
Inhibitory Effect and Mechanism of the Down-Regulation of TRIM32 in Colorectal Cancer


Abstract
1. Introduction
2. Results
2.1. TRIM32 Expression Is Up-Regulated in Human CRC Tissues
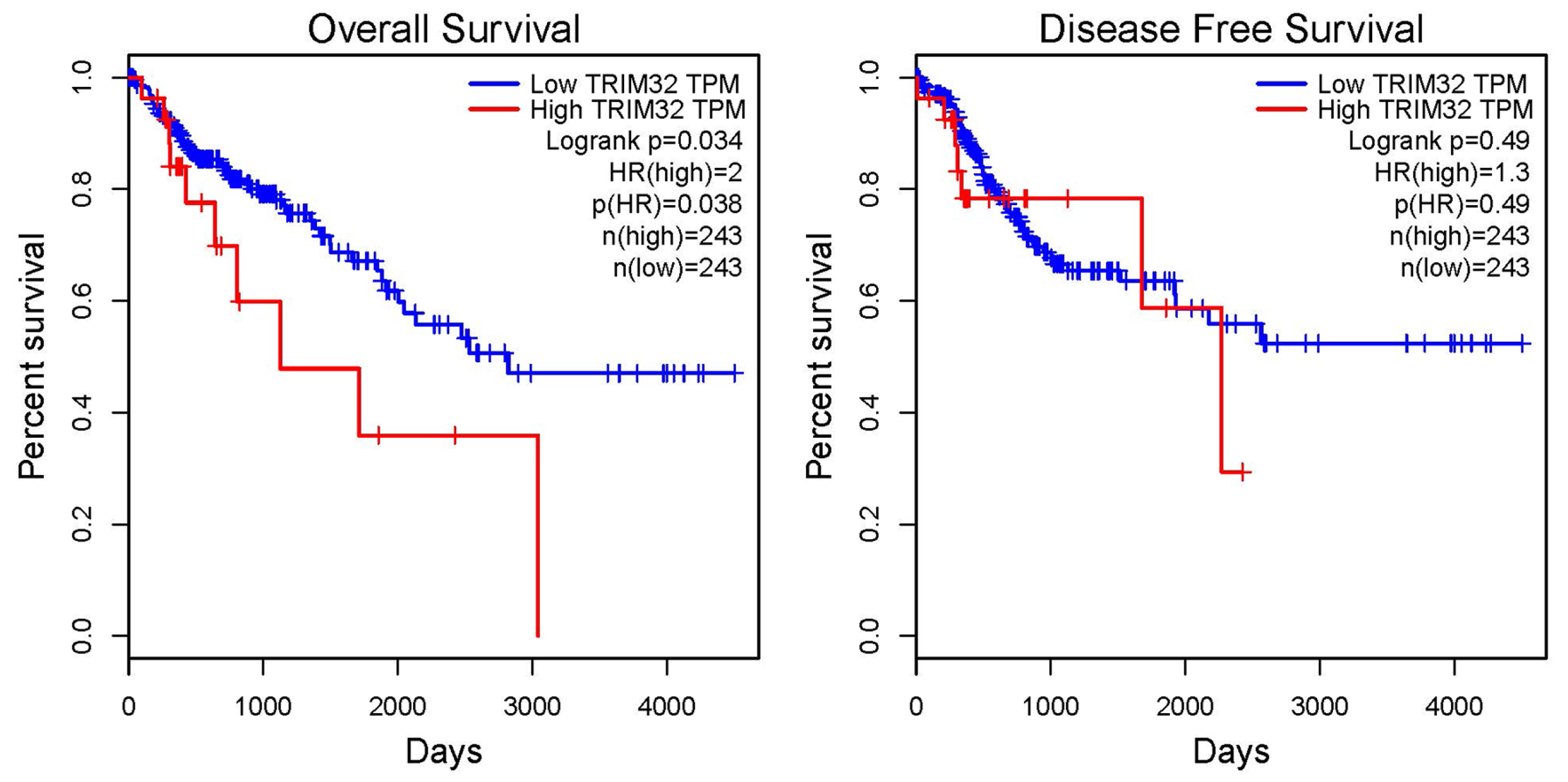
2.2. Correlation Analysis Between TRIM32 and the Prognosis of Patients with CRC
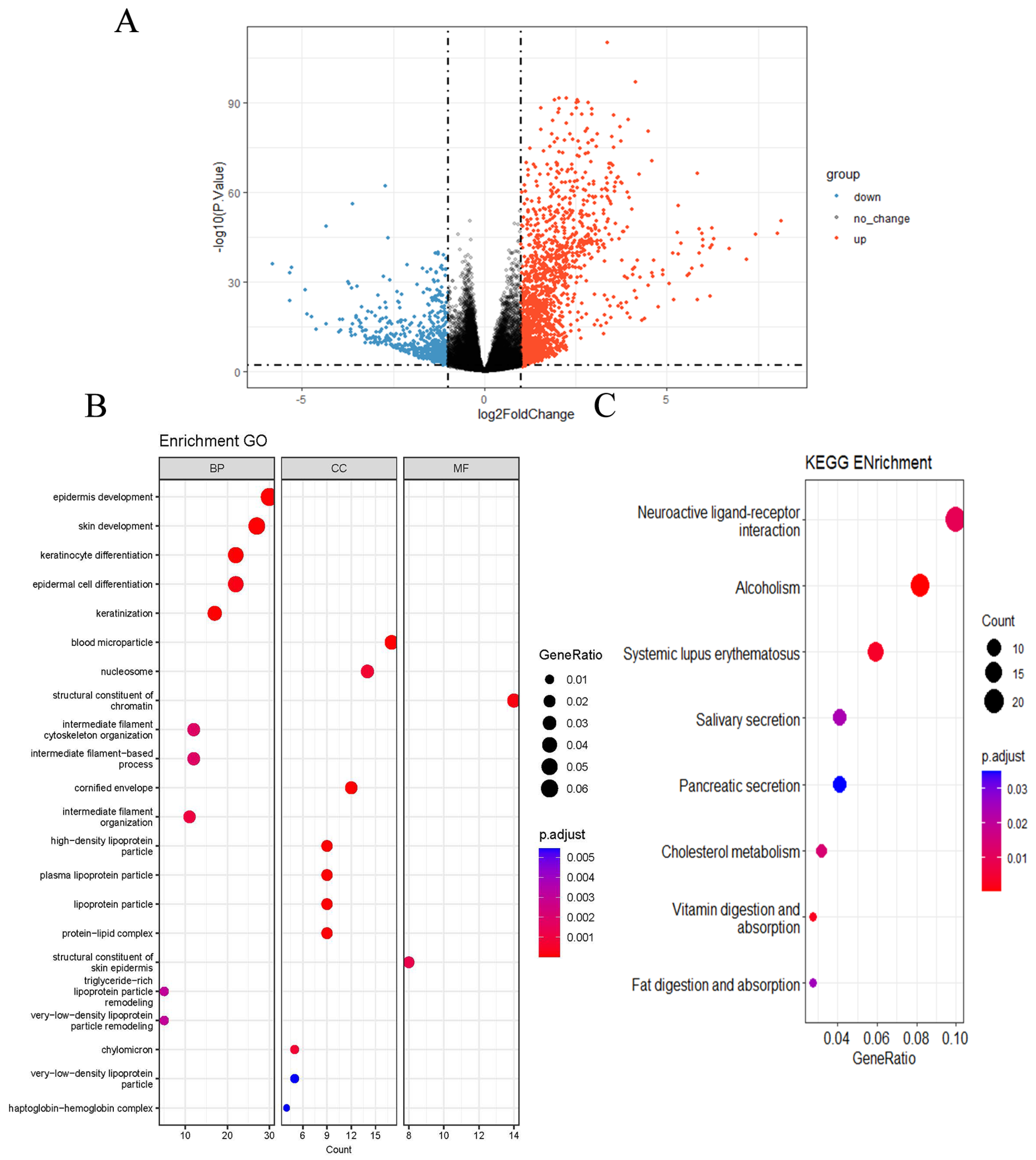
2.3. GO and KEGG Pathway Enrichment Analysis
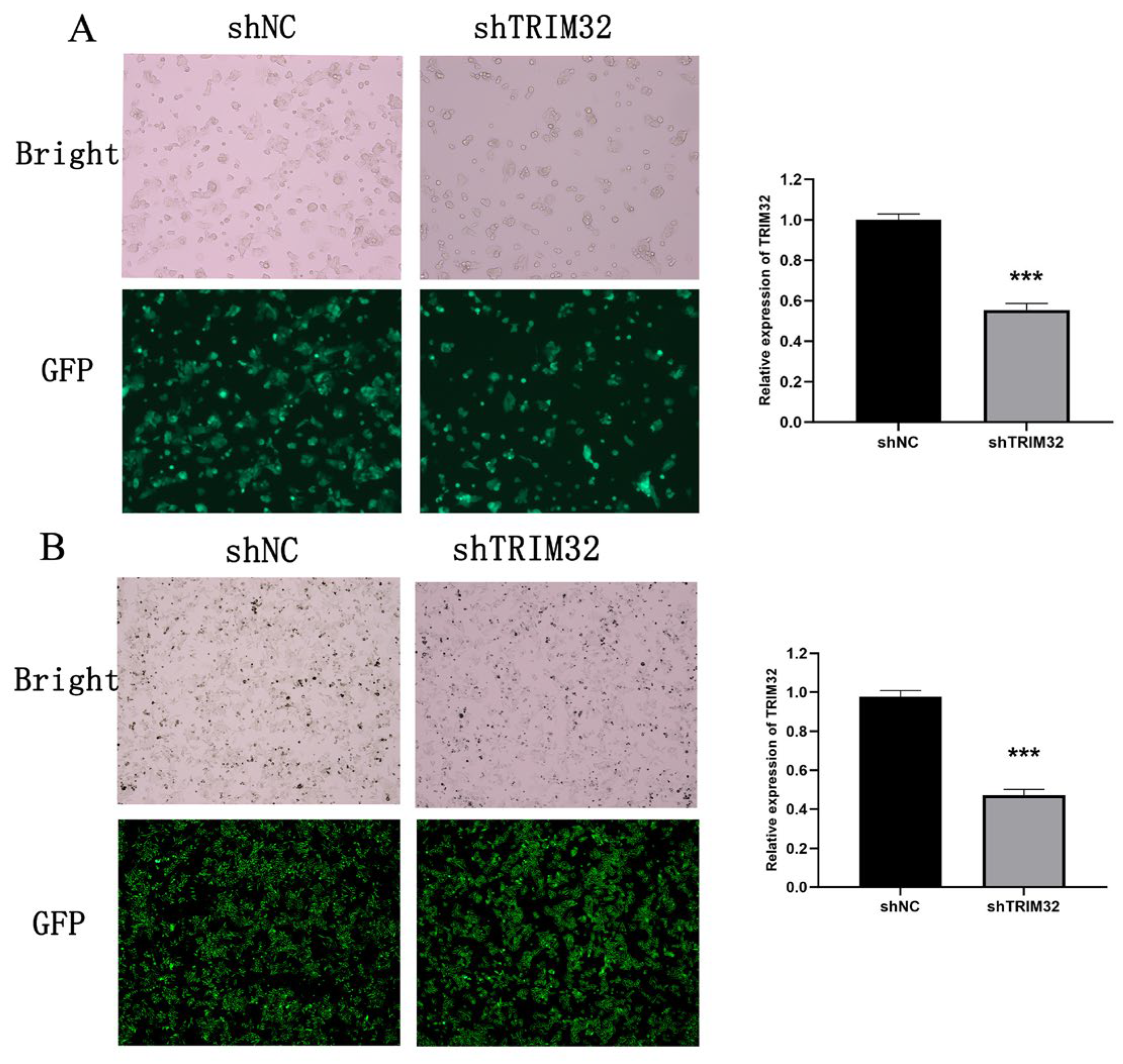
2.4. Verification of shRNA Knockdown Efficiency of TRIM32 in CRC Cells
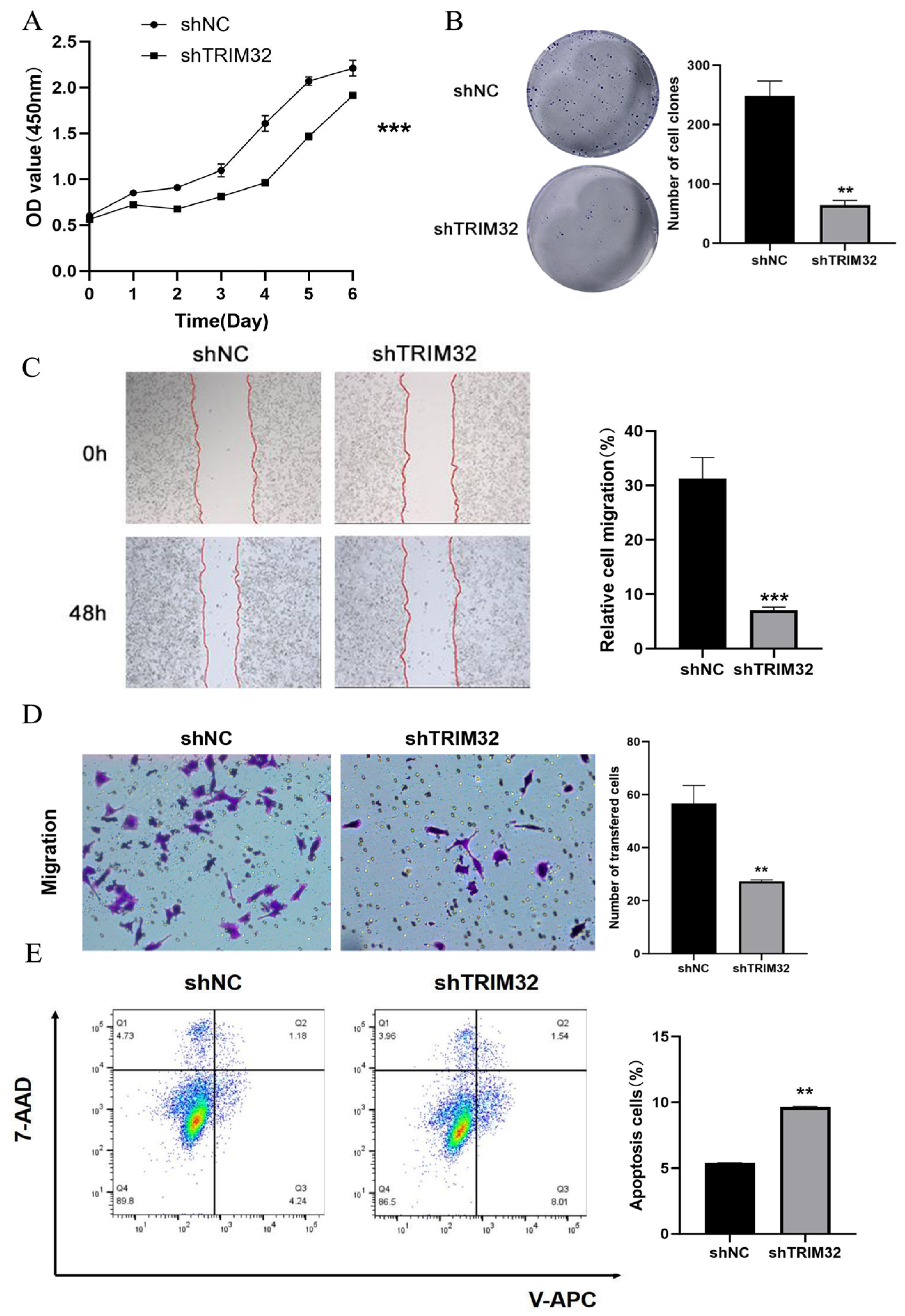
2.5. Effect of Knocking Down TRIM32 on the Proliferation, Migration, and Apoptosis of CRC Cells
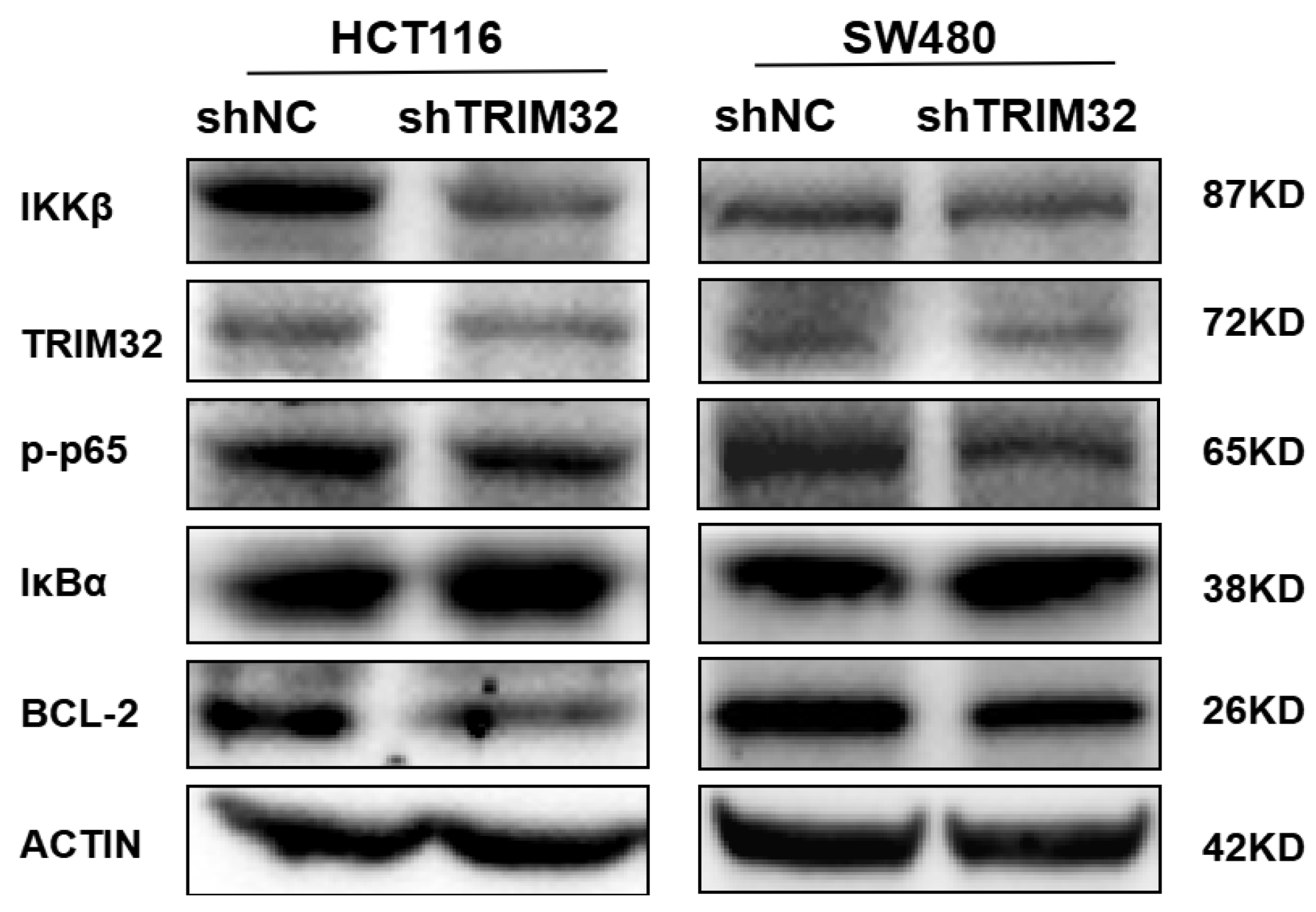
2.6. Knockdown of TRIM32 Inhibits the Growth of CRC Cells by Regulating the NF-κB Signaling Pathway
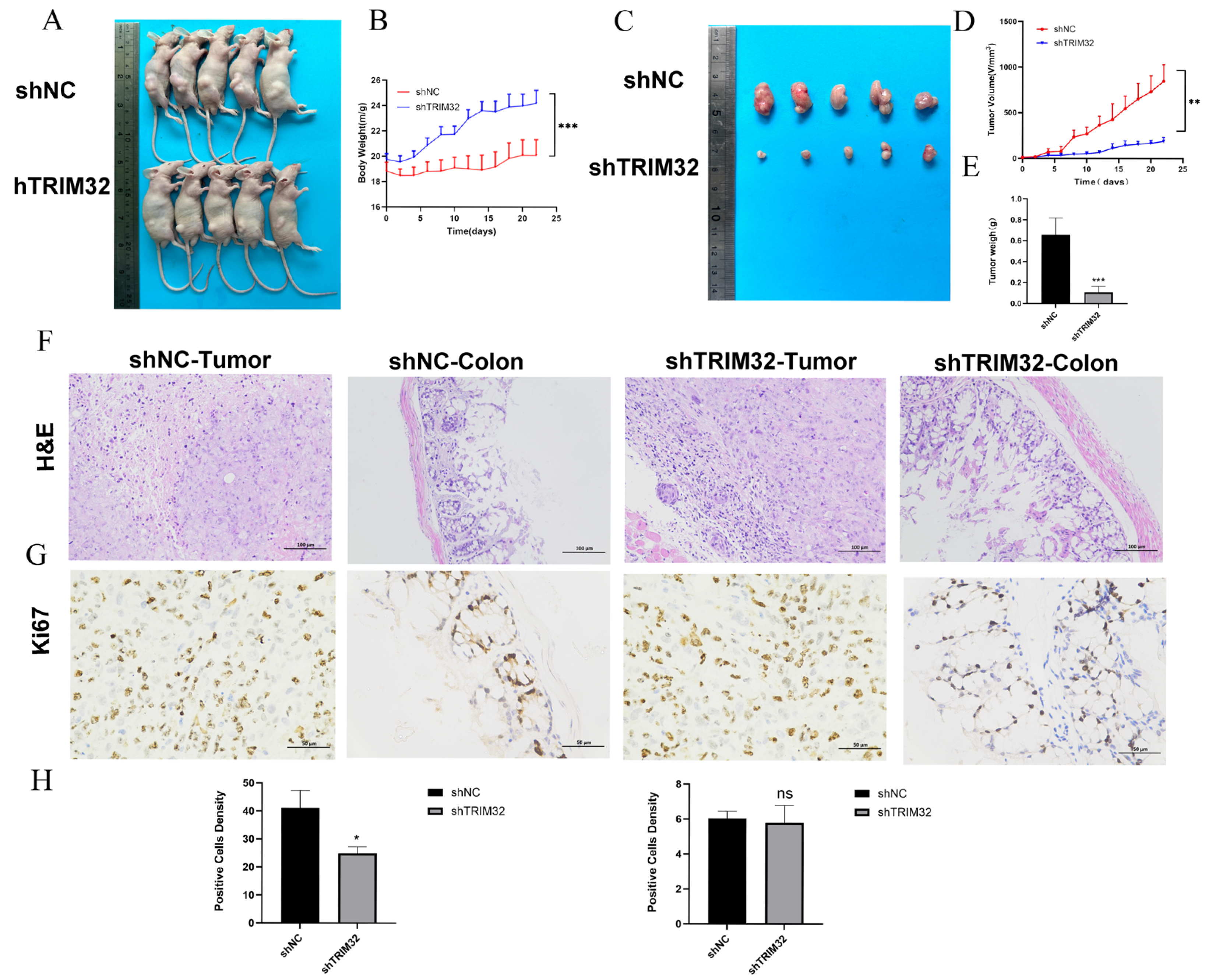
2.7. TRIM32 Knockdown Inhibits the Proliferation of CRC Cells In Vitro
3. Discussion
4. Materials and Methods
4.1. Analysis of TCGA and Gene Expression Omnibus (GEO) Database
4.2. Cell Culture
4.3. Construction of Stably Transformed Cell Line by Lentivirus Transfection
4.4. Cell Plate Clone Formation Experiment
4.5. Cell Scratch Test
4.6. Cell Counting Kit (CCK-8) Assay
4.7. Transwell MIGRATION Experiment
4.8. Apoptosis Assay
4.9. Detection of mRNA Expression by Fluorescence Quantitative PCR
4.10. Detection of Related Proteins by Western Blotting
4.11. Xenotransplantation Model
4.12. H&E Staining
4.13. Ki67 Immunohistochemistry
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Offermann, A.; Kang, D.; Watermann, C.; Weingart, A.; Hupe, M.C.; Saraji, A.; Stegmann-Frehse, J.; Kruper, R.; Schüle, R.; Pantel, K.; et al. Analysis of tripartite motif (TRIM) family gene expression in prostate cancer bone metastases. Carcinogenesis 2021, 42, 1475–1484. [Google Scholar] [CrossRef]
- Yanagi, T.; Watanabe, M.; Hata, H.; Kitamura, S.; Imafuku, K.; Yanagi, H.; Homma, A.; Wang, L.; Takahashi, H.; Shimizu, H.; et al. Loss of TRIM29 alters keratin distribution to promote cell invasion in squamous cell carcinoma. Cancer Res. 2018, 78, 6795–6806. [Google Scholar] [CrossRef]
- Zhan, W.; Han, T.; Zhang, C.; Xie, C.; Gan, M.; Deng, K.; Fu, M.; Wang, J.-B. TRIM59 promotes the proliferation and migration of non-small cell lung cancer cells by upregulating cell cycle related proteins. PLoS ONE 2015, 10, e0142596. [Google Scholar] [CrossRef]
- Horn, E.J.; Albor, A.; Liu, Y.; El-Hizawi, S.; Vanderbeek, G.E.; Babcock, M.; Bowden, G.T.; Hennings, H.; Lozano, G.; Weinberg, W.C.; et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 2004, 25, 157–167. [Google Scholar] [CrossRef]
- Zhao, T.T.; Jin, F.; Li, J.-G.; Xu, Y.-Y.; Dong, H.-T.; Liu, Q.; Xing, P.; Zhu, G.-L.; Xu, H.; Yin, S.-C.; et al. TRIM32 promotes proliferation and conferschemoresistance to breast cancer cells through activation of the NF-ĸB pathway. J. Cancer 2018, 9, 1349–1356. [Google Scholar] [CrossRef]
- Wang, C.; Xu, J.; Fu, H.; Zhang, Y.; Zhang, X.; Yang, D.; Zhu, Z.; Wei, Z.; Hu, Z.; Yan, R.; et al. TRIM32 promotes cell proliferation and invasion by activating β-catenin signalling in gastric cancer. J. Cell. Mol. Med. 2018, 22, 5020–5028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yan, Y.; Gu, Y.; Teng, F.; Lin, X.; Zhou, X.; Che, J.; Dong, X.; Zhou, L.; Lin, N. Inhibition of TRIM32 by ibr-7 treatmentsensitizes pancreatic cancer cells to gemcitabine via mTOR/p70S6K pathway. J. Cell. Mol. Med. 2022, 26, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Li, Z.; Chen, J.; Hu, X. Expression and the potential functions of TRIM32 in lung cancer tumorigenesis. J. Cell. Biochem. 2019, 120, 5232–5243. [Google Scholar] [CrossRef]
- Wang, J.; Fang, Y.; Liu, T. TRIM32 Promotes the Growth of Gastric Cancer Cells through Enhancing AKT Activity and Glucose Transportation. Biomed. Res. Int. 2020, 2020, 4027627. [Google Scholar] [CrossRef]
- Lazzari, E.; Meroni, G. TRIM32 ubiquitin E3 ligase, one enzyme for several pathologies: From muscular dystrophy to tumours. Int. J. Biochem. Cell Biol. 2016, 79, 469–477. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, W.; Du, B.; Zang, S.; Wang, X.; Mao, X.; Hu, Z. TRIM32 overexpression improves chemoresistance through regulation of mitochondrial function in non-small-cell lung cancers. OncoTargets Ther. 2018, 11, 7841–7852. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, H.; Chen, C.; Liu, L.; Ding, T.; Wang, Y.; Ma, D.; Ling, X.; Chen, X.; Li, J.; et al. TRIM32 promotes radioresistance by disrupting TC45-STAT3 interaction in triple-negative breast cancer. Oncogene 2022, 41, 1589–1599. [Google Scholar] [CrossRef]
- Ji, J.; Ding, K.; Luo, T.; Zhang, X.; Chen, A.; Zhang, D.; Li, G.; Thorsen, F.; Huang, B.; Li, X.; et al. TRIM22 activates NF-κB signaling in glioblastoma by accelerating the degradation of IκBα. Cell Death Differ. 2021, 28, 367–381. [Google Scholar] [CrossRef]
- Tan, P.; Cai, S.; Huang, Z.; Li, M.; Liu, S.; Chen, J.; Fu, W.; Zhao, L. E3 ubiquitin ligase FBXW11 as a novel inflammatory biomarker is associated with immune infiltration and NF-κB pathway activation in pancreatitis and pancreatic cancer. Cell. Signal. 2024, 116, 111033. [Google Scholar] [CrossRef]
- He, Y.; Shi, Q.; Ling, Y.; Guo, H.; Fei, Y.; Wu, R.; Tang, C.; Zhang, X.; Yao, L. ABLIM1, a novel ubiquitin E3 ligase, promotes growth and metastasis of colorectal cancer through targeting IĸBα ubiquitination and activating NF-ĸB signaling. Cell Death Differ. 2024, 31, 203–216. [Google Scholar] [CrossRef]
- Wong, D.; Teixeira, A.; Oikonomopoulos, S.; Humburg, P.; Lone, I.N.; Saliba, D.; Siggers, T.; Bulyk, M.; Angelov, D.; Dimitrov, S.; et al. Extensive characterization of NF-ĸB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 2011, 12, R70. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-ĸB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Macwan, R.S.; Ferrero, G.; Pardini, B.; Naccarati, A.; Kozlowski, P.B.; Papetti, M.J. TPM4 overexpression drives colon epithelial cell tumorigenesis by suppressing differentiation and promoting proliferation. Neoplasia 2025, 59, 101093. [Google Scholar] [CrossRef]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef]
- Cai, X.; Su, Y.; Ning, J.; Fan, X.; Shen, M. Research on the Effect and Mechanism of Selenium on Colorectal Cancer Through TRIM32. Biol. Trace Elem. Res. 2025, 203, 670–683. [Google Scholar] [CrossRef]
- Yige, L.; Dandan, Z. Progress on functional mechanisms of colorectal cancer causal SNPs in post-GWAS. Yi Chuan 2021, 43, 203–214. [Google Scholar] [CrossRef]
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9–14. [Google Scholar]
- Xu, X.; Qi, J.; Yang, J.; Pan, T.; Han, H.; Yang, M.; Han, Y. Up-Regulation of TRIM32 Associated with the Poor Prognosis of Acute Myeloid Leukemia by Integrated Bioinformatics Analysis with External Validation. Front. Oncol. 2022, 12, 848395. [Google Scholar] [CrossRef]
- Yang, X.; Ma, H.; Zhang, M.; Wang, R.; Li, X. TRIM32 promotes oral squamous cell carcinoma progression by enhancing FBP2 ubiquitination and degradation. Biochem. Biophys. Res. Commun. 2023, 678, 165–172. [Google Scholar] [CrossRef]
- Ito, M.; Migita, K.; Matsumoto, S.; Wakatsuki, K.; Tanaka, T.; Kunishige, T.; Nakade, H.; Nakatani, M.; Nakajima, Y. Overexpression of E3 ubiquitin ligase tripartite motif 32 correlates with a poor prognosis in patients with gastric cancer. Oncol. Lett. 2017, 13, 3131–3138. [Google Scholar] [CrossRef]
- Cui, X.; Lin, Z.; Chen, Y.; Mao, X.; Ni, W.; Liu, J.; Zhou, H.; Shan, X.; Chen, L.; Lv, J.; et al. Upregulated TRIM32 correlates with enhanced cell proliferation and poor prognosis in hepatocellular carcinoma. Mol. Cell. Biochem. 2016, 421, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xu-Monette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Gu, W.T.; Cheng, K.; Jia, P.F.; Li, F.; Wang, M.; Zhang, W.F.; Qiu, J.T.; Wu, Z.B.; Zhao, W.G. Knockdown of TRIM32 inhibits tumor growth and increases the therapeutic sensitivity to temozolomide in glioma in a p53-dependent and -independent manner. Biochem. Biophys. Res. Commun. 2021, 550, 134–141. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Miyake, H.; Hara, I.; Yamanaka, K.; Gohji, K.; Arakawa, S.; Kamidono, S. Overexpression of Bcl-2 enhances metastatic potential of human bladder cancer cells. Br. J. Cancer 1999, 79, 1651–1656. [Google Scholar] [CrossRef]
- Yu, J.; Dong, X.; Wang, L.; Ji, H.; Liu, A. Antitumor effects of seleno-β-lactoglobulin (Se-β-Lg) against human gastric cancer MGC-803 cells. Eur. J. Pharmacol. 2018, 833, 109–115. [Google Scholar] [CrossRef]
- Huang, Q.; Li, S.; Cheng, P.; Deng, M.; He, X.; Wang, Z.; Yang, C.-H.; Zhao, X.-Y.; Huang, J. High expression of anti-apoptotic protein Bcl-2 is a good prognostic factor in colorectal cancer: Result of a meta-analysis. World J. Gastroenterol. 2017, 23, 5018–5033. [Google Scholar] [CrossRef]
- Dai, X.Y.; Zhou, B.F.; Xie, Y.Y.; Lou, J.; Li, K.Q. Bufalin and 5-fluorouracil synergistically induce apoptosis in colorectal cancer cells. Oncol. Lett. 2018, 15, 8019–8026. [Google Scholar] [CrossRef]
- Jin, L.; Chen, Y.; Cheng, D.; He, Z.; Shi, X.; Du, B.; Xi, X.; Gao, Y.; Guo, Y. YAP inhibits autophagy and promotes progression of colorectal cancer via upregulating Bcl-2 expression. Cell Death Dis. 2021, 12, 457. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, X.; Guan, M.; Huo, C.; Yu, C.; Hu, B.; Cai, J. RASSF4 inhibits cell proliferation and increases drug sensitivity in colorectal cancer through YAP/Bcl-2 pathway. J. Cell. Mol. Med. 2022, 26, 3538–3547. [Google Scholar] [CrossRef]
- Chen, Z.J. Ubiquitin signalling in the NF-ĸB pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-ĸB by ubiquitination and degradation of the IĸBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Li, J.; Yadav, S.S.; Tewari, A.K. Recent insights into NF-ĸB signalling pathways and the link between inflammation and prostate cancer. BJU Int. 2014, 114, 168–176. [Google Scholar] [CrossRef]
- O’Dea, E.L.; Barken, D.; Peralta, R.Q.; Tran, K.T.; Werner, S.L.; Kearns, J.D.; Levchenko, A.; Hoffmann, A. A homeostatic model of IĸB metabolism to control constitutive NF-ĸB activity. Mol. Syst. Biol. 2007, 3, 111. [Google Scholar] [CrossRef]
- Fan, W.; Liu, X.; Zhang, J.; Qin, L.; Du, J.; Li, X.; Qian, S.; Chen, H.; Qian, P. TRIM67 Suppresses TNFalpha-Triggered NF-kB Activation by Competitively Binding β-TrCP to IkBa. Front. Immunol. 2022, 13, 793147. [Google Scholar] [CrossRef]
- Fang, R.; Wang, C.; Jiang, Q.; Lv, M.; Gao, P.; Yu, X.; Mu, P.; Zhang, R.; Bi, S.; Feng, J.-M.; et al. NEMO-IKKβ Are Essential for IRF3 and NF-ĸB Activation in the cGAS-STING Pathway. J. Immunol. 2007, 199, 3222–3233. [Google Scholar] [CrossRef]
- Liang, T.; Song, M.; Xu, K.; Guo, C.; Xu, H.; Zhang, H.; Xu, L. TRIM32 promotes inflammatory responses in rheumatoid arthritis fibroblast-like synoviocytes. Scand. J. Immunol. 2020, 91, e12876. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-ĸB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Solt, L.A.; May, M.J. The IĸB kinase complex: Master regulator of NF-ĸB signaling. Immunol. Res. 2008, 42, 3–18. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-ĸB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Naama, M.; Telpaz, S.; Awad, A.; Ben-Simon, S.; Harshuk-Shabso, S.; Modilevsky, S.; Rubin, E.; Sawaed, J.; Zelik, L.; Zigdon, M.; et al. Autophagy controls mucus secretion from intestinal goblet cells by alleviating ER stress. Cell Host Microbe 2023, 31, 433–446.e4. [Google Scholar] [CrossRef]
- Kobayashi, H.; Gieniec, K.A.; Lannagan, T.R.; Wang, T.; Asai, N.; Mizutani, Y.; Iida, T.; Ando, R.; Thomas, E.M.; Sakai, A.; et al. The Origin and Contribution of Cancer-Associated Fibroblasts in Colorectal Carcinogenesis. Gastroenterology 2002, 162, 890–906. [Google Scholar] [CrossRef]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Case Factors | Case Number | High Expression Cases (%) | p-Value | |
|---|---|---|---|---|
| Age | <60 | 147 | 57 (38.8) | 0.55 |
| ≥60 | 376 | 158 (42.0) | ||
| Gender | male | 276 | 122 (44.2) | 0.13 |
| female | 247 | 93 (37.6) | ||
| T stage | T1 + T2 | 103 | 45 (43.6) | 0.58 |
| T3 + T4 | 420 | 170 (40.5) | ||
| Lymph node metastasis | no | 308 | 138 (44.8) | 0.047 * |
| yes | 215 | 77 (35.8) | ||
| Distant metastasis | MX | 48 | 15 (31.3) | 0.34 |
| no | 398 | 168 (42.2) | ||
| yes | 77 | 32 (41.6) | ||
| TNM staging of tumor | I + II | 299 | 135 (45.2) | 0.031 * |
| III + IV | 224 | 80 (35.7) | ||
| Tumor site | ascending colon | 83 | 31 (37.3) | 0.99 |
| cecum | 87 | 35 (40.2) | ||
| colon | 108 | 47 (43.5) | ||
| descending colon | 17 | 6 (35.3) | ||
| hepatic flexure of the colon | 12 | 5 (41.7) | ||
| rectosigmoid junction | 5 | 2 (40.0) | ||
| rectum | 83 | 37 (44.6) | ||
| sigmoid colon | 109 | 44 (40.4) | ||
| transverse colon | 19 | 8 (42.1) | ||
| WHO pathological type | tubulovillous adenoma | 7 | 2 (28.6) | 0.17 |
| in adenocarcinoma | ||||
| mixed subtype adenocarcinoma | 3 | 2 (66.7) | ||
| adenocarcinoma NOS | 435 | 170 (39.1) | ||
| adenosquamous carcinoma | 1 | 1 (100) | ||
| carcinoma NOS | 1 | 1 (100) | ||
| mucinous adenocarcinoma | 68 | 36 (52.3) | ||
| papillary adenocarcinoma NOS | 2 | 0 (0) | ||
| tubular adenocarcinoma | 5 | 3 (100) | ||
| adenocarcinoma with | 1 | 0 (0) | ||
| neuroendocrine differentiation | ||||
| Fator | Univariate COX Regression Analysis | Multivariate COX Regression Analysis | ||
|---|---|---|---|---|
| HR (95%CI) | p-Value | HR (95%CI) | p-Value | |
| Age(<60 years vs. ≥60 years) | 1.70 (1.00–2.70) | <0.05 * | 2.25 (1.34–3.77) | <0.01 ** |
| Gender(Male vs. Female) | 0.93 (0.62–1.40) | >0.05 | ||
| Lymph node metastasis (absent vs. present) | 2.60 (1.70–4.00) | <0.001 *** | 0.53 (0.18–1.54) | >0.05 |
| Distant metastasis (absent vs. present) | 4.60 (3.00–7.0) | <0.001 *** | 2.96 (1.78–4.93) | <0.001 *** |
| T stage (T1 + T2 vs. T3 + T4) | 2.90 (1.30–6.20) | <0.01 ** | 1.84 (0.83–4.09) | >0.05 |
| TNM tumor stage (stage I + II vs. stage III + IV) | 3.00 (2.00–4.60) | <0.001 *** | 2.98 (1.79–4.96) | <0.05 * |
| TRIM32 (low expression vs. high expression) | 1.50 (1.00–2.30) | <0.05 * | 1.57 (1.05–2.34) | <0.05 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, J.; Cai, X.; Su, Y.; Fan, X.; Shen, M. Inhibitory Effect and Mechanism of the Down-Regulation of TRIM32 in Colorectal Cancer. Int. J. Mol. Sci. 2025, 26, 5047. https://doi.org/10.3390/ijms26115047
Ning J, Cai X, Su Y, Fan X, Shen M. Inhibitory Effect and Mechanism of the Down-Regulation of TRIM32 in Colorectal Cancer. International Journal of Molecular Sciences. 2025; 26(11):5047. https://doi.org/10.3390/ijms26115047
Chicago/Turabian StyleNing, Jiayu, Xiaohua Cai, Yintong Su, Xingxing Fan, and Mei Shen. 2025. "Inhibitory Effect and Mechanism of the Down-Regulation of TRIM32 in Colorectal Cancer" International Journal of Molecular Sciences 26, no. 11: 5047. https://doi.org/10.3390/ijms26115047
APA StyleNing, J., Cai, X., Su, Y., Fan, X., & Shen, M. (2025). Inhibitory Effect and Mechanism of the Down-Regulation of TRIM32 in Colorectal Cancer. International Journal of Molecular Sciences, 26(11), 5047. https://doi.org/10.3390/ijms26115047