
Photophysical Properties of a Chiral Iridium-Based Photosensitizer as an Efficient Photodynamic Therapy Agent: A Theoretical Investigation


Abstract
1. Introduction
2. Results and Discussion
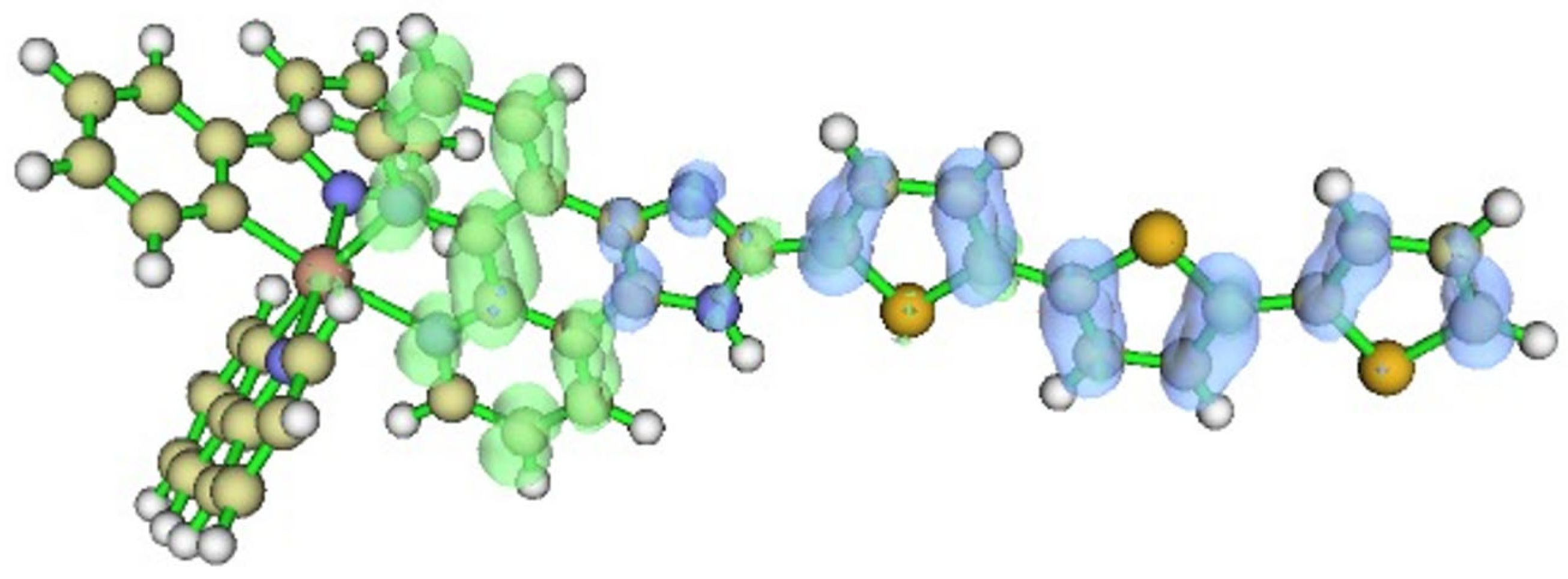
2.1. Structural Parameters
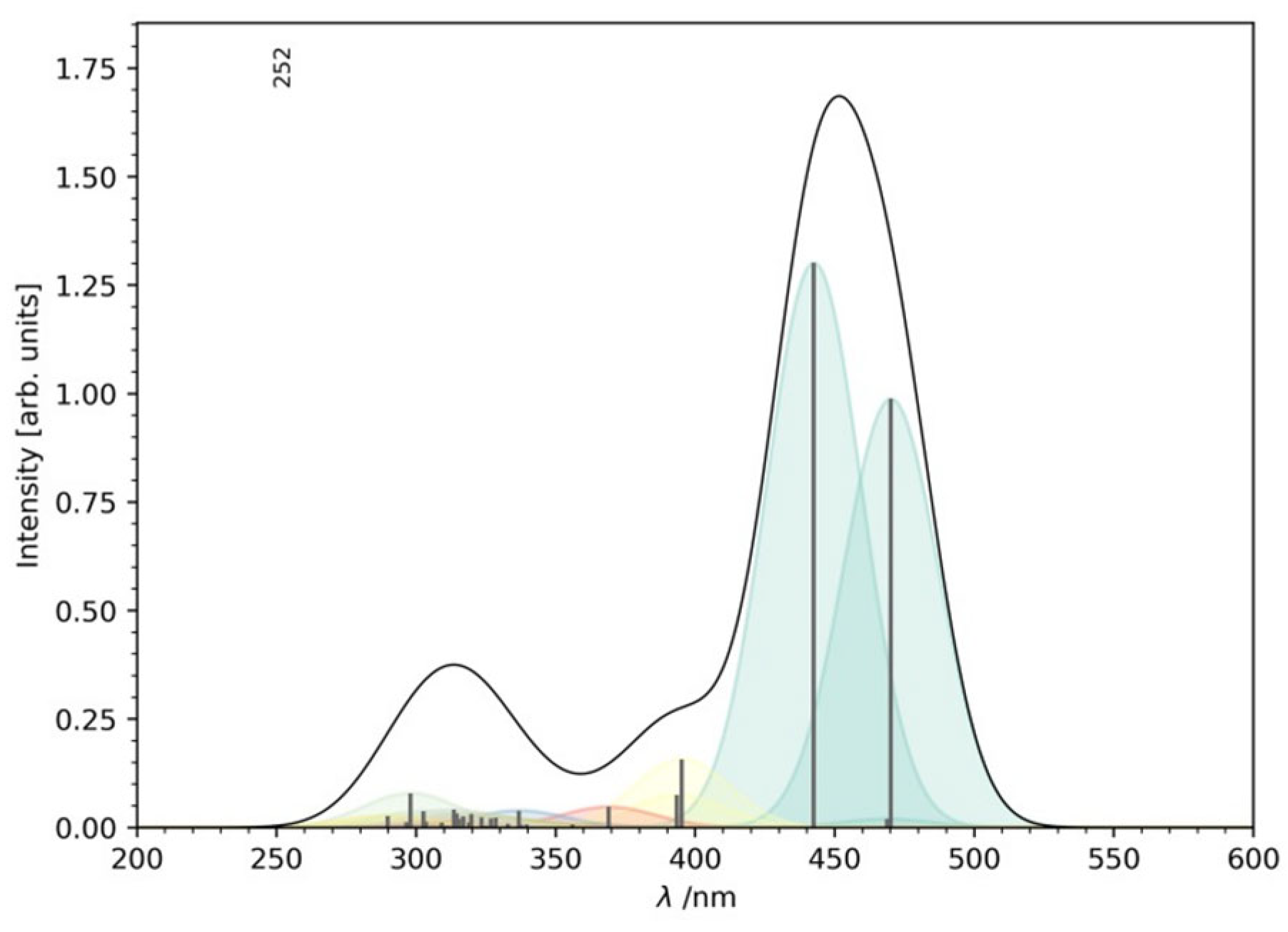
2.2. Excitation Energies and Absorption Spectra
2.3. Spin–Orbit Coupling and Intersystem Crossing
2.4. Type I Mechanism
3. Materials and Methods
4. Conclusions
- The optimized geometrical parameters, particularly the Ir-N bond lengths, are in good agreement with experimental X-ray crystallographic data for analogous iridium complexes, validating the computational approach.
- The calculated vertical excitation energies accurately reproduce the experimental absorption spectrum. Notably, the S1 absorption peak at ~470 nm is attributed to an intra-ligand charge transfer from the thiophene substituents to the imidazo-phenanthroline core, consistent with observed optical behavior.
- Spin–orbit coupling between singlet and triplet states, particularly in the S1–T2 and S1–T3 pathways, is sufficient to enable rapid intersystem crossing. This supports an efficient Type II PDT mechanism via singlet oxygen generation.
- Thermodynamic analysis indicates that electron transfer leading to superoxide formation is feasible under certain conditions—specifically when the photosensitizer is in the singlet excited state—suggesting that both Type I and Type II PDT mechanisms are accessible.
- These results position the chiral iridium complex as a versatile and promising candidate for PDT applications. Future work will focus on experimental validation, phototoxicity assessments, and structural optimization to enhance therapeutic performance and selectivity.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeRosa, M. Photosensitized Singlet Oxygen and Its Applications. Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA A Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, J.M.; Arnaut, L.G. Photodynamic Therapy (PDT) of Cancer: From Local to Systemic Treatment. Photochem. Photobiol. Sci. 2015, 14, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Hirohara, S.; Obata, M.; Hagiya, Y.; Ogura, S.; Ikeda, A.; Kataoka, H.; Tanaka, M.; Joh, T. Current States and Future Views in Photodynamic Therapy. J. Photochem. Photobiol. C. Photochem. Rev. 2011, 12, 46–67. [Google Scholar] [CrossRef]
- Spiegel, M. On the Photosensitizing Properties of Aloe-Emodin in Photodynamic Therapy: Insights from the Molecular Modeling. J. Phys. Chem. B 2025. [Google Scholar] [CrossRef]
- Sobhani, N.; Samadani, A.A. Implications of Photodynamic Cancer Therapy: An Overview of PDT Mechanisms Basically and Practically. J. Egypt Natl. Canc. Inst. 2021, 33, 34. [Google Scholar] [CrossRef]
- Correia, J.H.; Rodrigues, J.A.; Pimenta, S.; Dong, T.; Yang, Z. Photodynamic Therapy Review: Principles, Photosensitizers, Applications, and Future Directions. Pharmaceutics 2021, 13, 1332. [Google Scholar] [CrossRef]
- Spiegel, M.; Adamo, C. Tuning the Photophysical Properties of Ru(II) Photosensitizers for PDT by Protonation and Metallation: A DFT Study. J. Phys. Chem. A 2023, 127, 3625–3635. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Zhang, Y.; Shi, Y.; Zhang, R.; Chen, L.; Shi, Z.; Yuan, S.; Li, X.; He, C.; et al. Chiral Iridium-Based TLD-1433 Analogues: Exploration of Enantiomer-Dependent Behavior in Photodynamic Cancer Therapy. Inorg. Chem. 2024, 63, 7792–7798. [Google Scholar] [CrossRef]
- Roque, J.A.; Barrett, P.C.; Cole, H.D.; Lifshits, L.M.; Bradner, E.; Shi, G.; Von Dohlen, D.; Kim, S.; Russo, N.; Deep, G.; et al. Os(II) Oligothienyl Complexes as a Hypoxia-Active Photosensitizer Class for Photodynamic Therapy. Inorg. Chem. 2020, 59, 16341–16360. [Google Scholar] [CrossRef]
- Shi, G.; Monro, S.; Hennigar, R.; Colpitts, J.; Fong, J.; Kasimova, K.; Yin, H.; DeCoste, R.; Spencer, C.; Chamberlain, L.; et al. Ru(II) Dyads Derived from α-Oligothiophenes: A New Class of Potent and Versatile Photosensitizers for PDT. Coord. Chem. Rev. 2015, 282–283, 127–138. [Google Scholar] [CrossRef]
- Roque, J.A.; Barrett, P.C.; Cole, H.D.; Lifshits, L.M.; Shi, G.; Monro, S.; Von Dohlen, D.; Kim, S.; Russo, N.; Deep, G.; et al. Breaking the Barrier: An Osmium Photosensitizer with Unprecedented Hypoxic Phototoxicity for Real World Photodynamic Therapy. Chem. Sci. 2020, 11, 9784–9806. [Google Scholar] [CrossRef]
- Spiegel, M.; Russo, N. Understanding the Photophysical Properties of Pd and Pt Transition-Metal Isocorroles: A Theoretical Investigation. Chem. A Eur. J. 2025, 31, e202403725. [Google Scholar] [CrossRef]
- Alberto, M.E.; De Simone, B.C.; Liuzzi, S.; Marino, T.; Russo, N.; Toscano, M. Iodine Substituted Phosphorus Corrole Complexes as Possible Photosensitizers in Photodynamic Therapy: Insights from Theory. J. Comput. Chem. 2020, 41, 1395–1401. [Google Scholar] [CrossRef]
- Alberto, M.E.; Russo, N.; Adamo, C. Synergistic Effects of Metals in a Promising RuII −PtII Assembly for a Combined Anticancer Approach: Theoretical Exploration of the Photophysical Properties. Chem. A Eur. J. 2016, 22, 9162–9168. [Google Scholar] [CrossRef] [PubMed]
- Roque Iii, J.A.; Cole, H.D.; Barrett, P.C.; Lifshits, L.M.; Hodges, R.O.; Kim, S.; Deep, G.; Francés-Monerris, A.; Alberto, M.E.; Cameron, C.G.; et al. Intraligand Excited States Turn a Ruthenium Oligothiophene Complex into a Light-Triggered Ubertoxin with Anticancer Effects in Extreme Hypoxia. J. Am. Chem. Soc. 2022, 144, 8317–8336. [Google Scholar] [CrossRef]
- Graf, M.; Siegmund, D.; Gothe, Y.; Metzler-Nolte, N.; Sünkel, K. Metal and Substituent Influence on the Cytostatic Activity of Cationic Bis-cyclometallated Iridium and Rhodium Complexes with Substituted 1,10-Phenanthrolines as Ancillary Ligands. Z. Anorg. Allge. Chem. 2020, 646, 665–669. [Google Scholar] [CrossRef]
- Alberto, M.E.; Pirillo, J.; Russo, N.; Adamo, C. Theoretical Exploration of Type I/Type II Dual Photoreactivity of Promising Ru(II) Dyads for PDT Approach. Inorg. Chem. 2016, 55, 11185–11192. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Yu, H.; Dong, Y.; Tian, R.; Huang, G.; Boothman, D.A.; Sumer, B.D.; Gao, J. Photoactivation Switch from Type II to Type I Reactions by Electron-Rich Micelles for Improved Photodynamic Therapy of Cancer Cells under Hypoxia. J. Control. Release 2011, 156, 276–280. [Google Scholar] [CrossRef]
- Wu, Y.; Li, S.; Chen, Y.; He, W.; Guo, Z. Recent Advances in Noble Metal Complex Based Photodynamic Therapy. Chem. Sci. 2022, 13, 5085–5106. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Pollak, P.; Weigend, F. Segmented Contracted Error-Consistent Basis Sets of Double- and Triple-ζ Valence Quality for One- and Two-Component Relativistic All-Electron Calculations. J. Chem. Theory Comput. 2017, 13, 3696–3705. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Bursch, M.; Neugebauer, H.; Ehlert, S.; Grimme, S. Dispersion Corrected r2SCAN Based Global Hybrid Functionals: r2SCANh, r2SCAN0, and r2SCAN50. J. Chem. Phys. 2022, 156, 134105. [Google Scholar] [CrossRef]
- Lu, T. A Comprehensive Electron Wavefunction Analysis Toolbox for Chemists, Multiwfn. J. Chem. Phys. 2024, 161, 082503. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| State | D1 | D2 | D3 | D4 | D5 | D6 |
| S0 | 2.187 | 2.187 | 2.070 | 2.016 | 2.016 | 2.070 |
| S1 | 2.156 | 2.152 | 2.054 | 2.011 | 2.011 | 2.054 |
| T1 | 2.157 | 2.156 | 2.055 | 2.010 | 2.010 | 2.055 |
| State | E | λ | f | Transition (%) |
|---|---|---|---|---|
| T1 | 1.88 | 658.0 | 0.997 | H → L (63.6%) H → L + 1 (26.6%) |
| T2 | 2.50 | 495.6 | 0.997 | H → L (25.5%) H → L + 1 (52.5%) H − 2 → L (10.4%) |
| T3 | 2.61 | 474.3 | 0.997 | H − 1 → L (44.8%) H − 1 → L + 1 (41.6%) |
| S1 | 2.64 | 470.0 | 0.997 | H → L (86.6%) H → L + 1 (9.1%) |
| T | ΔE(S₁-Tⱼ) | SOC | kISC |
|---|---|---|---|
| T1 | 0.74 | 0.28 | 5.73 × 102 |
| T2 | 0.03 | 3.95 | 1.77 × 109 |
| T3 | −0.07 | 217.76 | 3.71 × 109 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiegel, M. Photophysical Properties of a Chiral Iridium-Based Photosensitizer as an Efficient Photodynamic Therapy Agent: A Theoretical Investigation. Int. J. Mol. Sci. 2025, 26, 5062. https://doi.org/10.3390/ijms26115062
Spiegel M. Photophysical Properties of a Chiral Iridium-Based Photosensitizer as an Efficient Photodynamic Therapy Agent: A Theoretical Investigation. International Journal of Molecular Sciences. 2025; 26(11):5062. https://doi.org/10.3390/ijms26115062
Chicago/Turabian StyleSpiegel, Maciej. 2025. "Photophysical Properties of a Chiral Iridium-Based Photosensitizer as an Efficient Photodynamic Therapy Agent: A Theoretical Investigation" International Journal of Molecular Sciences 26, no. 11: 5062. https://doi.org/10.3390/ijms26115062
APA StyleSpiegel, M. (2025). Photophysical Properties of a Chiral Iridium-Based Photosensitizer as an Efficient Photodynamic Therapy Agent: A Theoretical Investigation. International Journal of Molecular Sciences, 26(11), 5062. https://doi.org/10.3390/ijms26115062