
Evaluating Glial Fibrillary Acidic Protein and Neurofilament Light as Potential Biomarkers for Spinocerebellar Ataxia 7

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
2.1. Study Participants
2.2. Length of ATXN7 CAG Repeat Expansion Is Associated with Age of Ataxia Onset
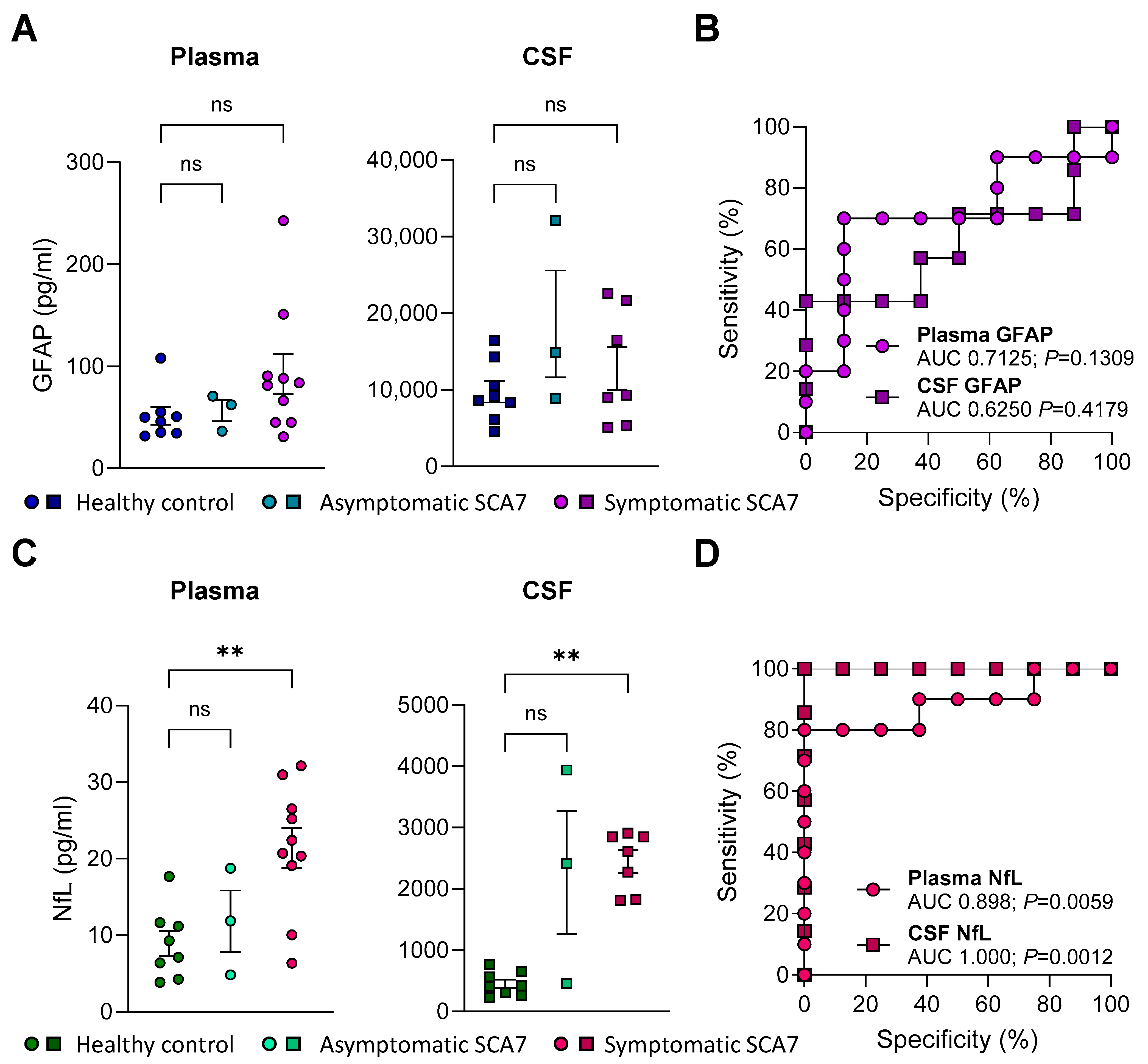
2.3. GFAP Concentrations in Plasma or CSF Do Not Distinguish Patients with SCA7 from Healthy Controls
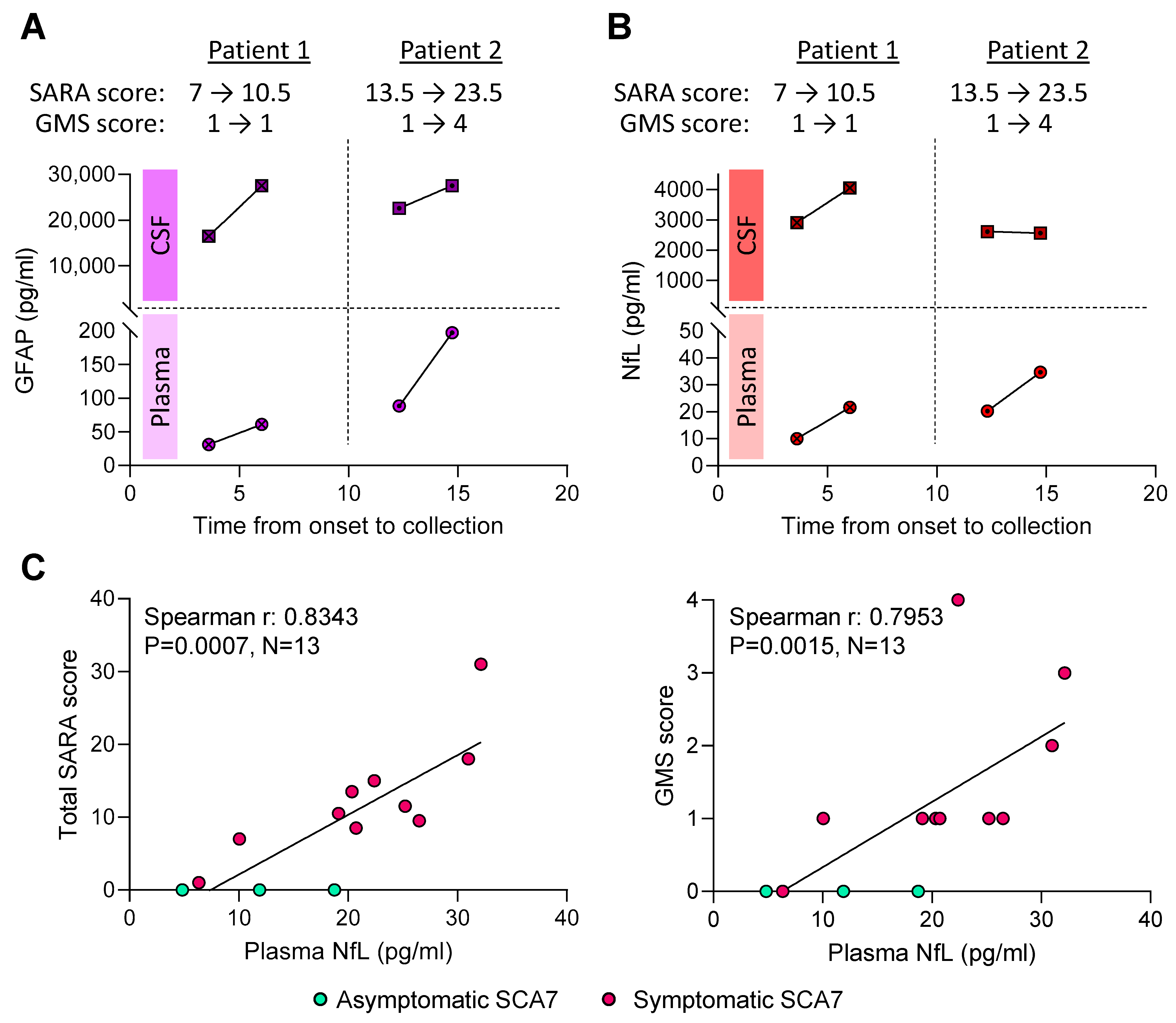
2.4. NfL Levels in Plasma and CSF Are Elevated in SCA7 Patients and Distinguishes Them from Healthy Controls
2.5. Higher Plasma NfL Levels Are Associated with Worse SARA Scores
3. Discussion
4. Materials and Methods
4.1. Standard Protocol Approvals, Registrations, and Patient Consents
4.2. Sample Collection and Preparation
4.3. GFAP and NfL Immunoassays
4.4. Genomic DNA Extraction
4.5. Repeat Expansion Assessment Using a Research Lab-Based Assay from Genomic DNA
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| ATXN7 | Ataxin 7 |
| CAG | Cytosine–adenine–guanine |
| CV | Coefficient of variation |
| CSF | Cerebrospinal fluid |
| GMS | Gait mobility scale |
| GFAP | Glial fibrillary acidic protein |
| IRB | Institutional Review Board |
| NfL | Neurofilament light |
| SARA | Scale for the assessment and rating of ataxia |
| STAGA | SPT3-TAFII31-GCN5L acetylase |
| SCA7 | Spinocerebellar ataxia type 7 |
References
- Fujioka, S.; Sundal, C.; Wszolek, Z.K. Autosomal dominant cerebellar ataxia type III: A review of the phenotypic and genotypic characteristics. Orphanet. J. Rare Dis. 2013, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Whaley, N.R.; Fujioka, S.; Wszolek, Z.K. Autosomal dominant cerebellar ataxia type I: A review of the phenotypic and genotypic characteristics. Orphanet. J. Rare Dis. 2011, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R. Spinocerebellar Ataxia Type 7. In GeneReviews ((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Cerecedo-Zapata, C.M.; Tapia-Guerrero, Y.S.; Ramírez-González, J.A.; Meza-Dorantes, A.; Tercero-Pérez, K.N.; Cortés, H.; Guerra-Grajeda, A.; Ortega-Ibarra, I.H.; Gatica-Ramos, G.; Poblete-Velazquez, A.; et al. Current Overview of Spinocerebellar Ataxia Type 7 in Mexican Population: Challenges in Specialized Care for a Rare Disease. Int. J. Mol. Sci. 2024, 25, 10750. [Google Scholar] [CrossRef]
- Rodríguez-Labrada, R.; Martins, A.C.; Magaña, J.J.; Vazquez-Mojena, Y.; Medrano-Montero, J.; Fernandez-Ruíz, J.; Cisneros, B.; Teive, H.; McFarland, K.N.; Saraiva-Pereira, M.L.; et al. Founder Effects of Spinocerebellar Ataxias in the American Continents and the Caribbean. Cerebellum 2020, 19, 446–458. [Google Scholar] [CrossRef]
- Smith, D.C.; Bryer, A.; Watson, L.M.; Greenberg, L.J. Inherited polyglutamine spinocerebellar ataxias in South Africa. S. Afr. Med. J. 2012, 102, 683–686. [Google Scholar] [CrossRef]
- Smith, D.C.; Greenberg, L.J.; Bryer, A. The hereditary ataxias: Where are we now? Four decades of local research. S. Afr. Med. J. 2016, 106, S38–S41. [Google Scholar]
- Magaña, J.J.; Gómez, R.; Maldonado-Rodríguez, M.; Velázquez-Pérez, L.; Tapia-Guerrero, Y.S.; Cortés, H.; Leyva-García, N.; Hernández-Hernández, O.; Cisneros, B. Origin of the spinocerebellar ataxia type 7 gene mutation in Mexican population. Cerebellum 2013, 12, 902–905. [Google Scholar] [CrossRef]
- Paradisi, I.; Ikonomu, V.; Arias, S. Spinocerebellar ataxias in Venezuela: Genetic epidemiology and their most likely ethnic descent. J. Hum. Genet. 2016, 61, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Michalík, A.; Del-Favero, J.; Mauger, C.; Löfgren, A.; Van Broeckhoven, C. Genomic organisation of the spinocerebellar ataxia type 7 (SCA7) gene responsible for autosomal dominant cerebellar ataxia with retinal degeneration. Hum. Genet. 1999, 105, 410–417. [Google Scholar] [CrossRef]
- David, G.; Abbas, N.; Stevanin, G.; Dürr, A.; Yvert, G.; Cancel, G.; Weber, C.; Imbert, G.; Saudou, F.; Antoniou, E.; et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997, 17, 65–70. [Google Scholar] [CrossRef]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [PubMed]
- Bonsor, M.; Ammar, O.; Schnoegl, S.; Wanker, E.E.; Ramos, E.S. Polyglutamine disease proteins: Commonalities and differences in interaction profiles and pathological effects. Proteomics 2024, 24, e2300114. [Google Scholar] [CrossRef] [PubMed]
- Atadzhanov, M.; Smith, D.C.; Mwaba, M.H.; Siddiqi, O.K.; Bryer, A.; Greenberg, L.J. Clinical and genetic analysis of spinocerebellar ataxia type 7 (SCA7) in Zambian families. Cerebellum Ataxias 2017, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Bah, M.G.; Rodriguez, D.; Cazeneuve, C.; Mochel, F.; Devos, D.; Suppiej, A.; Roubertie, A.; Meunier, I.; Gitiaux, C.; Curie, A.; et al. Deciphering the natural history of SCA7 in children. Eur. J. Neurol. 2020, 27, 2267–2276. [Google Scholar] [CrossRef]
- To, K.W.; Adamian, M.; Jakobiec, F.A.; Berson, E.L. Olivopontocerebellar atrophy with retinal degeneration. An electroretinographic and histopathologic investigation. Ophthalmology 1993, 100, 15–23. [Google Scholar] [CrossRef]
- Aleman, T.S.; Cideciyan, A.V.; Volpe, N.J.; Stevanin, G.; Brice, A.; Jacobson, S.G. Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp. Eye Res. 2002, 74, 737–745. [Google Scholar] [CrossRef]
- Ahn, J.K.; Seo, J.-M.; Chung, H.; Yu, H.G. Anatomical and functional characteristics in atrophic maculopathy associated with spinocerebellar ataxia type 7. Am. J. Ophthalmol. 2005, 139, 923–925. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.; Cerecedo-Zapata, C.M.; Hernández-Hernández, O.; Martínez-Cruz, E.; Tapia-Guerrero, Y.S.; González-Piña, R.; Salas-Vargas, J.; Rodríguez-Labrada, R.; Gurrola-Betancourth, R.; Leyva-García, N.; et al. A comprehensive clinical and genetic study of a large Mexican population with spinocerebellar ataxia type 7. Neurogenetics 2015, 16, 11–21. [Google Scholar] [CrossRef]
- Chatterjee, P.; Pedrini, S.; Stoops, E.; Goozee, K.; Villemagne, V.L.; Asih, P.R.; Verberk, I.M.W.; Dave, P.; Taddei, K.; Sohrabi, H.R.; et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl. Psychiatry 2021, 11, 27. [Google Scholar] [CrossRef]
- Abdelhak, A.; Foschi, M.; Abu-Rumeileh, S.; Yue, J.K.; D’anna, L.; Huss, A.; Oeckl, P.; Ludolph, A.C.; Kuhle, J.; Petzold, A.; et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat. Rev. Neurol. 2022, 18, 158–172. [Google Scholar] [CrossRef]
- Ganne, A.; Balasubramaniam, M.; Griffin, W.S.T.; Reis, R.J.S.; Ayyadevara, S. Glial Fibrillary Acidic Protein: A Biomarker and Drug Target for Alzheimer’s Disease. Pharmaceutics 2022, 14, 1354. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Pedrini, S.; Ashton, N.J.; Tegg, M.; Goozee, K.; Singh, A.K.; Karikari, T.K.; Simrén, J.; Vanmechelen, E.; Armstrong, N.J.; et al. Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer’s disease. Alzheimers Dement. 2022, 18, 1141–1154. [Google Scholar] [CrossRef]
- Aamodt, W.W.; Waligorska, T.; Shen, J.; Tropea, T.F.; Siderowf, A.; Weintraub, D.; Grossman, M.; Irwin, D.; Wolk, D.A.; Xie, S.X.; et al. Neurofilament Light Chain as a Biomarker for Cognitive Decline in Parkinson Disease. Mov. Disord. 2021, 36, 2945–2950. [Google Scholar] [CrossRef]
- Pareyson, D.; Shy, M.E. Neurofilament light, biomarkers, and Charcot-Marie-Tooth disease. Neurology 2018, 90, 257–259. [Google Scholar] [CrossRef]
- Benkert, P.; Meier, S.; Schaedelin, S.; Manouchehrinia, A.; Yaldizli, Ö.; Maceski, A.; Oechtering, J.; Achtnichts, L.; Conen, D.; Derfuss, T.; et al. Serum neurofilament light chain for individual prognostication of disease activity in people with multiple sclerosis: A retrospective modelling and validation study. Lancet Neurol. 2022, 21, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.S.N.; Zimmerman, K.A.; Moro, F.; Heslegrave, A.; Maillard, S.A.; Bernini, A.; Miroz, J.-P.; Donat, C.K.; Lopez, M.Y.; Bourke, N.; et al. Axonal marker neurofilament light predicts long-term outcomes and progressive neurodegeneration after traumatic brain injury. Sci. Transl. Med. 2021, 13, eabg9922. [Google Scholar] [CrossRef]
- Chelban, V.; Nikram, E.; Perez-Soriano, A.; Wilke, C.; Foubert-Samier, A.; Vijiaratnam, N.; Guo, T.; Jabbari, E.; Olufodun, S.; Gonzalez, M.; et al. Neurofilament light levels predict clinical progression and death in multiple system atrophy. Brain 2022, 145, 4398–4408. [Google Scholar] [CrossRef]
- Gendron, T.F.; Heckman, M.G.; White, L.J.; Veire, A.M.; Pedraza, O.; Burch, A.R.; Bozoki, A.C.; Dickerson, B.C.; Domoto-Reilly, K.; Foroud, T.; et al. Comprehensive cross-sectional and longitudinal analyses of plasma neurofilament light across FTD spectrum disorders. Cell Rep. Med. 2022, 3, 100607. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.; Bender, F.; Hayer, S.N.; Brockmann, K.; Schöls, L.; Kuhle, J.; Synofzik, M. Serum neurofilament light is increased in multiple system atrophy of cerebellar type and in repeat-expansion spinocerebellar ataxias: A pilot study. J. Neurol. 2018, 265, 1618–1624. [Google Scholar] [CrossRef]
- Wilke, C.; Haas, E.; Reetz, K.; Faber, J.; Garcia-Moreno, H.; Santana, M.M.; van de Warrenburg, B.; Hengel, H.; Lima, M.; Filla, A.; et al. Neurofilaments in spinocerebellar ataxia type 3: Blood biomarkers at the preataxic and ataxic stage in humans and mice. EMBO Mol. Med. 2020, 12, e11803. [Google Scholar] [CrossRef]
- Peng, L.; Wang, S.; Chen, Z.; Peng, Y.; Wang, C.; Long, Z.; Peng, H.; Shi, Y.; Hou, X.; Lei, L.; et al. Blood Neurofilament Light Chain in Genetic Ataxia: A Meta-Analysis. Mov. Disord. 2022, 37, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Koike, Y.; Jansen-West, K.R.; Al-Shaikh, R.H.; Carlomagno, Y.; Song, Y.; Dunmore, J.A.; LeDoux, M.S.; Friedman, J.H.; Pena, A.B.; Uitti, R.J.; et al. Urine levels of the polyglutamine ataxin-3 protein are elevated in patients with spinocerebellar ataxia type 3. Parkinsonism Relat. Disord. 2021, 89, 151–154. [Google Scholar] [CrossRef]
- Prudencio, M.; Garcia-Moreno, H.; Jansen-West, K.R.; Al-Shaikh, R.H.; Gendron, T.F.; Heckman, M.G.; Spiegel, M.R.; Carlomagno, Y.; Daughrity, L.M.; Song, Y.; et al. Toward allele-specific targeting therapy and pharmacodynamic marker for spinocerebellar ataxia type 3. Sci. Transl. Med. 2020, 12, eabb7086. [Google Scholar] [CrossRef]
- Garcia-Moreno, H.; Prudencio, M.; Thomas-Black, G.; Solanky, N.; Jansen-West, K.R.; Al-Shaikh, R.H.; Heslegrave, A.; Zetterberg, H.; Santana, M.M.; de Almeida, L.P.; et al. Tau and neurofilament light-chain as fluid biomarkers in spinocerebellar ataxia type 3. Eur. J. Neurol. 2022, 29, 2439–2452. [Google Scholar] [CrossRef]
- Coarelli, G.; Darios, F.; Petit, E.; Dorgham, K.; Adanyeguh, I.; Petit, E.; Brice, A.; Mochel, F.; Durr, A. Plasma neurofilament light chain predicts cerebellar atrophy and clinical progression in spinocerebellar ataxia. Neurobiol. Dis. 2021, 153, 105311. [Google Scholar] [CrossRef] [PubMed]
- Nassisi, M.; Coarelli, G.; Blanchard, B.; Dubec-Fleury, C.; Drine, K.; Kitic, N.; Sancho, S.; Hilab, R.; du Montcel, S.T.; Junge, C.; et al. ATXN7-Related Cone-Rod Dystrophy: The Integrated Functional Evaluation of the Cerebellum (CERMOI) Study. JAMA Ophthalmol. 2024, 142, 301–308. [Google Scholar] [CrossRef]
- Coarelli, G.; Dubec-Fleury, C.; Petit, E.; Sayah, S.; Fischer, C.; Nassisi, M.; Gatignol, P.; Dorgham, K.; Daghsen, L.; Daye, P.; et al. Longitudinal Changes of Clinical, Imaging, and Fluid Biomarkers in Preataxic and Early Ataxic Spinocerebellar Ataxia Type 2 and 7 Carriers. Neurology 2024, 103, e209749. [Google Scholar] [CrossRef]
- Dulski, J.; Al-Shaikh, R.H.; Prudencio, M.; Petrucelli, L.; Sulek, A.; Bernatowicz, K.; Sławek, J.; Wszolek, Z.K. First families with spinocerebellar ataxia type 7 in Poland. Neurol. Neurochir. Pol. 2023, 57, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Hubsch, T.; Du Montcel, S.T.; Baliko, L.; Berciano, J.; Boesch, S.; Depondt, C.; Giunti, P.; Globas, C.; Infante, J.; Kang, J.S.; et al. Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology 2006, 66, 1717–1720. [Google Scholar] [CrossRef]
- Chirino, A.; Hernandez-Castillo, C.R.; Galvez, V.; Contreras, A.; Diaz, R.; Beltran-Parrazal, L.; Fernandez-Ruiz, J. Motor and cognitive impairments in spinocerebellar ataxia type 7 and its correlations with cortical volumes. Eur. J. Neurosci. 2018, 48, 3199–3211. [Google Scholar] [CrossRef]
- Giunti, P.; Stevanin, G.; Worth, P.; David, G.; Brice, A.; Wood, N. Molecular and clinical study of 18 families with ADCA type II: Evidence for genetic heterogeneity and de novo mutation. Am. J. Hum. Genet. 1999, 64, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Dakna, M.; Kruse, N.; Galasko, D.; Foroud, T.; Zetterberg, H.; Schade, S.; Gera, R.G.; Wang, W.; Gao, F.; et al. Validation of Serum Neurofilament Light Chain as a Biomarker of Parkinson’s Disease Progression. Mov. Disord. 2020, 35, 1999–2008. [Google Scholar] [CrossRef]
- Buhmann, C.; Magnus, T.; Choe, C.-U. Blood neurofilament light chain in Parkinson’s disease. J. Neural Transm. 2023, 130, 755–762. [Google Scholar] [CrossRef]
- Paulson, H.L.; Bonini, N.M.; Roth, K.A. Polyglutamine disease and neuronal cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 12957–12958. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A. Intranuclear neuronal inclusions: A common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 1997, 19, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- orres-Ramos, Y.; Montoya-Estrada, A.; Cisneros, B.; Tercero-Pérez, K.; León-Reyes, G.; Leyva-García, N.; Hernández-Hernández, O.; Magaña, J.J. Oxidative Stress in Spinocerebellar Ataxia Type 7 Is Associated with Disease Severity. Cerebellum 2018, 17, 601–609. [Google Scholar] [CrossRef]
- Heimfarth, L.; Passos, F.R.S.; Monteiro, B.S.; Araújo, A.A.d.S.; Júnior, L.J.Q.; Quintans, J.d.S.S. Serum glial fibrillary acidic protein is a body fluid biomarker: A valuable prognostic for neurological disease—A systematic review. Int. Immunopharmacol. 2022, 107, 108624. [Google Scholar] [CrossRef]
- Brouillette, A.M.; Öz, G.; Gomez, C.M. Cerebrospinal Fluid Biomarkers in Spinocerebellar Ataxia: A Pilot Study. Dis. Markers 2015, 2015, 413098. [Google Scholar] [CrossRef]
- Öz, G.; Hutter, D.; Tkáč, I.; Clark, H.B.; Gross, M.D.; Jiang, H.; Eberly, L.E.; Bushara, K.O.; Gomez, C.M. Neurochemical alterations in spinocerebellar ataxia type 1 and their correlations with clinical status. Mov. Disord. 2010, 25, 1253–1261. [Google Scholar] [CrossRef]
- Zeitlberger, A.M.; Thomas-Black, G.; Garcia-Moreno, H.; Foiani, M.; Heslegrave, A.J.; Zetterberg, H.; Giunti, P. Plasma Markers of Neurodegeneration Are Raised in Friedreich’s Ataxia. Front. Cell. Neurosci. 2018, 12, 366. [Google Scholar] [CrossRef]
- Petzold, A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J. Neurol. Sci. 2005, 233, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Gaiottino, J.; Norgren, N.; Dobson, R.; Topping, J.; Nissim, A.; Malaspina, A.; Bestwick, J.P.; Monsch, A.U.; Regeniter, A.; Lindberg, R.L.; et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS ONE 2013, 8, e75091. [Google Scholar] [CrossRef]
- Rosengren, L.E.; Karlsson, J.; Persson, L.I.; Wikkelsø, C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J. Neurochem. 1996, 67, 2013–2018. [Google Scholar] [CrossRef]
- Gendron, T.F.; Badi, M.K.; Heckman, M.G.; Jansen-West, K.R.; Vilanilam, G.K.; Johnson, P.W.; Burch, A.R.; Walton, R.L.; Ross, O.A.; Brott, T.G.; et al. Plasma neurofilament light predicts mortality in patients with stroke. Sci. Transl. Med. 2020, 12, eaay1913. [Google Scholar] [CrossRef]
- Meeter, L.H.; Gendron, T.F.; Sias, A.C.; Jiskoot, L.C.; Russo, S.P.; Donker Kaat, L.; Papma, J.M.; Panman, J.L.; van der Ende, E.L.; Dopper, E.G.; et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann. Clin. Transl. Neurol. 2018, 5, 583–597. [Google Scholar] [CrossRef]
- Prudencio, M.; Erben, Y.; Marquez, C.P.; Jansen-West, K.R.; Franco-Mesa, C.; Heckman, M.G.; White, L.J.; Dunmore, J.A.; Cook, C.N.; Lilley, M.T.; et al. Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID-19. Sci. Transl. Med. 2021, 13, eabi7643. [Google Scholar] [CrossRef] [PubMed]
- Erben, Y.; Prudencio, M.; Marquez, C.P.; Jansen-West, K.R.; Heckman, M.G.; White, L.J.; Dunmore, J.A.; Cook, C.N.; Lilley, M.T.; Qosja, N.; et al. Neurofilament light chain and vaccination status associate with clinical outcomes in severe COVID-19. iScience 2022, 25, 105272. [Google Scholar] [CrossRef]
- Rojas, J.C.; Wang, P.; Staffaroni, A.M.; Heller, C.; Cobigo, Y.; Wolf, A.; Goh, S.-Y.M.; A Ljubenkov, P.; Heuer, H.W.; Fong, J.C.; et al. Plasma Neurofilament Light for Prediction of Disease Progression in Familial Frontotemporal Lobar Degeneration. Neurology 2021, 96, e2296–e2312. [Google Scholar] [CrossRef]
- Alcolea, D.; Vilaplana, E.; Suárez-Calvet, M.; Illán-Gala, I.; Blesa, R.; Clarimón, J.; Lladó, A.; Sánchez-Valle, R.; Molinuevo, J.L.; García-Ribas, G.; et al. CSF sAPPbeta, YKL-40, and neurofilament light in frontotemporal lobar degeneration. Neurology 2017, 89, 178–188. [Google Scholar] [CrossRef]
- Kuhle, J.; Kropshofer, H.; Haering, D.A.; Kundu, U.; Meinert, R.; Barro, C.; Dahlke, F.; Tomic, D.; Leppert, D.; Kappos, L. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology 2019, 92, e1007–e1015. [Google Scholar] [CrossRef] [PubMed]
- Barro, C.; Healy, B.C.; Liu, Y.; Saxena, S.; Paul, A.; Polgar-Turcsanyi, M.; Guttmann, C.R.; Bakshi, R.; Kropshofer, H.; Weiner, H.L.; et al. Serum GFAP and NfL Levels Differentiate Subsequent Progression and Disease Activity in Patients with Progressive Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2023, 10, e200052. [Google Scholar] [CrossRef] [PubMed]
- Alagaratnam, J.; von Widekind, S.; De Francesco, D.; Underwood, J.; Edison, P.; Winston, A.; Zetterberg, H.; Fidler, S. Correlation between CSF and blood neurofilament light chain protein: A systematic review and meta-analysis. BMJ Neurol. Open 2021, 3, e000143. [Google Scholar] [CrossRef]
- Andersson, E.; Janelidze, S.; Lampinen, B.; Nilsson, M.; Leuzy, A.; Stomrud, E.; Blennow, K.; Zetterberg, H.; Hansson, O. Blood and cerebrospinal fluid neurofilament light differentially detect neurodegeneration in early Alzheimer’s disease. Neurobiol. Aging 2020, 95, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Stocker, H.; Beyer, L.; Trares, K.; Perna, L.; Rujescu, D.; Holleczek, B.; Beyreuther, K.; Gerwert, K.; Schöttker, B.; Brenner, H. Association of Kidney Function with Development of Alzheimer Disease and Other Dementias and Dementia-Related Blood Biomarkers. JAMA Netw. Open 2023, 6, e2252387. [Google Scholar] [CrossRef]
- Manouchehrinia, A.; Piehl, F.; Hillert, J.; Kuhle, J.; Alfredsson, L.; Olsson, T.; Kockum, I. Confounding effect of blood volume and body mass index on blood neurofilament light chain levels. Ann. Clin. Transl. Neurol. 2020, 7, 139–143. [Google Scholar] [CrossRef]
- La Spada, A.R.; Fu, Y.H.; Sopher, B.L.; Libby, R.T.; Wang, X.; Li, L.Y.; Einum, D.D.; Huang, J.; Possin, D.E.; Smith, A.C.; et al. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 2001, 31, 913–927. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Variable | Healthy Controls (N = 8) | Asymptomatic SCA7 (N = 3) | Symptomatic SCA7 (N = 10 Plasma, N = 7 CSF) |
|---|---|---|---|
| Age at ataxia onset (years) | N/A | N/A | 30 (7, 55) |
| Sex (female) | 5 (62.50%) | 2 (66.67%) | 5 (50.00.%) |
| Age at plasma collection (years) | 48.09 (31.74, 62.13) | 37.75 (23.87, 54.52) | 39.72 (10.12, 66.85) |
| Age at CSF collection (years) | 48.09 (31.74, 62.13) | 37.75 (23.87, 54.52) | 40.61 (32.61, 65.32) |
| Disease duration of ataxia at plasma collection (years) | N/A | N/A | 8.73 (3.12, 18.82) |
| Disease duration of ataxia at CSF collection (years) | N/A | N/A | 8.61 (3.61, 17.54) |
| ATXN7 CAG-repeat length | 5.0 (5, 8) | 31 (30, 34) | 39 (34, 43) |
| GMS | |||
| 0 = Asymptomatic | 8 (100.0%) | 3 (100.0%) | 1 (10.0%) |
| 1 = Impaired gait (no assistance) | 0 (0.0%) | 0 (0.0%) | 6 (60.0%) |
| 2 = Impaired gait (require cane) | 0 (0.0%) | 0 (0.0%) | 1 (10.0%) |
| 3 = Requires a walker | 0 (0.0%) | 0 (0.0%) | 1 (10.0%) |
| 4 = Wheelchair bound | 0 (0.0%) | 0 (0.0%) | 1 (10.0%) |
| 5 = Bedridden | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| Total SARA | 0 (0, 1) | 0 (0, 0) | 11.5 (3.0, 31.0) |
| Plasma GFAP (pg/mL) | 48.0 (31.8, 108.0) | 62.4 (36.6, 71.0) | 82.7 (31.2, 242.9) |
| CSF GFAP (pg/mL) | 8906 (4537, 16,433) | 14,881 (8888, 32,098) | 9318 (5093, 22,573) |
| Plasma NfL (pg/mL) | 8.2 (3.8, 17.7) | 11.9 (4.8, 18.7) | 21.6 (6.3, 32.2) |
| CSF NfL (pg/mL) | 414.6 (223.5, 769.3) | 2412.0 (458.0, 3942.0) | 2615.0 (1814.5, 2911.0) |
| Unadjusted Analysis | Multilinear Regression Model | ||||
|---|---|---|---|---|---|
| Variable | N | β (95% CI) | p-Value | β (95% CI) | p-Value |
| Association with plasma GFAP | |||||
| SARA | 13 | 0.010 (−0.007, 0.027) | 0.2067 | 0.006 (−0.017, 0.029) | 0.5691 |
| GMS | 13 | 0.051 (−0.078, 0.180) | 0.4014 | −0.002 (−0.162, 0.158) | 0.9782 |
| Association with CSF GFAP | |||||
| SARA | 10 | −0.011 (−0.044, 0.021) | 0.4398 | −0.031 (−0.082, 0.021) | 0.1892 |
| GMS | 10 | 0.001 (−0.185, 0.185) | 0.9893 | 0.069 (−0.225, 0.363) | 0.5744 |
| Association with plasma NfL | |||||
| SARA | 13 | 0.021 (0.008, 0.035) | 0.0048 | 0.013 (−0.003, 0.029) | 0.0932 |
| GMS | 13 | 0.129 (0.016, 0.241) | 0.0288 | 0.074 (−0.042, 0.191) | 0.1784 |
| Association with CSF NfL | |||||
| SARA | 10 | 0.012 (−0.019, 0.042) | 0.4026 | −0.022 (−0.051, 0.007) | 0.1139 |
| GMS | 10 | 0.058 (−0.109, 0.226) | 0.4465 | 0.004 (−0.182, 0.190) | 0.9576 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanna Al-Shaikh, R.; Jansen-West, K.; Strongosky, A.; Parrales, Z.; Dunmore, J.A.; Song, Y.; Gendron, T.F.; Guevara, J.C.; Teive, H.A.G.; Dulski, J.; et al. Evaluating Glial Fibrillary Acidic Protein and Neurofilament Light as Potential Biomarkers for Spinocerebellar Ataxia 7. Int. J. Mol. Sci. 2025, 26, 5070. https://doi.org/10.3390/ijms26115070
Hanna Al-Shaikh R, Jansen-West K, Strongosky A, Parrales Z, Dunmore JA, Song Y, Gendron TF, Guevara JC, Teive HAG, Dulski J, et al. Evaluating Glial Fibrillary Acidic Protein and Neurofilament Light as Potential Biomarkers for Spinocerebellar Ataxia 7. International Journal of Molecular Sciences. 2025; 26(11):5070. https://doi.org/10.3390/ijms26115070
Chicago/Turabian StyleHanna Al-Shaikh, Rana, Karen Jansen-West, Audrey Strongosky, Zoe Parrales, Judith A. Dunmore, Yuping Song, Tania F. Gendron, Juan C. Guevara, Helio A. G. Teive, Jarosław Dulski, and et al. 2025. "Evaluating Glial Fibrillary Acidic Protein and Neurofilament Light as Potential Biomarkers for Spinocerebellar Ataxia 7" International Journal of Molecular Sciences 26, no. 11: 5070. https://doi.org/10.3390/ijms26115070
APA StyleHanna Al-Shaikh, R., Jansen-West, K., Strongosky, A., Parrales, Z., Dunmore, J. A., Song, Y., Gendron, T. F., Guevara, J. C., Teive, H. A. G., Dulski, J., Sławek, J., Petrucelli, L., Wszolek, Z. K., & Prudencio, M. (2025). Evaluating Glial Fibrillary Acidic Protein and Neurofilament Light as Potential Biomarkers for Spinocerebellar Ataxia 7. International Journal of Molecular Sciences, 26(11), 5070. https://doi.org/10.3390/ijms26115070