

MAPK15 Prevents IFNB1 Expression by Suppressing Oxidative Stress-Dependent Activation of the JNK-JUN Pathway

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
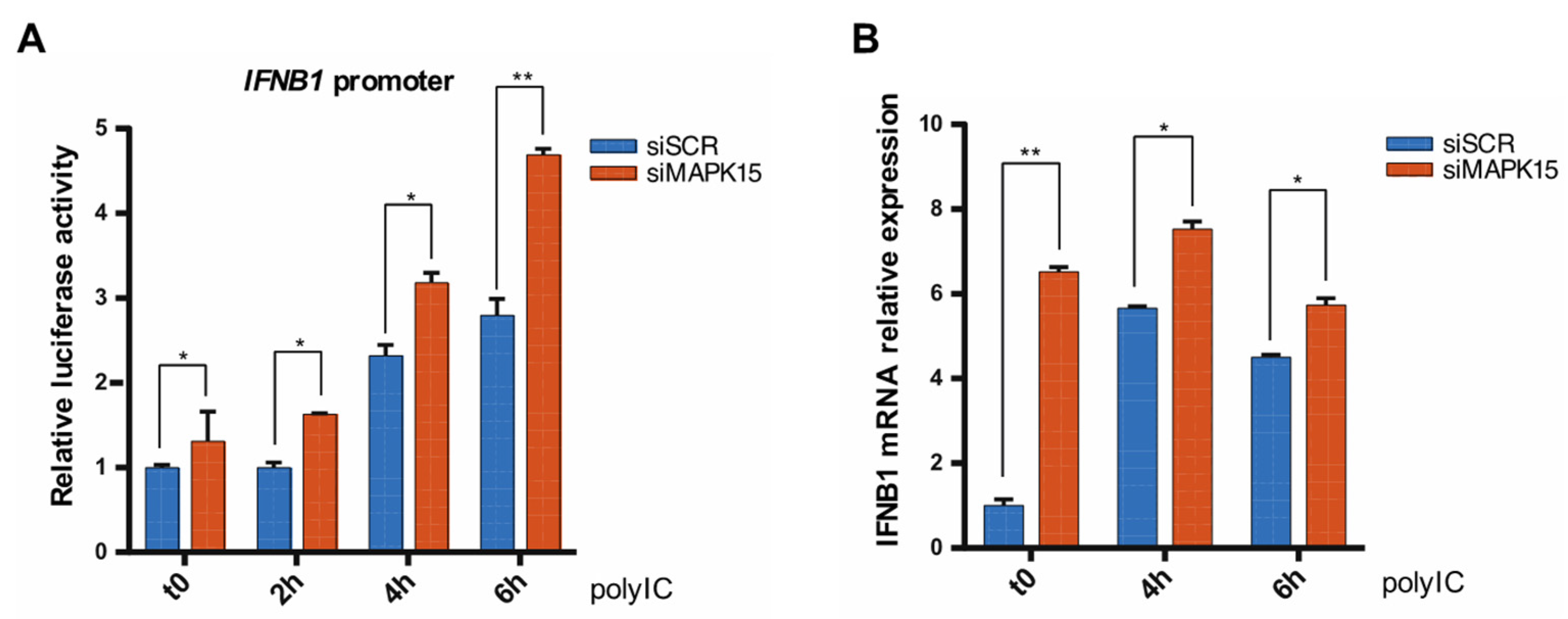
2.1. Loss of MAPK15 Results in Increased IFNB1 Response
2.2. Loss of MAPK15 Impairs Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-kB) Activity
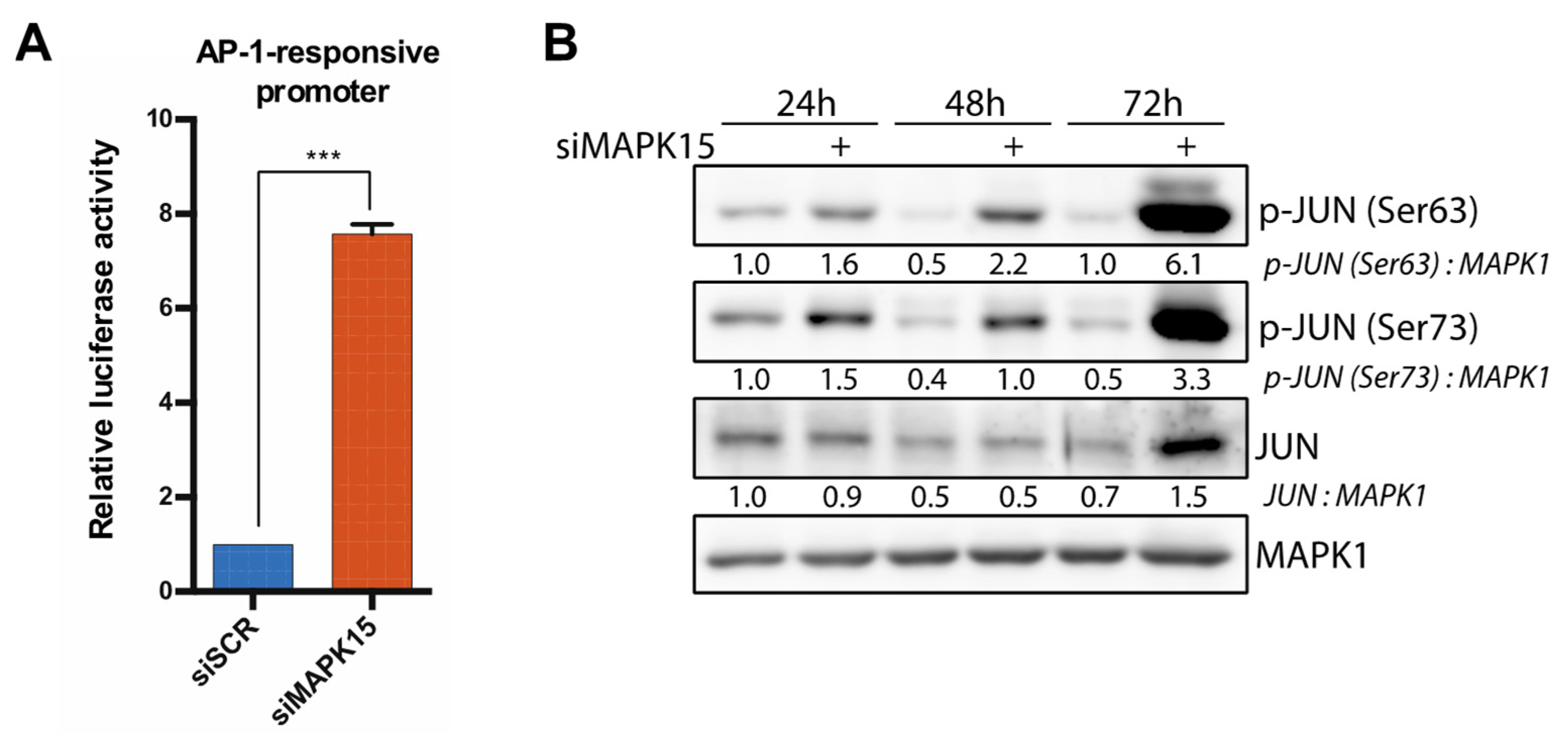
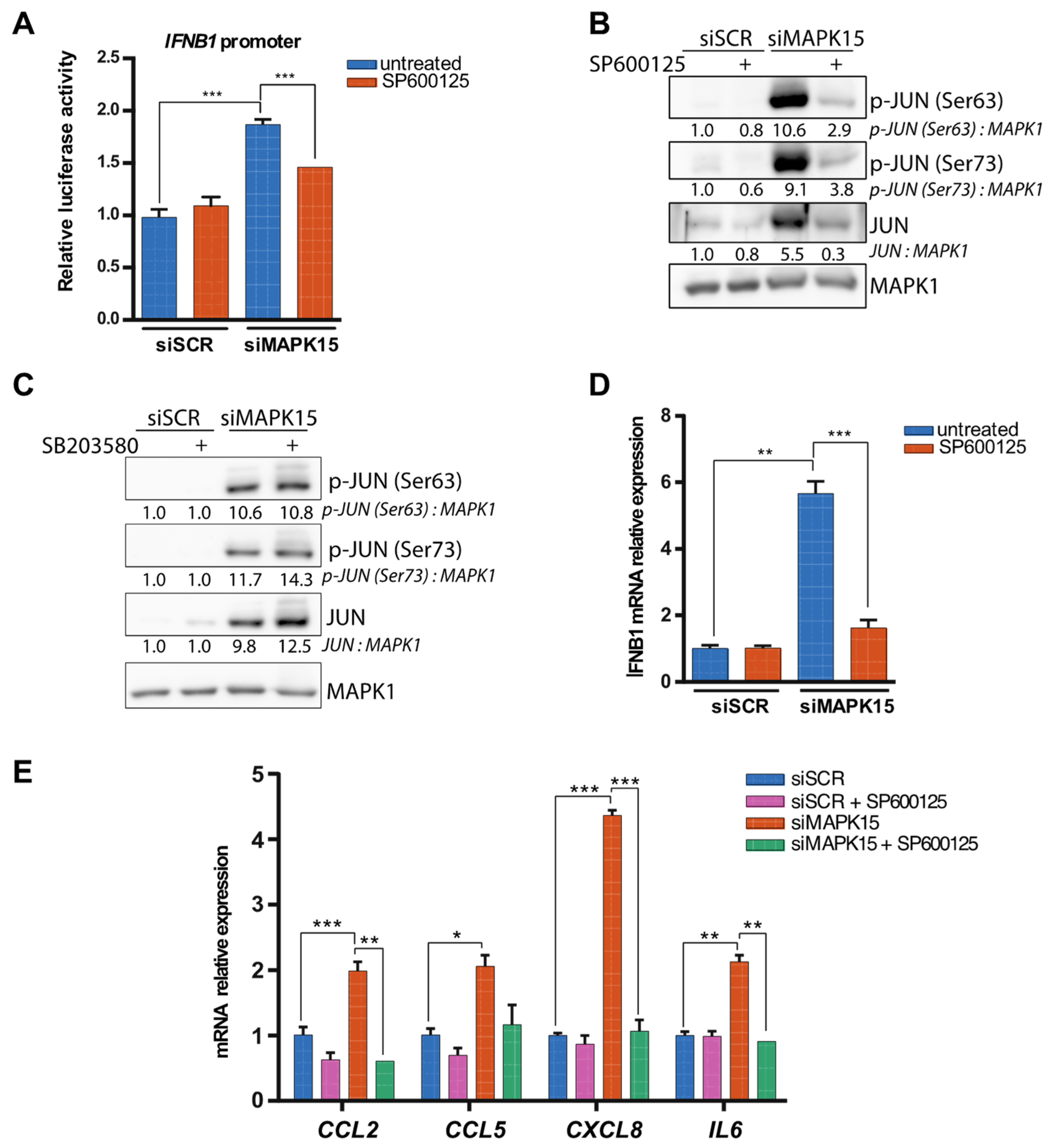
2.3. Reduced MAPK15 Activity Leads to JUN Activation
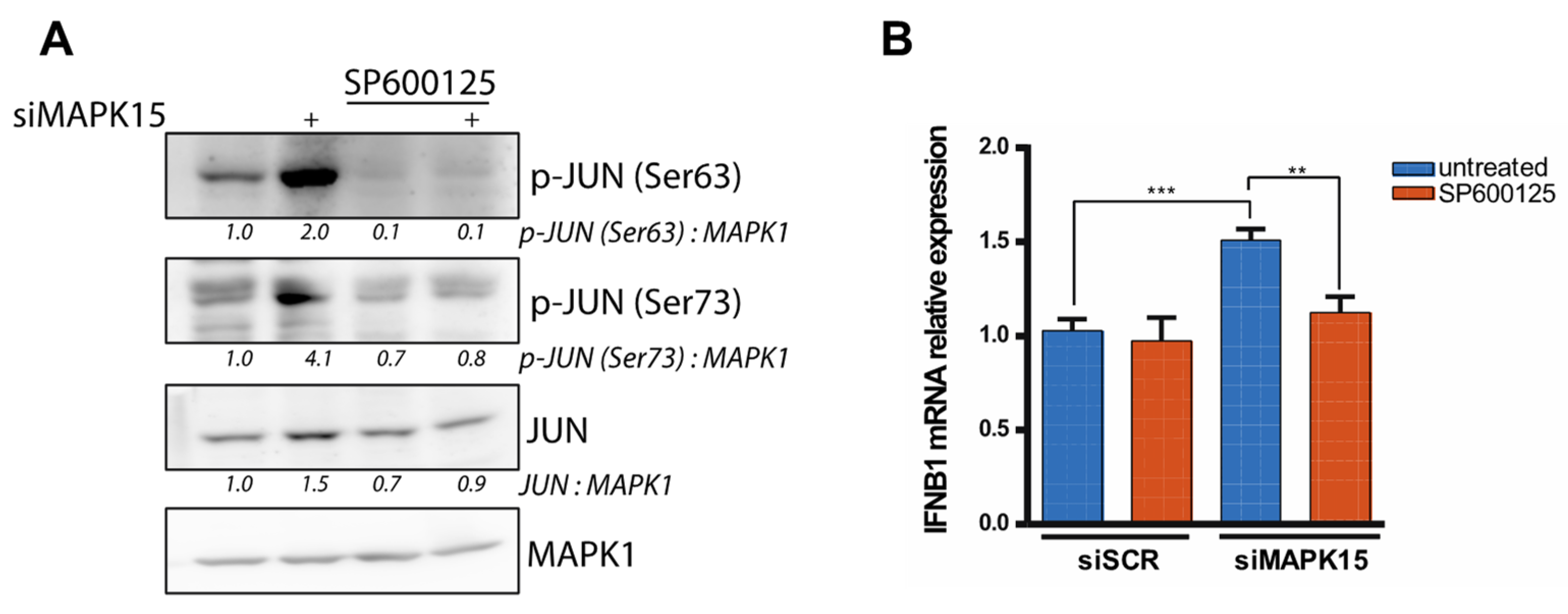
2.4. The Antioxidant Molecule NACET Restores IFNB1 Levels
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Plasmids
4.3. Cell Culture
4.4. MAPK15 Silencing
4.5. Luciferase Assays
4.6. RT-qPCR
4.7. ELISA Assay
4.8. Western Blots
4.9. ROS Measurement
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| siSCR | siRNA scrambled |
| siMAPK15 | siRNA for MAPK15 |
| poly (I:C) | polyinosinic:polycytidylic acid |
| NACET | N-acetylcysteine ethyl ester |
| JNKs | c-Jun N-terminal Kinases |
| IFNs | Interferons |
| MAPKs | Mitogen activated protein kinases |
| ROS | Reactive oxygen species |
| SASP | Senescent-associated secretory phenotype |
| ELISA | Enzyme-Linked Immunosorbent Assay |
References
- Taniguchi, T.; Mantei, N.; Schwarzstein, M.; Nagata, S.; Muramatsu, M.; Weissmann, C. Human Leukocyte and Fibroblast Interferons Are Structurally Related. Nature 1980, 285, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, Interferon-like Cytokines, and Their Receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Li, T.; Chen, H.; Yang, Y.; Lu, E.; Liu, J.; Qiao, W.; Chen, H. The Crucial Regulatory Role of Type I Interferon in Inflammatory Diseases. Cell Biosci. 2023, 13, 230. [Google Scholar] [CrossRef] [PubMed]
- Hata, N.; Sato, M.; Takaoka, A.; Asagiri, M.; Tanaka, N.; Taniguchi, T. Constitutive IFN-α/β Signal for Efficient IFN-α/β Gene Induction by Virus. Biochem. Biophys. Res. Commun. 2001, 285, 518–525. [Google Scholar] [CrossRef]
- Gough, D.J.; Messina, N.L.; Clarke, C.J.P.; Johnstone, R.W.; Levy, D.E. Constitutive Type I Interferon Modulates Homeostatic Balance through Tonic Signaling. Immunity 2012, 36, 166–174. [Google Scholar] [CrossRef]
- Taniguchi, T.; Takaoka, A. A Weak Signal for Strong Responses: Interferon-α/β Revisited. Nat. Rev. Mol. Cell Biol. 2001, 2, 378–386. [Google Scholar] [CrossRef]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical Roles of the Immune System during Cancer Development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 496. [Google Scholar] [CrossRef]
- Sulaimani, M.N.; Ahmed, S.; Anjum, F.; Mohammad, T.; Shamsi, A.; Dohare, R.; Hassan, M.I. Structure-Guided Identification of Mitogen-Activated Protein Kinase-1 Inhibitors towards Anticancer Therapeutics. PLoS ONE 2025, 20, e0311954. [Google Scholar] [CrossRef]
- Abe, M.K.; Saelzler, M.P.; Espinosa, R., III; Kahle, K.T.; Hershenson, M.B.; Le Beau, M.M.; Rosner, M.R. ERK8, a New Member of the Mitogen-Activated Protein Kinase Family. J. Biol. Chem. 2002, 277, 16733–16743. [Google Scholar] [CrossRef]
- Klevernic, I.V.; Martin, N.M.B.; Cohen, P. Regulation of the Activity and Expression of ERK8 by DNA Damage. FEBS Lett. 2009, 583, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Colecchia, D.; Ilardi, G.; Acunzo, M.; Nigita, G.; Sasdelli, F.; Celetti, A.; Strambi, A.; Staibano, S.; Croce, C.M.; et al. MAPK15 Upregulation Promotes Cell Proliferation and Prevents DNA Damage in Male Germ Cell Tumors. Oncotarget 2016, 7, 20981–20998. [Google Scholar] [CrossRef] [PubMed]
- Groehler, A.L.; Lannigan, D.A. A Chromatin-Bound Kinase, ERK8, Protects Genomic Integrity by Inhibiting HDM2-Mediated Degradation of the DNA Clamp PCNA. J. Cell Biol. 2010, 190, 575–586. [Google Scholar] [CrossRef]
- Cerone, M.A.; Burgess, D.J.; Naceur-Lombardelli, C.; Lord, C.J.; Ashworth, A. High-Throughput RNAi Screening Reveals Novel Regulators of Telomerase. Cancer Res. 2011, 71, 3328–3340. [Google Scholar] [CrossRef]
- Colecchia, D.; Strambi, A.; Sanzone, S.; Iavarone, C.; Rossi, M.; Dall’Armi, C.; Piccioni, F.; Verrotti Di Pianella, A.; Chiariello, M. MAPK15/ERK8 Stimulates Autophagy by Interacting with LC3 and GABARAP Proteins. Autophagy 2012, 8, 1724–1740. [Google Scholar] [CrossRef]
- Colecchia, D.; Dapporto, F.; Tronnolone, S.; Salvini, L.; Chiariello, M. MAPK15 Is Part of the ULK Complex and Controls Its Activity to Regulate Early Phases of the Autophagic Process. J. Biol. Chem. 2018, 293, 15962–15976. [Google Scholar] [CrossRef]
- Franci, L.; Tubita, A.; Bertolino, F.M.; Palma, A.; Cannino, G.; Settembre, C.; Rasola, A.; Rovida, E.; Chiariello, M. MAPK15 Protects from Oxidative Stress-Dependent Cellular Senescence by Inducing the Mitophagic Process. Aging Cell 2022, 21, e13620. [Google Scholar] [CrossRef]
- Franci, L.; Vallini, G.; Bertolino, F.M.; Cicaloni, V.; Inzalaco, G.; Cicogni, M.; Tinti, L.; Calabrese, L.; Barone, V.; Salvini, L.; et al. MAPK15 Controls Cellular Responses to Oxidative Stress by Regulating NRF2 Activity and Expression of Its Downstream Target Genes. Redox Biol. 2024, 72, 103131. [Google Scholar] [CrossRef]
- Erlandsson, L.; Blumenthal, R.; Eloranta, M.L.; Engel, H.; Alm, G.; Weisst, S.; Leanderson, T. Interferon-β Is Required for Interferon-α Production in Mouse Fibroblasts. Curr. Biol. 1998, 8, 223–226. [Google Scholar] [CrossRef]
- Takaoka, A.; Mitani, Y.; Suemori, H.; Sato, M.; Yokochi, T.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Cross Talk between Interferon-γ and -α/β Signaling Components in Caveolar Membrane Domains. Science 2000, 288, 2357–2360. [Google Scholar] [CrossRef]
- Bals, R.; Hiemstra, P.S. Innate Immunity in the Lung: How Epithelial Cells Fight against Respiratory Pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Vareille, M.; Kieninger, E.; Edwards, M.R.; Regamey, N. The Airway Epithelium: Soldier in the Fight against Respiratory Viruses. Clin. Microbiol. Rev. 2011, 24, 210–229. [Google Scholar] [CrossRef] [PubMed]
- Gentili, M.; Kowal, J.; Tkach, M.; Satoh, T.; Lahaye, X.; Conrad, C.; Boyron, M.; Lombard, B.; Durand, S.; Kroemer, G.; et al. Transmission of Innate Immune Signaling by Packaging of CGAMP in Viral Particles. Science 2015, 349, 1232–1236. [Google Scholar] [CrossRef]
- Colecchia, D.; Rossi, M.; Sasdelli, F.; Sanzone, S.; Strambi, A.; Chiariello, M. MAPK15 Mediates BCR-ABL1-Induced Autophagy and Regulates Oncogene-Dependent Cell Proliferation and Tumor Formation. Autophagy 2015, 11, 1790–1802. [Google Scholar] [CrossRef]
- Longhi, M.P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic Cells Require a Systemic Type I Interferon Response to Mature and Induce CD4+ Th1 Immunity with Poly IC as Adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef]
- Lau, A.T.Y.; Xu, Y.M. Regulation of Human Mitogen-Activated Protein Kinase 15 (Extracellular Signal-Regulated Kinase 7/8) and Its Functions: A Recent Update. J. Cell. Physiol. 2018, 234, 75–88. [Google Scholar] [CrossRef]
- Wu, D.D.; Lau, A.T.Y.; Yu, F.Y.; Cai, N.L.; Dai, L.J.; Kim, M.O.; Jin, D.Y.; Xu, Y.M. Extracellular Signal-Regulated Kinase 8-Mediated NF-ΚB Activation Increases Sensitivity of Human Lung Cancer Cells to Arsenic Trioxide. Oncotarget 2017, 8, 49144–49155. [Google Scholar] [CrossRef]
- Wu, D.D.; Dai, L.J.; Tan, H.W.; Zhao, X.Y.; Wei, Q.Y.; Zhong, Q.H.; Ji, Y.C.; Yin, X.H.; Yu, F.Y.; Jin, D.Y.; et al. Transcriptional Upregulation of MAPK15 by NF-ΚB Signaling Boosts the Efficacy of Combination Therapy with Cisplatin and TNF-α. iScience 2022, 25, 105459. [Google Scholar] [CrossRef]
- Smeal, T.; Binetruy, B.; Mercola, D.A.; Birrer, M.; Karin, M. Oncogenic and Transcriptional Cooperation with Ha-Ras Requires Phosphorylation of c-Jun on Serines 63 and 73. Nature 1991, 354, 494–496. [Google Scholar] [CrossRef]
- Pulverer, B.J.; Kyriakis, J.M.; Avruch, J.; Nikolakaki, E.; Woodgett, J.R. Phosphorylation of C-Jun Mediated by MAP Kinases. Nature 1991, 353, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Humar, M.; Loop, T.; Schmidt, R.; Hoetzel, A.; Roesslein, M.; Andriopoulos, N.; Pahl, H.L.; Geiger, K.K.; Pannen, B.H.J. The Mitogen-Activated Protein Kinase P38 Regulates Activator Protein 1 by Direct Phosphorylation of c-Jun. Int. J. Biochem. Cell Biol. 2007, 39, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an Anthrapyrazolone Inhibitor of Jun N-Terminal Kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Novoszel, P.; Holcmann, M.; Stulnig, G.; De Sa Fernandes, C.; Zyulina, V.; Borek, I.; Linder, M.; Bogusch, A.; Drobits, B.; Bauer, T.; et al. Psoriatic Skin Inflammation Is Promoted by C-Jun/AP-1-dependent CCL2 and IL-23 Expression in Dendritic Cells. EMBO Mol. Med. 2021, 13, e12409. [Google Scholar] [CrossRef]
- Oltmanns, U.; Issa, R.; Sukkar, M.B.; John, M.; Chung, K.F. Role of C-Jun N-Terminal Kinase in the Induced Release of GM-CSF, RANTES and IL-8 from Human Airway Smooth Muscle Cells. Br. J. Pharmacol. 2003, 139, 1228–1234. [Google Scholar] [CrossRef]
- Khalaf, H.; Jass, J.; Olsson, P.E. Differential Cytokine Regulation by NF-ΚB and AP-1 in Jurkat T-Cells. BMC Immunol. 2010, 11, 26. [Google Scholar] [CrossRef]
- Dendorfer, U.; Oettgen, P.; Libermann, T.A. Multiple Regulatory Elements in the Interleukin-6 Gene Mediate Induction by Prostaglandins, Cyclic AMP, and Lipopolysaccharide. Mol. Cell. Biol. 1994, 14, 4443–4454. [Google Scholar] [CrossRef]
- Nakano, M.; Fujii, T.; Hashimoto, M.; Yukawa, N.; Yoshifuji, H.; Ohmura, K.; Nakaizumi, A.; Mimori, T. Type I Interferon Induces CX3CL1 (Fractalkine) and CCL5 (RANTES) Production in Human Pulmonary Vascular Endothelial Cells. Clin. Exp. Immunol. 2012, 170, 94–100. [Google Scholar] [CrossRef]
- Lehmann, M.H.; Torres-Domínguez, L.E.; Price, P.J.R.; Brandmüller, C.; Kirschning, C.J.; Sutter, G. CCL2 Expression Is Mediated by Type I IFN Receptor and Recruits NK and T Cells to the Lung during MVA Infection. J. Leukoc. Biol. 2016, 99, 1057–1064. [Google Scholar] [CrossRef]
- Lo, Y.Y.C.; Wong, J.M.S.; Cruz, T.F. Reactive Oxygen Species Mediate Cytokine Activation of C-Jun NH2- Terminal Kinases. J. Biol. Chem. 1996, 271, 15703–15707. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, F. Reactive Oxygen Species (ROS), Troublemakers between Nuclear Factor-ΚB (NF-ΚB) and c-Jun NH2-Terminal Kinase (JNK). Cancer Res. 2004, 64, 1902–1905. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Liu, Z.G. JNK Signaling Pathway Is a Key Modulator in Cell Death Mediated by Reactive Oxygen and Nitrogen Species. Free Radic. Biol. Med. 2006, 40, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK Signaling by GSTp. EMBO J. 1999, 18, 1321–1334. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial Dysfunction and Reactive Oxygen Species Imbalance Promote Breast Cancer Cell Motility through a CXCL14-Mediated Mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Ugbode, C.; Garnham, N.; Fort-Aznar, L.; Evans, G.J.O.; Chawla, S.; Sweeney, S.T. JNK Signalling Regulates Antioxidant Responses in Neurons. Redox Biol. 2020, 37, 101712. [Google Scholar] [CrossRef]
- Tosi, G.M.; Giustarini, D.; Franci, L.; Minetti, A.; Imperatore, F.; Caldi, E.; Fiorenzani, P.; Aloisi, A.M.; Sparatore, A.; Rossi, R.; et al. Superior Properties of N-Acetylcysteine Ethyl Ester over n-Acetyl Cysteine to Prevent Retinal Pigment Epithelial Cells Oxidative Damage. Int. J. Mol. Sci. 2021, 22, 600. [Google Scholar] [CrossRef]
- Yu, F.Y.; Xu, Q.; Zhao, X.Y.; Mo, H.Y.; Zhong, Q.H.; Luo, L.; Lau, A.T.Y.; Xu, Y.M. The Atypical MAP Kinase MAPK15 Is Required for Lung Adenocarcinoma Metastasis via Its Interaction with NF-ΚB P50 Subunit and Transcriptional Regulation of Prostaglandin E2 Receptor EP3 Subtype. Cancers 2023, 15, 1398. [Google Scholar] [CrossRef]
- Xu, Y.M.; Zhu, F.; Cho, Y.Y.; Carper, A.; Peng, C.; Zheng, D.; Yao, K.; Lau, A.T.Y.; Zykova, T.A.; Kim, H.G.; et al. Extracellular Signal-Regulated Kinase 8-Mediated c-Jun Phosphorylation Increases Tumorigenesis of Human Colon Cancer. Cancer Res. 2010, 70, 3218–3227. [Google Scholar] [CrossRef]
- Jin, D.H.; Lee, J.; Kim, K.M.; Kim, S.; Kim, D.H.; Park, J. Overexpression of MAPK15 in Gastric Cancer Is Associated with Copy Number Gain and Contributes to the Stability of C-Jun. Oncotarget 2015, 6, 20190–20203. [Google Scholar] [CrossRef]
- Cui, L.; Chen, S.Y.; Lerbs, T.; Lee, J.W.; Domizi, P.; Gordon, S.; Kim, Y.-H.; Nolan, G.; Betancur, P.; Wernig, G. Activation of JUN in Fibroblasts Promotes Pro-Fibrotic Programme and Modulates Protective Immunity. Nat. Commun. 2020, 11, 2795. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Katiyar, S.; Willmarth, N.E.; Liu, M.; Ma, X.; Flomenberg, N.; Lisanti, M.P.; Pestell, R.G. C-Jun Induces Mammary Epithelial Cellular Invasion and Breast Cancer Stem Cell Expansion. J. Biol. Chem. 2010, 285, 8218–8226. [Google Scholar] [CrossRef] [PubMed]
- Jouanguy, E.; Zhang, S.Y.; Chapgier, A.; Sancho-Shimizu, V.; Puel, A.; Picard, C.; Boisson-Dupuis, S.; Abel, L.; Casanova, J.L. Human Primary Immunodeficiencies of Type I Interferons. Biochimie 2007, 89, 878–883. [Google Scholar] [CrossRef]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Type i Interferons in Autoimmune Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 369–393. [Google Scholar] [CrossRef]
- Tripathi, S.; Sharma, Y.; Kumar, D. Unveiling the Link between Chronic Inflammation and Cancer. Metabol. Open 2025, 25, 100347. [Google Scholar] [CrossRef]
- Iavarone, C.; Acunzo, M.; Carlomagno, F.; Catania, A.; Melillo, R.M.; Carlomagno, S.M.; Santoro, M.; Chiariello, M. Activation of the Erk8 Mitogen-Activated Protein (MAP) Kinase by RET/PTC3, a Constitutively Active Form of the RET Proto-Oncogene. J. Biol. Chem. 2006, 281, 10567–10576. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taranta, M.; Panepinto, S.; Galvagni, F.; Franci, L.; Chiariello, M. MAPK15 Prevents IFNB1 Expression by Suppressing Oxidative Stress-Dependent Activation of the JNK-JUN Pathway. Int. J. Mol. Sci. 2025, 26, 5148. https://doi.org/10.3390/ijms26115148
Taranta M, Panepinto S, Galvagni F, Franci L, Chiariello M. MAPK15 Prevents IFNB1 Expression by Suppressing Oxidative Stress-Dependent Activation of the JNK-JUN Pathway. International Journal of Molecular Sciences. 2025; 26(11):5148. https://doi.org/10.3390/ijms26115148
Chicago/Turabian StyleTaranta, Monia, Sara Panepinto, Federico Galvagni, Lorenzo Franci, and Mario Chiariello. 2025. "MAPK15 Prevents IFNB1 Expression by Suppressing Oxidative Stress-Dependent Activation of the JNK-JUN Pathway" International Journal of Molecular Sciences 26, no. 11: 5148. https://doi.org/10.3390/ijms26115148
APA StyleTaranta, M., Panepinto, S., Galvagni, F., Franci, L., & Chiariello, M. (2025). MAPK15 Prevents IFNB1 Expression by Suppressing Oxidative Stress-Dependent Activation of the JNK-JUN Pathway. International Journal of Molecular Sciences, 26(11), 5148. https://doi.org/10.3390/ijms26115148