTGATGA | ||
| MYC | F | CGGTTCAGGTACTCAGTCATCC | 162 |
| R | TTGGACGGACAGGATGTATGC | ||
| CDKN1A | F | AGGTGGACCTGGAGACTCTCAG | 95 |
| R | TCCTCTTGGAGAAGATCAGCCG | ||
| SQSTM1 (p62) | F | AAGCCGGGTGGGAATGTTG | 116 |
| R | CCTGAACAGTTATCCGACTCCAT | ||
| RUNX1 | F | CTTGTCTCCACTGAGGCACA | 133 |
| R | CTGTGTAGGGGAGCCACATT | ||
| TXNIP | F | ATATGGGTGTGTAGACTACTGGG | 103 |
| R | GACATCCACCAGATCCACTACT | ||
| PLK1 | F | TGACTCAACACGCCTCATCC | 155 |
| R | GCTCGCTCATGTAATTGCGG | ||
| UHRF1 | F | AGGTGGTCATGCTCAACTACA | 116 |
| R | CACGTTGGCGTAGAGTTCCC | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, P.-S.; Yen, J.-H.; Chen, P.-Y.; Wu, M.-J. Molecular Mechanisms of Biochanin A in AML Cells: Apoptosis Induction and Pathway-Specific Regulation in U937 and THP-1. Int. J. Mol. Sci. 2025, 26, 5317. https://doi.org/10.3390/ijms26115317
Wu P-S, Yen J-H, Chen P-Y, Wu M-J. Molecular Mechanisms of Biochanin A in AML Cells: Apoptosis Induction and Pathway-Specific Regulation in U937 and THP-1. International Journal of Molecular Sciences. 2025; 26(11):5317. https://doi.org/10.3390/ijms26115317
Chicago/Turabian StyleWu, Pei-Shan, Jui-Hung Yen, Pei-Yi Chen, and Ming-Jiuan Wu. 2025. "Molecular Mechanisms of Biochanin A in AML Cells: Apoptosis Induction and Pathway-Specific Regulation in U937 and THP-1" International Journal of Molecular Sciences 26, no. 11: 5317. https://doi.org/10.3390/ijms26115317
APA StyleWu, P.-S., Yen, J.-H., Chen, P.-Y., & Wu, M.-J. (2025). Molecular Mechanisms of Biochanin A in AML Cells: Apoptosis Induction and Pathway-Specific Regulation in U937 and THP-1. International Journal of Molecular Sciences, 26(11), 5317. https://doi.org/10.3390/ijms26115317