
Genome-Wide Identification and Characterization of AGO, DCL, and RDR Gene Families in Siraitia grosvenorii

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Identification and Structural Analysis of SgAGO, SgDCL, and SgRDR Genes in S. grosvenorii
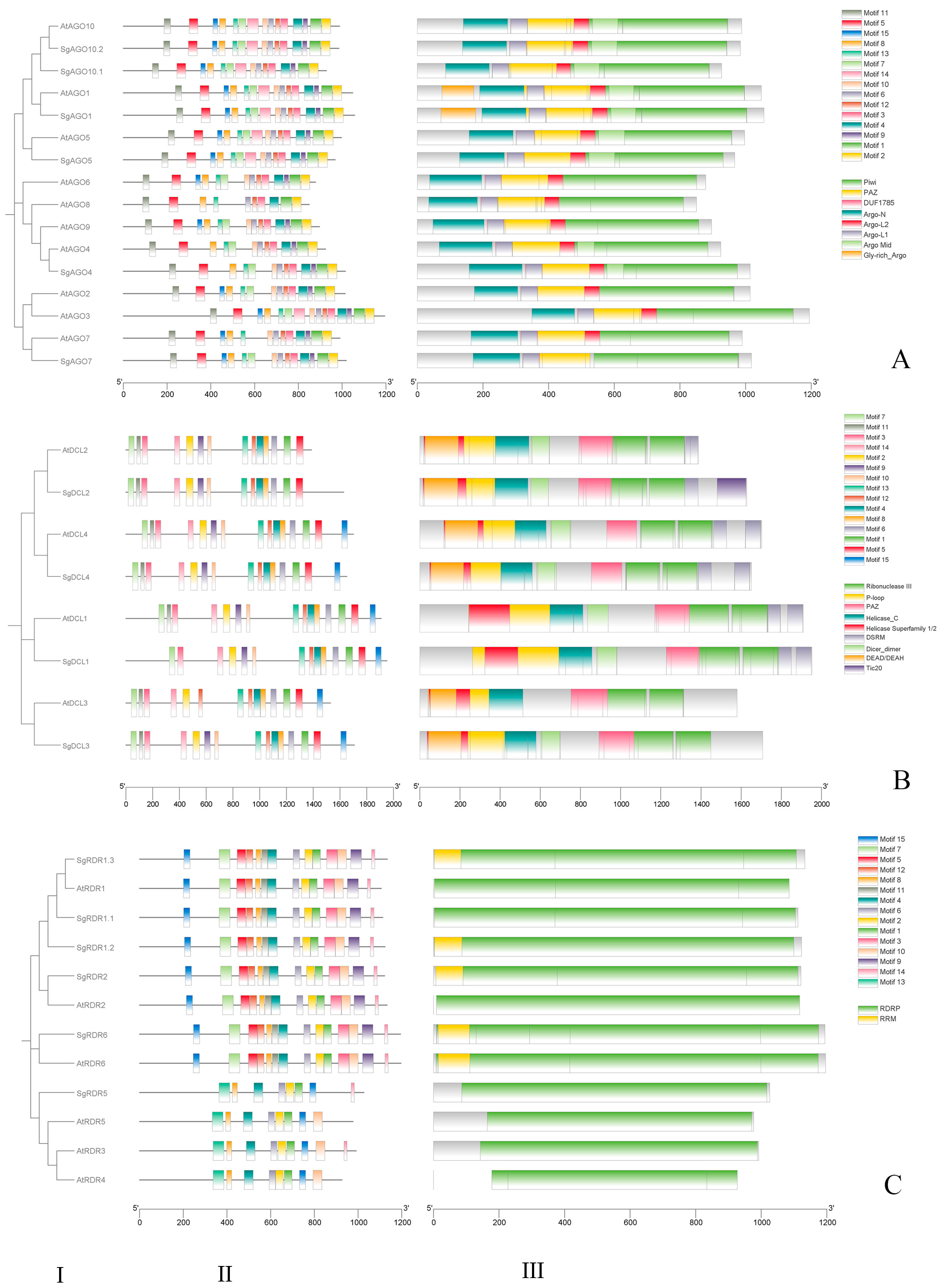
2.2. Motif and Domain Analyses of SgAGO, SgDCL, and SgRDR in S. grosvenorii
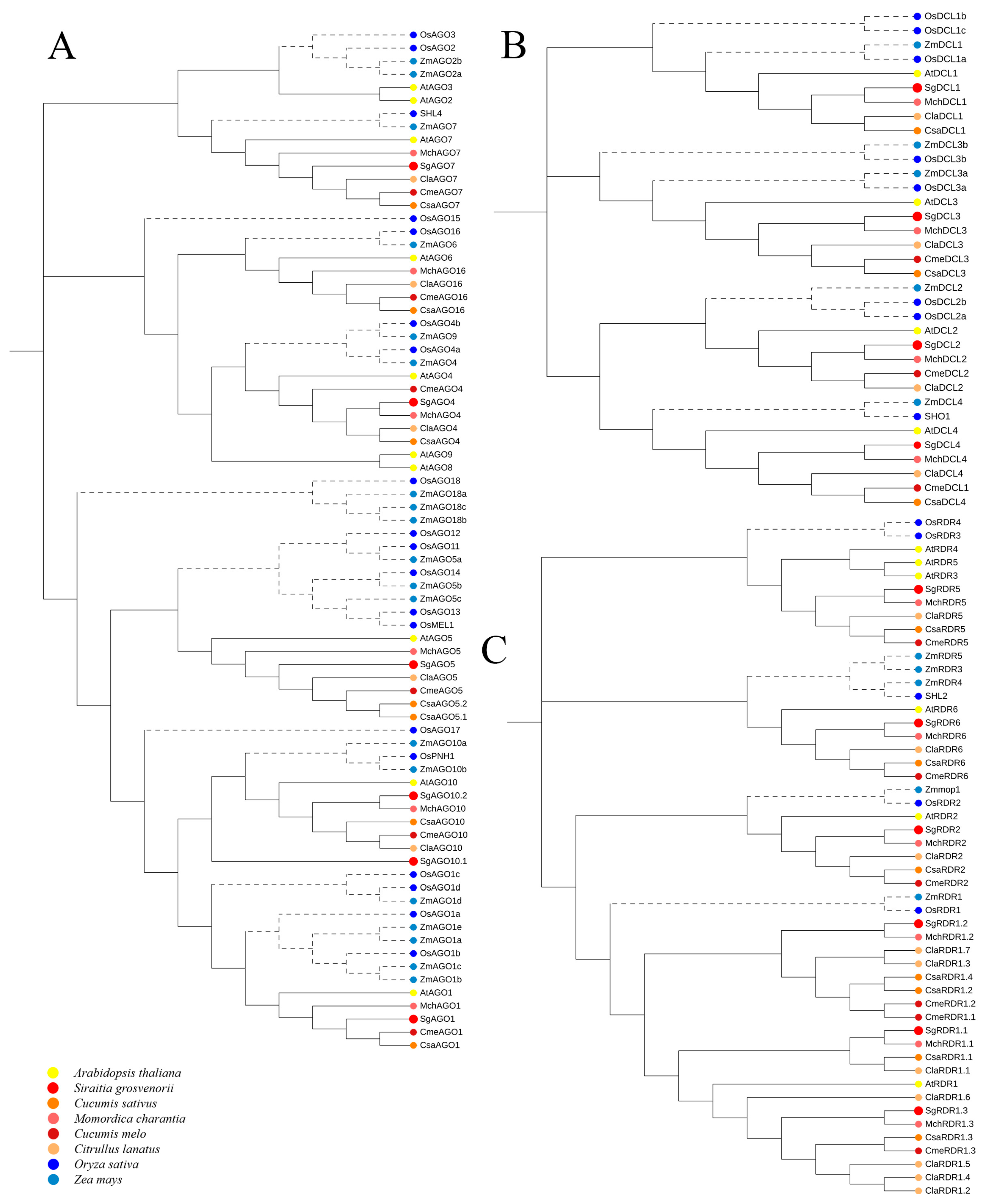
2.3. Phylogenetic Analysis of SgAGO, SgDCL, and SgRDR in S. grosvenorii and Other Plant Species
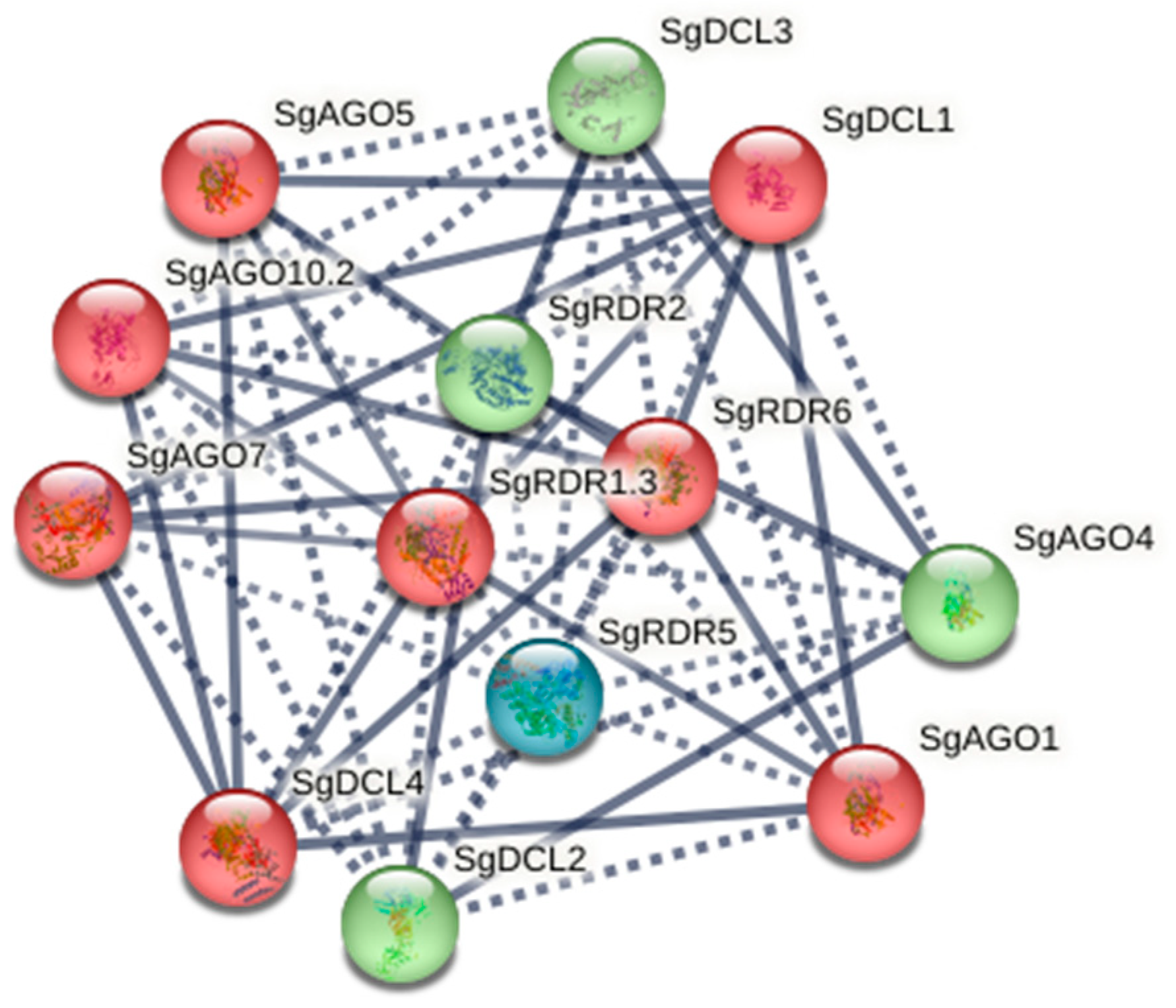
2.4. Protein–Protein Interactions of SgAGO, SgDCL, and SgRDR
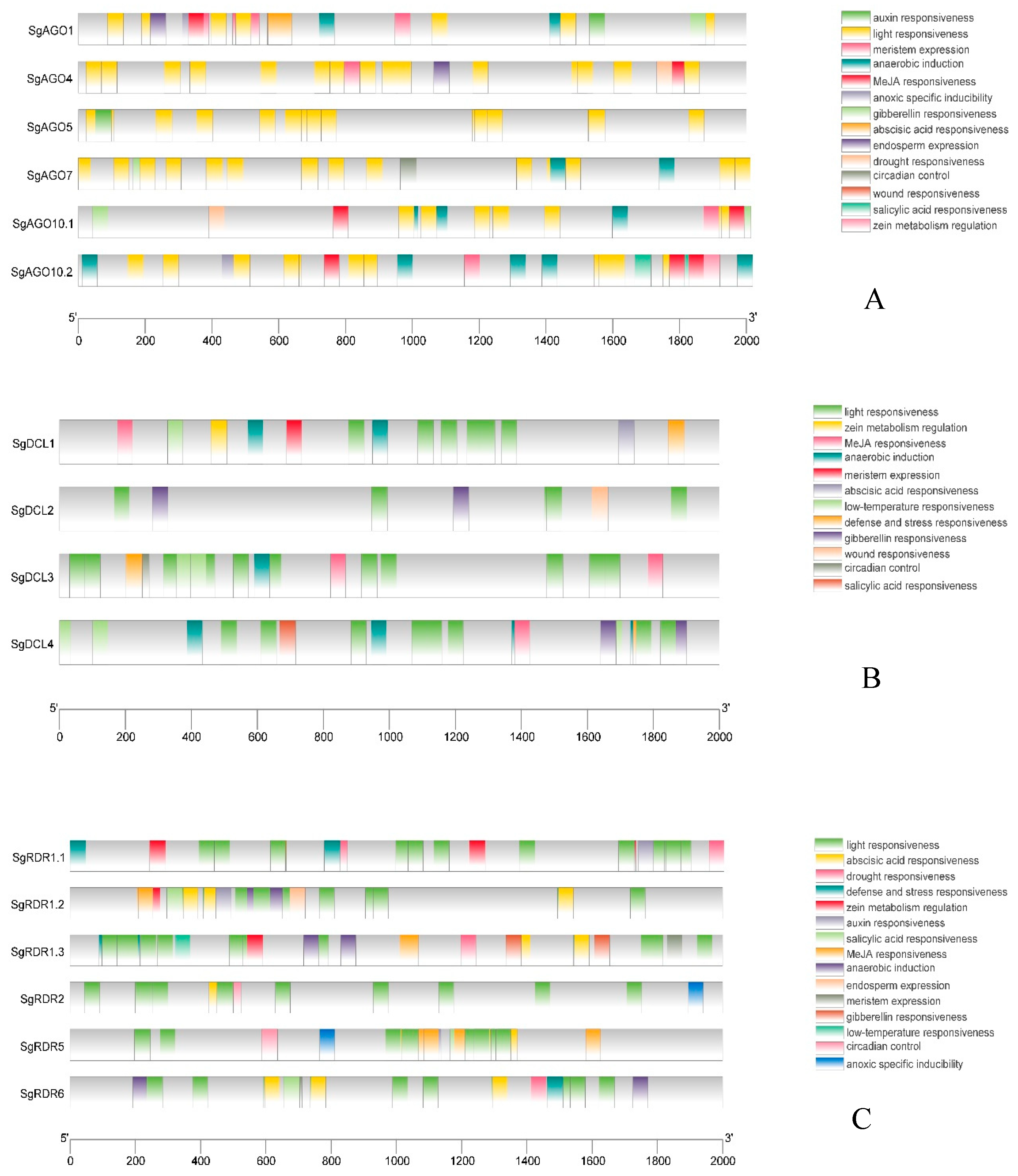
2.5. Cis-Acting Element Analysis in SgAGO, SgDCL, and SgRDR Genes
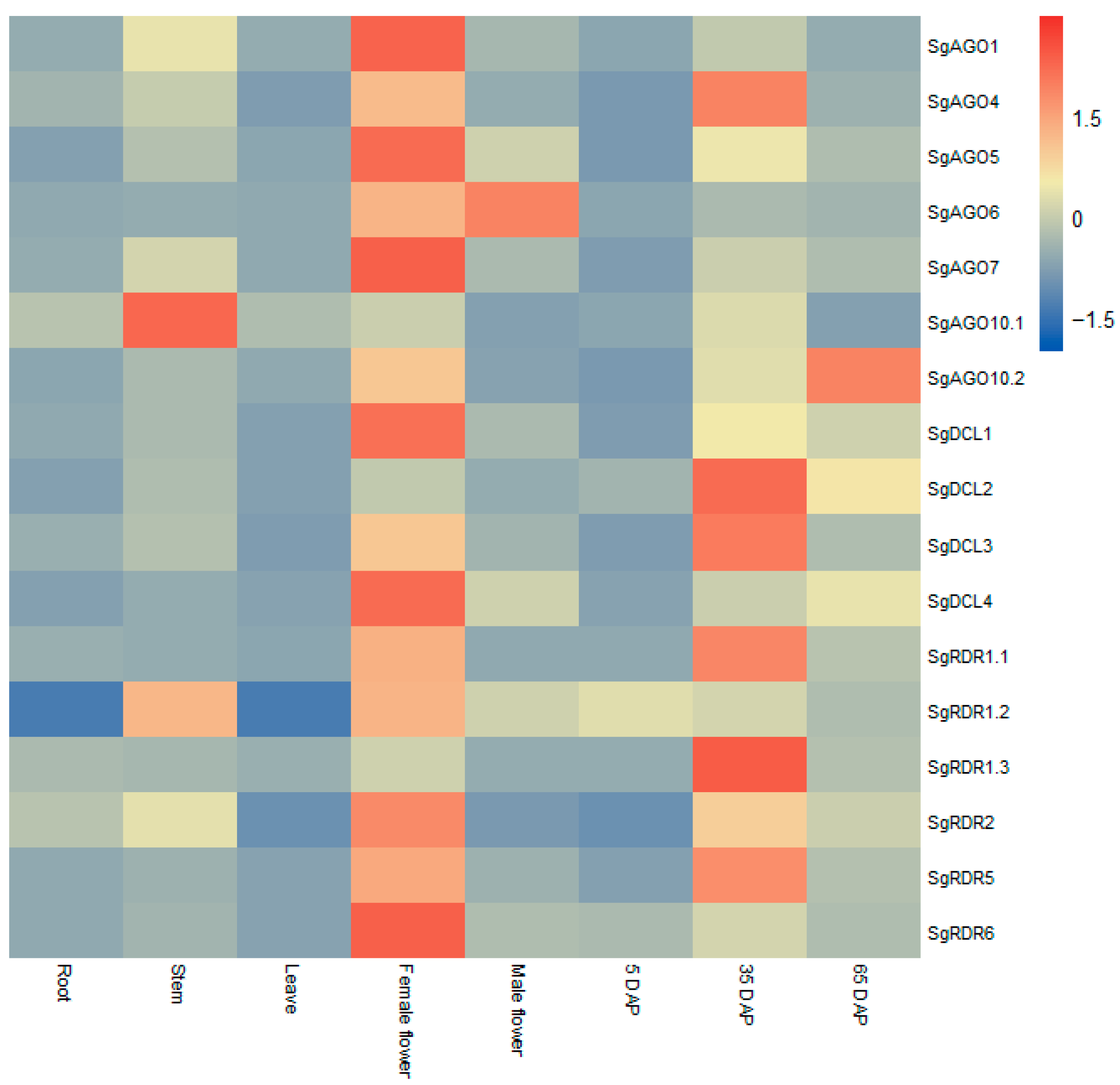
2.6. Tissue-Specific Expression Patterns of SgAGO, SgDCL, and SgRDR Genes
3. Discussion
3.1. Evolution and Functional Divergence Mechanisms of RNAi Pathway Gene Families
3.2. Cis-Acting Element Analysis Reveals Hormonal and Stress Response Regulatory Mechanisms of RNAi Core Genes in S.grosvenorii
3.3. Functional Divergence of AGO, DCL, and RDR Gene Families in Tissue-Specific Expression and Developmental Regulation in S. grosvenorii
4. Materials and Methods
4.1. Plant Material
4.2. Data Collection and Identification of AGO, DCL, and RDR in S. grosvenorii
4.3. Phylogenetic Tree Construction
4.4. Conserved Motif and Gene Structure Analysis
4.5. Gene Expression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABA | Abscisic Acid |
| AGO | Argonaute |
| DAP | Days After Pollination |
| DCL | Dicer-like |
| DSRM | Double-stranded RNA-binding domain |
| dsRNA | Double-stranded RNA |
| GA | Gibberellin |
| GRAVY | grand average of hydropathicity |
| ICL | Isocitrate lyase |
| MeJA | Methyl Jasmonate |
| MEME | Multiple Em for Motif Elicitation |
| ORF | open reading frame |
| PPI | Protein–protein interaction |
| qRT-PCR | Quantitative real-time PCR |
| RDR | RNA-dependent RNA polymerase |
| RdDM | RNA-directed DNA methylation |
| RNAi | RNA interference |
| RISC | RNA-induced silencing complex |
| SA | Salicylic Acid |
| sRNAs | small RNAs |
| siRNAs | small interfering RNAs |
| UBQ | Ubiquitin |
References
- Bologna, N.G.; Voinnet, O. The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu. Rev. Plant Biol. 2014, 65, 473–503. [Google Scholar] [CrossRef]
- Wilson, R.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Matzke, M.A. The small RNA world. J. Cell Sci. 2003, 116, 4689–4693. [Google Scholar] [CrossRef]
- Duan, C.-G.; Zhu, J.-K. Plant Argonaute Proteins; Springer: New York, NY, USA, 2017; pp. 129–135. [Google Scholar]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef]
- Moazed, D. Small RNAs in transcriptional gene silencing and genome defence. Nature 2009, 457, 413–420. [Google Scholar] [CrossRef]
- Ahmed, F.F.; Hossen, I.; Sarkar, A.R.; Konak, J.N.; Zohra, F.T. Genome-wide identification of DCL, AGO and RDR gene families and their associated functional regulatory elements analyses in banana (Musa acuminata). PLoS ONE 2021, 16, e0256873. [Google Scholar] [CrossRef]
- Höck, J.; Meister, G. The Argonaute protein family. Genome Biol. 2008, 9, 210. [Google Scholar] [CrossRef]
- Singh, J.; Mishra, V.; Wang, F.; Huang, H.-Y.; Pikaard, C.S. Reaction mechanisms of Pol IV, RDR2 and DCL3 drive RNA channeling in the siRNA-directed DNA methylation pathway. Mol. Cell 2019, 75, 576–589.e5. [Google Scholar] [CrossRef]
- Deleris, A.; Gallego-Bartolome, J.; Bao, J.; Kasschau, K.D.; Carrington, J.C.; Voinnet, O. Hierarchical action and inhibition of plant dicer-like proteins in antiviral defense. Science 2006, 313, 68–71. [Google Scholar] [CrossRef]
- Bouché, N.; Lauressergues, D.; Gasciolli, V.; Vaucheret, H. An antagonistic function for Arabidopsis DCL2 in development and a new function for DCL4 in generating viral siRNAs. EMBO J. 2006, 25, 3347–3356. [Google Scholar] [CrossRef]
- Margis, R.; Fusaro, A.F.; Smith, N.A.; Curtin, S.J.; Watson, J.M.; Finnegan, E.J.; Waterhouse, P.M. The evolution and diversification of Dicers in plants. FEBS Lett. 2006, 580, 2442–2450. [Google Scholar] [CrossRef]
- Vrbsky, J.; Akimcheva, S.; Watson, J.M.; Turner, T.L.; Daxinger, L.; Vyskot, B.; Aufsatz, W.; Riha, K. siRNA–mediated methylation of Arabidopsis telomeres. PLoS Genet. 2010, 6, e1000986. [Google Scholar] [CrossRef]
- Chan, S.W.-L.; Zilberman, D.; Xie, Z.; Johansen, L.K.; Carrington, J.C.; Jacobsen, S.E. RNA silencing genes control de novo DNA methylation. Science 2004, 303, 1336. [Google Scholar] [CrossRef]
- Fang, Y.; Spector, D.L. Identification of nuclear dicing bodies containing proteins for microRNA biogenesis in living Arabidopsis plants. Curr. Biol. 2007, 17, 818–823. [Google Scholar] [CrossRef]
- Qin, L.; Mo, N.; Muhammad, T.; Liang, Y. Genome-wide analysis of DCL, AGO, and RDR gene families in pepper (Capsicum annuum L.). Int. J. Mol. Sci. 2018, 19, 1038. [Google Scholar] [CrossRef]
- Podder, A.; Ahmed, F.F.; Suman, Z.H.; Mim, A.Y.; Hasan, K. Genome-wide identification of DCL, AGO and RDR gene families and their associated functional regulatory element analyses in sunflower (Helianthus annuus). PLoS ONE 2023, 18, e0286994. [Google Scholar] [CrossRef]
- Song, L.; Han, M.-H.; Lesicka, J.; Fedoroff, N. Arabidopsis primary microRNA processing proteins HYL1 and DCL1 define a nuclear body distinct from the Cajal body. Proc. Natl. Acad. Sci. USA 2007, 104, 5437–5442. [Google Scholar] [CrossRef]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004, 2, e104. [Google Scholar] [CrossRef]
- Kapoor, M.; Arora, R.; Lama, T.; Nijhawan, A.; Khurana, J.P.; Tyagi, A.K.; Kapoor, S. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genom. 2008, 9, 451. [Google Scholar] [CrossRef]
- Qian, Y.; Cheng, Y.; Cheng, X.; Jiang, H.; Zhu, S.; Cheng, B. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep. 2011, 30, 1347–1363. [Google Scholar] [CrossRef]
- Mosharaf, P.; Rahman, H.; Ahsan, A.; Akond, Z.; Ahmed, F.F.; Islam, M.; Moni, M.A.; Mollah, N.H. In silico identification and characterization of AGO, DCL and RDR gene families and their associated regulatory elements in sweet orange (Citrus sinensis L.). PLoS ONE 2020, 15, e0228233. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Xu, L.; Huai, X.; Zhang, H.; Zhao, F.; Qiao, Y. Genome-wide identification and characterization of argonaute, dicer-like and RNA-dependent RNA polymerase gene families and their expression analyses in Fragaria spp. Genes 2023, 14, 121. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, K.; Wang, J.; Chen, X.; Chen, Z.; Cai, R.; Xiang, Y. Comprehensive Analysis of Dicer-Like, Argonaute, and RNA-dependent RNA Polymerase Gene Families in Grapevine (Vitis vinifera). J. Plant Growth Regul. 2015, 34, 108–121. [Google Scholar] [CrossRef]
- Belal, M.; Ntini, C.; Sylvia, C.; Wassie, M.; Magdy, M.; Ogutu, C.; Ezzat, M.; Mollah, M.D.A.; Cao, Y.; Zhang, W.; et al. Genome-wide identification of AGO, DCL, and RDR genes and their expression analysis in response to drought stress in peach. Horticulturae 2024, 10, 1228. [Google Scholar] [CrossRef]
- Gan, D.; Zhan, M.; Yang, F.; Zhang, Q.; Hu, K.; Xu, W.; Lu, Q.; Zhang, L.; Liang, D. Expression analysis of argonaute, Dicer-like, and RNA-dependent RNA polymerase genes in cucumber (Cucumis sativus L.) in response to abiotic stress. J. Genet. 2017, 96, 235–249. [Google Scholar] [CrossRef]
- Wu, J.; Jian, Y.; Wang, H.; Huang, H.; Gong, L.; Liu, G.; Yang, Y.; Wang, W. A review of the phytochemistry and pharmacology of the fruit of Siraitia grosvenorii (Swingle): A traditional Chinese medicinal food. Molecules 2022, 27, 6618. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Koonin, E.V.; Aravind, L. Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct. Biol. 2003, 3, 1. [Google Scholar] [CrossRef]
- Matsumoto, S.; Jin, M.; Dewa, Y.; Nishimura, J.; Moto, M.; Murata, Y.; Shibutani, M.; Mitsumori, K. Suppressive effect of Siraitia grosvenorii extract on dicyclanil-promoted hepatocellular proliferative lesions in male mice. J. Toxicol. Sci. 2009, 34, 109–118. [Google Scholar] [CrossRef]
- Qiao, J.; Luo, Z.; Gu, Z.; Zhang, Y.; Zhang, X.; Ma, X. Identification of a novel specific cucurbitadienol synthase allele in Siraitia grosvenorii correlates with high catalytic efficiency. Molecules 2019, 24, 627. [Google Scholar] [CrossRef]
- Voinnet, O. Origin, biogenesis, and activity of plant MicroRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.-H. Evolution of Gene Duplication in Plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [PubMed]
- Hepler, N.K.; Bowman, A.; Carey, R.E.; Cosgrove, D.J. Expansin gene loss is a common occurrence during adaptation to an aquatic environment. Plant J. 2020, 101, 666–680. [Google Scholar] [CrossRef] [PubMed]
- Krishnatreya, D.B. Genome-wide identification, evolutionary relationship and expression analysis of AGO, DCL and RDR family genes in tea. Sci. Rep. 2021, 11, 8679. [Google Scholar] [CrossRef]
- Großhans, H.; Filipowicz, W. The expanding world of small RNAs. Nature 2008, 451, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Lu, S. Identification, molecular cloning and expression analysis of five RNA-dependent RNA polymerase genes in Salvia miltiorrhiza. PLoS ONE 2014, 9, e95117. [Google Scholar] [CrossRef]
- Yang, J.H.; Seo, H.H.; Han, S.J.; Yoon, E.K.; Yang, M.S.; Lee, W.S. Phytohormone abscisic acid control RNA-dependent RNA polymerase 6 gene expression and post-transcriptional gene silencing in rice cells. Nucleic Acids Res. 2007, 36, 1220–1226. [Google Scholar] [CrossRef]
- Li, Z.; Li, D.; Li, B.; Liu, Y.; Niu, X.; Aslam, M.; Cai, H.; Su, Z.; Qin, Y. Genome-wide identification, characterization of RDR genes and their expression analysis during reproductive development and stress in pineapple. Trop. Plant Biol. 2020, 13, 13–22. [Google Scholar] [CrossRef]
- Zhang, K. The Effects of Methyl Jasmonate on the Biosynthetic Pathway of Mogrosides and the Preliminary Screening of bHLH Transcription Factors in Siraitia grosvenorii Fruits; Peking Union Medical College: Beijing, China, 2016. [Google Scholar]
- Cui, D.-L. Genome-wide DCL, AGO and RDR gene families in Saccharum spontaneum. Sci. Rep. 2020, 10, 13202. [Google Scholar] [CrossRef]
- Gong, M.; Wang, Y.; Zhang, J.; Zhao, Y.; Wan, J.; Shang, J.; Yang, R.; Wu, Y.; Li, Y.; Tan, Q.; et al. Chilling stress triggers VvAgo1-mediated miRNA-Like RNA biogenesis in Volvariella volvacea. Front. Microbiol. 2020, 11, 523593. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, Y.-C.; Zhang, D.-W.; Yang, C.-P. Cloning and stress tolerance analysis of an LbGRP gene in Limonium bicolor: Cloning and stress tolerance analysis of an LbGRP gene in Limonium bicolor. Hered. Beijing 2010, 32, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Wang, Y.; Dong, L.; Gu, Y.; Jia, F.; Jiang, T.; Zhou, B. Function analysis of the transcription factor PsnbZIP1 of Populus simonii × P. nigra in response to salt stress. Bull. Bot. Res. 2023, 43, 288–299. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, F.; Wang, R.; Zhou, X.; Sze, S.-H.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef]
- Zhang, H. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 18, 489–506. [Google Scholar] [CrossRef]
- Henderson, I.R.; Zhang, X.; Lu, C.; Johnson, L.; Meyers, B.C.; Green, P.J.; Jacobsen, S.E. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Genet. 2006, 38, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Hong, L.; Fitzpatrick, K.E.; Amasino, R.M. DICER-LIKE 1 and DICER-LIKE 3 redundantly act to promote flowering via repression of FLOWERING LOCUS C in Arabidopsis thaliana. Genetics 2007, 176, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Leibman, D.; Pashkovsky, E.; Shnaider, Y.; Shtarkman, M.; Gaba, V.; Gal-On, A. Analysis of the RNA-dependent RNA polymerase 1 (RDR1) gene family in melon. Plants 2022, 11, 1795. [Google Scholar] [CrossRef]
- Olmedo-Monfil, V.; Durán-Figueroa, N.; Arteaga-Vandázquez, M.; Demesa-Arévalo, E.; Autran, D.; Grimanelli, D.; Slotkin, K.; Martienssen, R.A.; Vielle-Calzada, J.-P. Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature 2010, 464, 628–632. [Google Scholar] [CrossRef]
- Ron, M.; Alandete Saez, M.; Eshed Williams, L.; Fletcher, J.C.; McCormick, S. Proper regulation of a sperm-specific cis -nat-siRNA is essential for double fertilization in Arabidopsis. Genes Dev. 2010, 24, 1010–1021. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Guo, Q.; Ma, X.; Bai, L.; Pan, L.; Feng, S.; Mo, C. Screening of reference genes in Siraitia grosvenorii and spatio-temporal expression analysis of 3-hydroxy-3-methylglutaryl coenzyme A. Chin. Tradit. Herb. Drugs 2014, 45, 2224–2229. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene Locus | Number of Amino Acids | Molecular Weight (kDa) | Theoretical pI | Grand Average of Hydropathicity (GRAVY) | Predicted Subcellular Localization |
|---|---|---|---|---|---|---|
| SgAGO1 | Chr09.g17302 | 1056 | 117.11 | 9.31 | −0.50 | Nucleus |
| SgAGO4 | Chr06.g10665 | 1014 | 113.60 | 8.79 | −0.32 | Cell membrane, chloroplast, and nucleus |
| SgAGO5 | Chr09.g16238 | 967 | 107.54 | 9.48 | −0.41 | Nucleus |
| SgAGO7 | Chr10.g18565 | 1018 | 115.58 | 9.37 | −0.40 | Chloroplast and nucleus |
| SgAGO10.1 | Chr08.g15010 | 927 | 105.33 | 9.22 | −0.39 | Cell membrane and nucleus |
| SgAGO10.2 | Chr03.g06089 | 984 | 110.63 | 9.33 | −0.44 | Nucleus |
| SgDCL1 | Chr03.g06642 | 1952 | 219.66 | 5.87 | −0.44 | Nucleus |
| SgDCL2 | Chr08.g14552 | 1628 | 183.60 | 8.55 | −0.060 | Nucleus |
| SgDCL3 | Chr02.g03104 | 1708 | 191.74 | 6.39 | −0.16 | Nucleus |
| SgDCL4 | Chr08.g15125 | 1651 | 186.62 | 6.30 | −0.27 | Nucleus |
| SgRDR1.1 | Chr10.g17889 | 1112 | 127.79 | 7.89 | −0.32 | Chloroplast |
| SgRDR1.2 | Chr01.g01240 | 1123 | 128.68 | 8.11 | −0.38 | Chloroplast and nucleus |
| SgRDR1.3 | Chr10.g17890 | 1133 | 129.16 | 8.30 | −0.29 | Chloroplast |
| SgRDR2 | Chr08.g15576 | 1121 | 128.81 | 6.72 | −0.29 | Nucleus |
| SgRDR5 | Chr08.g14932 | 1026 | 116.35 | 7.89 | −0.30 | Chloroplast |
| SgRDR6 | Chr02.g03034 | 1194 | 135.98 | 6.97 | −0.30 | Chloroplast and nucleus |
| Query Index | Query Item | Preferred Name | Identity (%) | Bitscore |
|---|---|---|---|---|
| 1 | SgAGO1 | AtAGO1 | 82.8 | 1565.8 |
| 2 | SgAGO4 | AtAGO4 | 72.9 | 1357.8 |
| 3 | SgAGO5 | AtAGO5 | 67 | 1164.1 |
| 4 | SgAGO7 | AtAGO7 | 70.2 | 1269.6 |
| 5 | SgAGO10.1 | AtAGO10 | 74.5 | 1348.6 |
| 6 | SgAGO10.2 | AtAGO10 | 82.6 | 1661.4 |
| 7 | SgDCL1 | AtDCL1 | 71.7 | 2708.3 |
| 8 | SgDCL2 | AtDCL2 | 57.8 | 1550 |
| 9 | SgDCL3 | AtDCL3 | 47.8 | 1435.6 |
| 10 | SgDCL4 | AtDCL4 | 55.4 | 1721.8 |
| 11 | SgRDR1.1 | AtRDR1 | 61.2 | 1370.5 |
| 12 | SgRDR1.2 | AtRDR1 | 58.6 | 1266.9 |
| 13 | SgRDR1.3 | AtRDR1 | 63.7 | 1444.9 |
| 14 | SgRDR2 | AtRDR2 | 61.2 | 1403.7 |
| 15 | SgRDR5 | AtRDR5 | 51.3 | 866.3 |
| 16 | SgRDR6 | AtRDR6 | 67 | 1674.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zang, Y.; Wang, C.; Su, J.; Mo, C.; Xie, L.; Luo, Z.; Ma, X. Genome-Wide Identification and Characterization of AGO, DCL, and RDR Gene Families in Siraitia grosvenorii. Int. J. Mol. Sci. 2025, 26, 5301. https://doi.org/10.3390/ijms26115301
Zang Y, Wang C, Su J, Mo C, Xie L, Luo Z, Ma X. Genome-Wide Identification and Characterization of AGO, DCL, and RDR Gene Families in Siraitia grosvenorii. International Journal of Molecular Sciences. 2025; 26(11):5301. https://doi.org/10.3390/ijms26115301
Chicago/Turabian StyleZang, Yimei, Chongnan Wang, Jiaxian Su, Changming Mo, Lei Xie, Zuliang Luo, and Xiaojun Ma. 2025. "Genome-Wide Identification and Characterization of AGO, DCL, and RDR Gene Families in Siraitia grosvenorii" International Journal of Molecular Sciences 26, no. 11: 5301. https://doi.org/10.3390/ijms26115301
APA StyleZang, Y., Wang, C., Su, J., Mo, C., Xie, L., Luo, Z., & Ma, X. (2025). Genome-Wide Identification and Characterization of AGO, DCL, and RDR Gene Families in Siraitia grosvenorii. International Journal of Molecular Sciences, 26(11), 5301. https://doi.org/10.3390/ijms26115301