
Transcriptome Analysis Reveals Co-Expression Regulation of Sugar Transport and Signaling Networks in Initiating Stolon-to-Tuber Transition in Potato

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
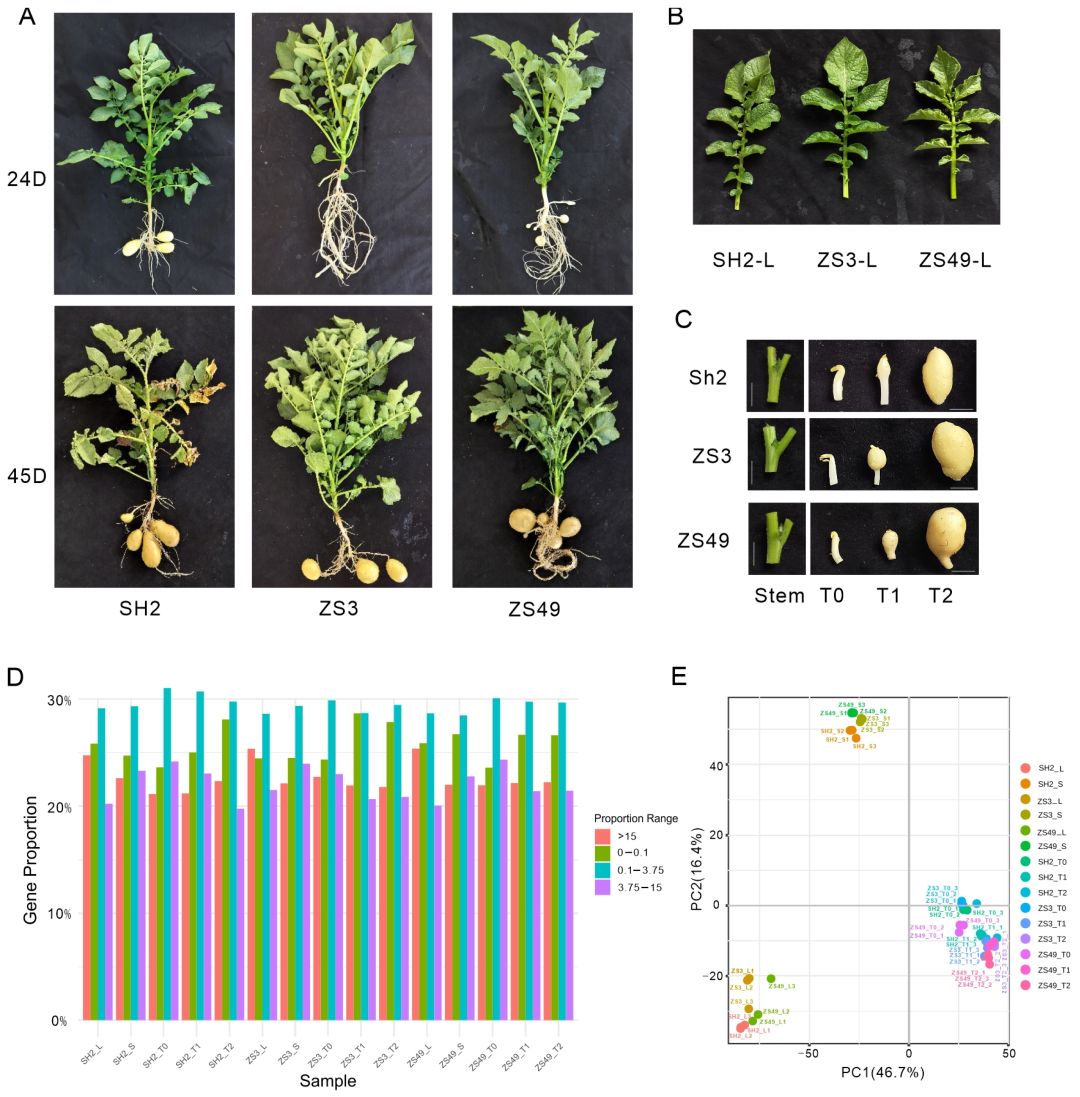
2.1. Analysis of Transcriptome Sequencing
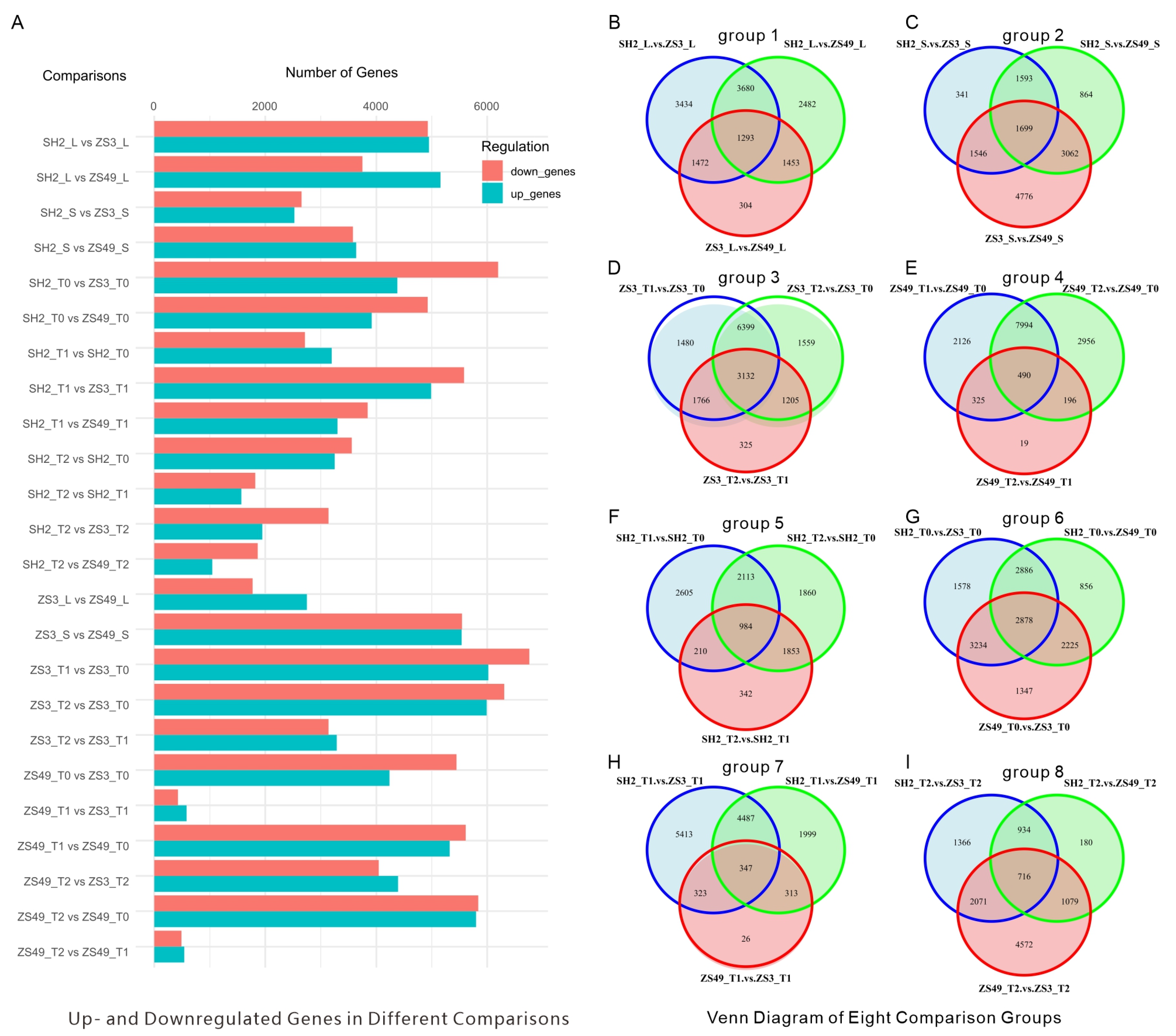
2.2. Tissue-Specific DEGs Highlight Key Metabolic and Signaling Pathways
2.3. Tissue-Specific Expression Patterns of Key Genes Involved in Regulating Potato Tuber Initiation
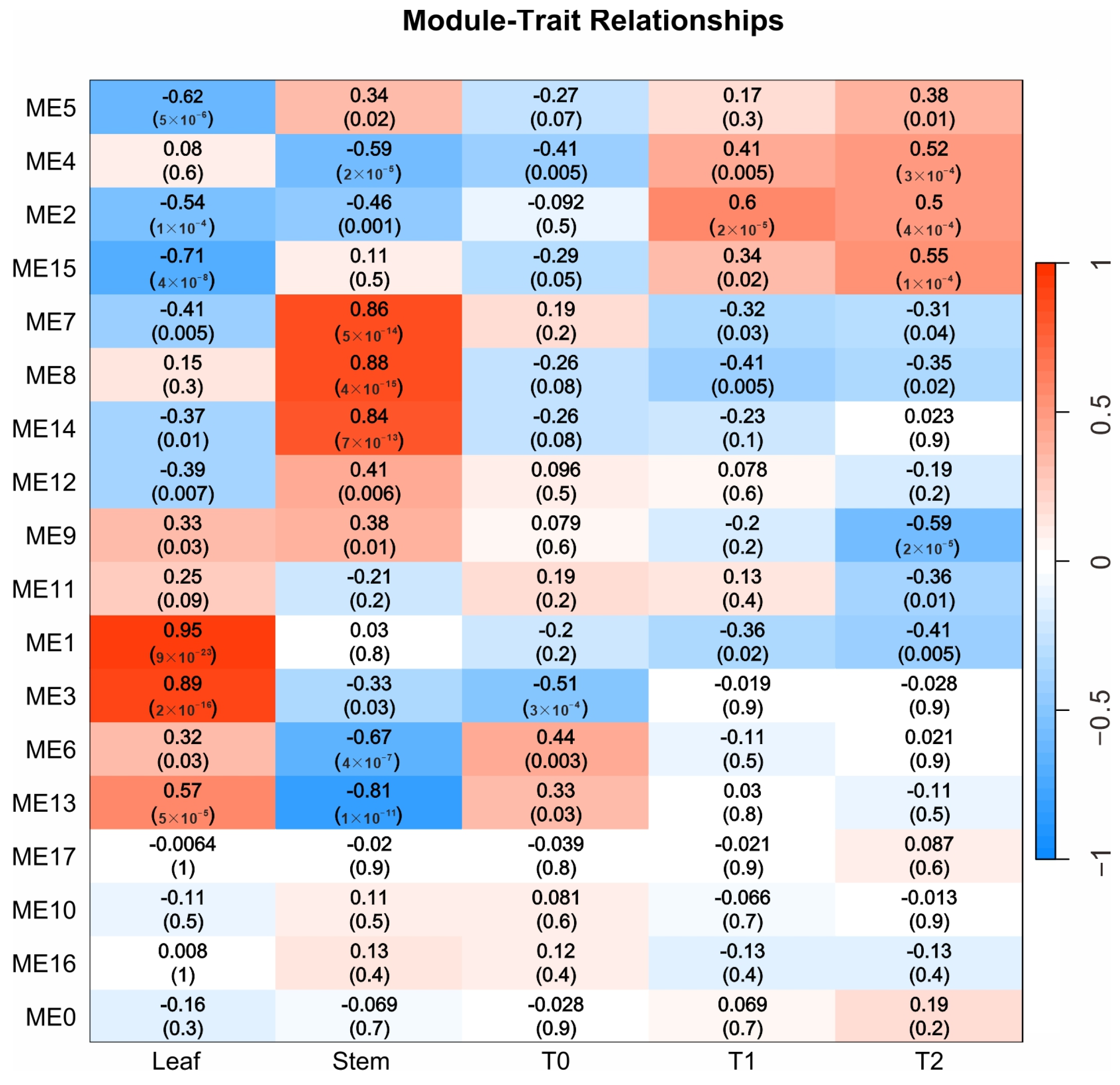
2.4. Weighted Gene Co-Expression Network Analysis Identifies Tissue-Specific Modules in Potato
2.5. Co-Expression Network Reveals the Key Role of Sugar Transport and Metabolism in Conjunction with Tuberization Signaling-Associated Genes from Leaves and Stems to Tubers During Tuber Initiation
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and cDNA Library Construction
4.3. RNA Sequencing and Data Analysis
4.4. Weighted Gene Co-Expression Network Analysis
4.5. Validation by Quantitative PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hui, Z.; Xu, J.; Jian, Y.; Bian, C.; Duan, S.; Hu, J.; Li, G.; Jin, L. Identification of Long-Distance Transport Signal Molecules Associated with Plant Maturity in Tetraploid Cultivated Potatoes (Solanum tuberosum L.). Plants 2022, 11, 1707. [Google Scholar] [CrossRef] [PubMed]
- Zierer, W.; Rüscher, D.; Sonnewald, U.; Sonnewald, S. Tuber and Tuberous Root Development. Annu. Rev. Plant Biol. 2021, 72, 551–580. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhu, Y.; Li, G.; Liu, L. Regulation of storage organ formation by long-distance tuberigen signals in potato. Hortic. Res. 2025, 12, uhae360. [Google Scholar] [CrossRef]
- Qu, L.; Huang, X.; Su, X.; Zhu, G.; Zheng, L.; Lin, J.; Wang, J.; Xue, H. Potato: From functional genomics to genetic improvement. Mol. Hortic. 2024, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Li, G.; Jian, Y.; Duan, S.; Liu, J.; Xu, J.; Jin, L. Genes related to circadian rhythm are involved in regulating tuberization time in potato. Hortic. Plant J. 2022, 8, 369–380. [Google Scholar] [CrossRef]
- Singh, P.; Arif, Y.; Siddiqui, H.; Upadhyaya, C.P.; Pichtel, J.; Hayat, S. Critical Factors Responsible for Potato Tuberization. Bot. Rev. 2023, 89, 421–437. [Google Scholar] [CrossRef]
- Romero, J.M.; Serrano-Bueno, G.; Camacho-Fernández, C.; Vicente, M.H.; Ruiz, M.T.; Pérez-Castiñeira, J.R.; Pérez-Hormaeche, J.; Nogueira, F.T.S.; Valverde, F. CONSTANS, a HUB for all seasons: How photoperiod pervades plant physiology regulatory circuits. Plant Cell 2024, 36, 2086–2102. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.; Jiang, P.; Sun, X.; Yu, L.; Wang, E.; Qin, J.; Zhang, F.; Prat, S.; Song, B. Long-distance control of potato storage organ formation by SELF PRUNING 3D and FLOWERING LOCUS T-like 1. Plant Commun. 2023, 4, 100547. [Google Scholar] [CrossRef]
- Wang, E.; Zhou, T.; Jing, S.; Dong, L.; Sun, X.; Fan, Y.; Shen, Y.; Liu, T.; Song, B. Leaves and stolons transcriptomic analysis provide insight into the role of phytochrome F in potato flowering and tuberization. Plant J. 2023, 113, 402–415. [Google Scholar] [CrossRef]
- Abelenda, J.A.; Cruz-Oró, E.; Franco-Zorrilla, J.M.; Prat, S. Potato StCONSTANS-like1 Suppresses Storage Organ Formation by Directly Activating the FT-like StSP5G Repressor. Curr. Biol. 2016, 26, 872–881. [Google Scholar] [CrossRef]
- Abelenda, J.A.; Navarro, C.; Prat, S. From the model to the crop: Genes controlling tuber formation in potato. Curr. Opin. Biotechnol. 2011, 22, 287–292. [Google Scholar] [CrossRef]
- Teo, C.-J.; Takahashi, K.; Shimizu, K.; Shimamoto, K.; Taoka, K.-i. Potato Tuber Induction is Regulated by Interactions Between Components of a Tuberigen Complex. Plant Cell Physiol. 2016, 58, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, E.; Yu, L.; Liu, S.; Liu, T.; Qin, J.; Jiang, P.; He, S.; Cai, X.; Jing, S.; et al. TCP transcription factor StAST1 represses potato tuberization by regulating tuberigen complex activity. Plant Physiol. 2024, 195, 1347–1364. [Google Scholar] [CrossRef]
- Kondhare, K.R.; Vetal, P.V.; Kalsi, H.S.; Banerjee, A.K. BEL1-like protein (StBEL5) regulates CYCLING DOF FACTOR1 (StCDF1) through tandem TGAC core motifs in potato. J. Plant Physiol. 2019, 241, 153014. [Google Scholar] [CrossRef] [PubMed]
- Hannapel, D.J.; Sharma, P.; Lin, T.; Banerjee, A.K. The Multiple Signals That Control Tuber Formation. Plant Physiol. 2017, 174, 845–856. [Google Scholar] [CrossRef]
- Bizuayehu, D.; Getachew, A. The Role of Cytokinin and Gibberellin in Potato Tuber Sprouting. Innovare J. Agric. Sci. 2024, 12, 1–9. [Google Scholar] [CrossRef]
- Kolachevskaya, O.O.; Lomin, S.N.; Arkhipov, D.V.; Romanov, G.A. Auxins in potato: Molecular aspects and emerging roles in tuber formation and stress resistance. Plant Cell Rep. 2019, 38, 681–698. [Google Scholar] [CrossRef]
- Jing, S.; Sun, X.; Yu, L.; Wang, E.; Cheng, Z.; Liu, H.; Jiang, P.; Qin, J.; Begum, S.; Song, B. Transcription factor StABI5-like 1 binding to the FLOWERING LOCUS T homologs promotes early maturity in potato. Plant Physiol. 2022, 189, 1677–1693. [Google Scholar] [CrossRef]
- Yoon, J.; Cho, L.-H.; Tun, W.; Jeon, J.-S.; An, G. Sucrose signaling in higher plants. Plant Sci. 2021, 302, 110703. [Google Scholar] [CrossRef]
- Hu, J.; Duan, Y.; Hu, J.; Zhang, S.; Li, G. Phylogenetic and Expression Analysis of the Sucrose Synthase and Sucrose Phosphate Synthase Gene Family in Potatoes. Metabolites 2024, 14, 70. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, T.; Li, X.; Song, C.-P.; Zhu, J.-K.; Chen, L.; Zhao, Y. Phosphorylation of SWEET sucrose transporters regulates plant root: Shoot ratio under drought. Nat. Plants 2022, 8, 68–77. [Google Scholar] [CrossRef]
- Li, M.; Xie, H.; He, M.; Su, W.; Yang, Y.; Wang, J.; Ye, G.; Zhou, Y. Genome-wide identification and expression analysis of the StSWEET family genes in potato (Solanum tuberosum L.). Genes Genom. 2020, 42, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Abelenda, J.A.; Bergonzi, S.; Oortwijn, M.; Sonnewald, S.; Du, M.; Visser, R.G.F.; Sonnewald, U.; Bachem, C.W.B. Source-Sink Regulation is Mediated by Interaction of an FT Homolog with a SWEET Protein in Potato. Curr. Biol. 2019, 29, 1178–1186.e6. [Google Scholar] [CrossRef]
- Lauschke, A.; Maibaum, L.; Engel, M.; Eisengräber, L.; Bayer, S.; Hackel, A.; Kühn, C. The potato sugar transporter SWEET1g affects apoplasmic sugar ratio and phloem-mobile tuber- and flower-inducing signals. Plant Physiol. 2024, 197, kiae602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jian, H.; Shang, L.; Wang, K.; Wen, S.; Li, Z.; Liu, R.; Jia, L.; Huang, Z.; Lyu, D. Transcriptome Analysis Reveals Novel Genes Potentially Involved in Tuberization in Potato. Plants 2024, 13, 795. [Google Scholar] [CrossRef]
- Ding, K.; Shan, Y.; Wang, L.; Zhang, Y.; Tian, G. Transcriptomics combined with physiological analysis and metabolomics revealed the response of potato tuber formation to nitrogen. BMC Plant Biol. 2024, 24, 1109. [Google Scholar] [CrossRef]
- Dutta, M.; Mali, S.; Raturi, V.; Zinta, G. Transcriptional and Post-transcriptional Regulation of Tuberization in Potato (Solanum tuberosum L.). J. Plant Growth Regul. 2024, 43, 1–24. [Google Scholar] [CrossRef]
- Zhou, T.; Song, B.; Liu, T.; Shen, Y.; Dong, L.; Jing, S.; Xie, C.; Liu, J. Phytochrome F plays critical roles in potato photoperiodic tuberization. Plant J. 2019, 98, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; He, X.; Tang, Y.; Chen, Z.; Zhou, L.; Li, X.; Zhang, C.; Huang, X.; Yang, Y.; Zhang, W.; et al. Photoperiod controls plant seed size in a CONSTANS-dependent manner. Nat. Plants 2023, 9, 343–354. [Google Scholar] [CrossRef]
- Yu, B.; Hu, Y.; Hou, X. More than flowering: CONSTANS plays multifaceted roles in plant development and stress responses. J. Integr. Plant Biol. 2025, 67, 425–439. [Google Scholar] [CrossRef]
- Shirani-Bidabadi, M.; Nazarian-Firouzabadi, F.; Sorkheh, K.; Ismaili, A. Transcriptomic analysis of potato (Solanum tuberosum L.) tuber development reveals new insights into starch biosynthesis. PLoS ONE 2024, 19, e0297334. [Google Scholar] [CrossRef] [PubMed]
- Dinant, S.; Le Hir, R. Delving deeper into the link between sugar transport, sugar signaling, and vascular system development. Physiol. Plant. 2022, 174, e13684. [Google Scholar] [CrossRef] [PubMed]
- Lugassi, N.; Stein, O.; Egbaria, A.; Belausov, E.; Zemach, H.; Arad, T.; Granot, D.; Carmi, N. Sucrose Synthase and Fructokinase Are Required for Proper Meristematic and Vascular Development. Plants 2022, 11, 1035. [Google Scholar] [CrossRef]
- Dong, X.; Chen, L.; Yang, H.; Tian, L.; Dong, F.; Chai, Y.; Qu, L.Q. Pho1 cooperates with DPE1 to control short maltooligosaccharide mobilization during starch synthesis initiation in rice endosperm. Theor. Appl. Genet. 2023, 136, 47. [Google Scholar] [CrossRef]
- Dong, X.; Yang, H.; Chai, Y.; Han, B.; Liu, J.; Tian, L.; Cui, S.; Xiong, S.; Zhong, M.; Fu, B.; et al. Simultaneous knockout of cytosolic and plastidial disproportionating enzymes disrupts grain setting and filling in rice. Plant Physiol. 2024, 196, 1391–1406. [Google Scholar] [CrossRef]
- Yu, J.; Wang, K.; Beckles, D.M. Starch branching enzymes as putative determinants of postharvest quality in horticultural crops. BMC Plant Biol. 2021, 21, 479. [Google Scholar] [CrossRef]
- Zhu, Y.; Tian, Y.; Han, S.; Wang, J.; Liu, Y.; Yin, J. Structure, evolution, and roles of SWEET proteins in growth and stress responses in plants. Int. J. Biol. Macromol. 2024, 263, 130441. [Google Scholar] [CrossRef] [PubMed]
- Isoda, R.; Palmai, Z.; Yoshinari, A.; Chen, L.-Q.; Tama, F.; Frommer, W.B.; Nakamura, M. SWEET13 transport of sucrose, but not gibberellin, restores male fertility in Arabidopsis. Proc. Natl. Acad. Sci. USA 2022, 119, e2207558119. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, L.; Zhang, P.; Liu, J.; Li, L.; Li, H.; Wang, X.; Bai, Y.; Jiang, G.; Qin, P. Comparative transcriptomes and WGCNA reveal hub genes for spike germination in different quinoa lines. BMC Genom. 2024, 25, 1231. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, J.; Bian, C.; Duan, S.; Hu, J.; Qin, J.; Wu, H.; He, M.; Jian, Y.; Duan, Y.; et al. Natural Variation of StNADC Regulates Plant Senescence in Tetraploid Potatoes (Solanum tuberosum L.). Int. J. Mol. Sci. 2025, 26, 4389. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Hu, J.; Duan, S.; Xiang, C.; Duan, Y.; Zhang, S.; Li, G. Transcriptome Analysis Reveals Co-Expression Regulation of Sugar Transport and Signaling Networks in Initiating Stolon-to-Tuber Transition in Potato. Int. J. Mol. Sci. 2025, 26, 5278. https://doi.org/10.3390/ijms26115278
Hu J, Hu J, Duan S, Xiang C, Duan Y, Zhang S, Li G. Transcriptome Analysis Reveals Co-Expression Regulation of Sugar Transport and Signaling Networks in Initiating Stolon-to-Tuber Transition in Potato. International Journal of Molecular Sciences. 2025; 26(11):5278. https://doi.org/10.3390/ijms26115278
Chicago/Turabian StyleHu, Jun, Jinxue Hu, Shaoguang Duan, Congchao Xiang, Yanfeng Duan, Shuqing Zhang, and Guangcun Li. 2025. "Transcriptome Analysis Reveals Co-Expression Regulation of Sugar Transport and Signaling Networks in Initiating Stolon-to-Tuber Transition in Potato" International Journal of Molecular Sciences 26, no. 11: 5278. https://doi.org/10.3390/ijms26115278
APA StyleHu, J., Hu, J., Duan, S., Xiang, C., Duan, Y., Zhang, S., & Li, G. (2025). Transcriptome Analysis Reveals Co-Expression Regulation of Sugar Transport and Signaling Networks in Initiating Stolon-to-Tuber Transition in Potato. International Journal of Molecular Sciences, 26(11), 5278. https://doi.org/10.3390/ijms26115278