
Activating Transcription Factor 3 (ATF3) Regulates Cellular Senescence and Osteoclastogenesis via STAT3/ERK and p65/AP-1 Pathways in Human Periodontal Ligament Cells

Abstract
1. Introduction
2. Results
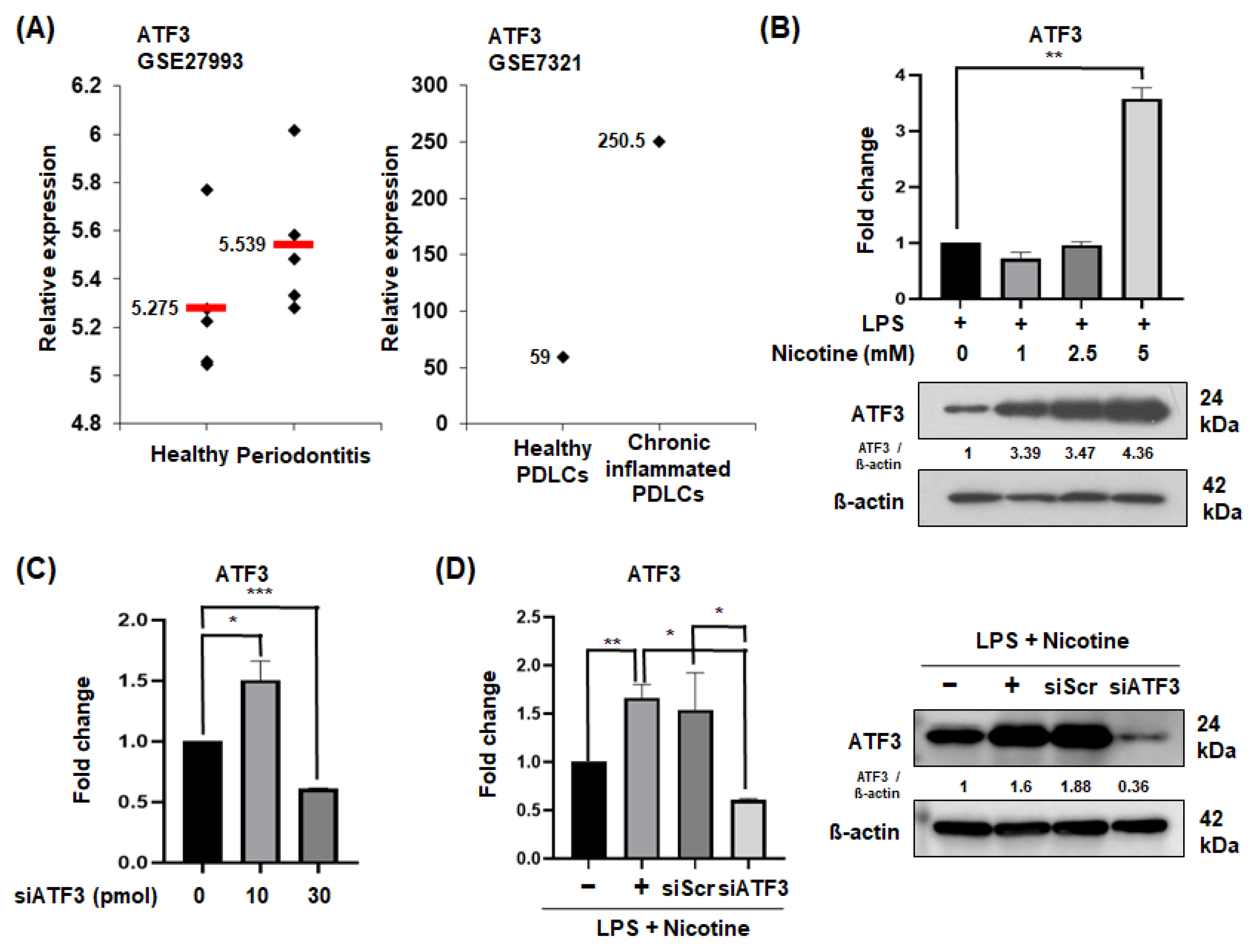
2.1. ATF3 Expression in Periodontitis Patients and Senescent PDLCs
2.2. Silencing of ATF3 Reduces SASP Factors in Senescent PDLCs
2.3. CM from ATF3-Silenced PDLCs Inhibits Osteoclast Differentiation and Bone Resorption
2.4. ATF3 Knockdown Inhibits STAT3 and ERK Phosphorylation and p65 and AP-1 Translocation in PDLCs
3. Discussion
4. Materials and Methods
4.1. Gene Expression Profiling
4.2. Cell Culture and Preparation of Condition Medium (CM)
4.3. ATF3 Knockdown
4.4. Assessment of SASP Factors
4.5. Osteoclast Differentiation and Bone Resorption Assay
4.6. Western Blotting
4.7. Immunofluorescence Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, X.; Luo, J.; Bao, T.; Wang, S.; Wu, X. Molecular mechanisms of aging and anti-aging strategies. Cell Commun. Signal. 2024, 22, 285. [Google Scholar] [CrossRef] [PubMed]
- Baechle, J.J.; Chen, N.; Makhijani, P.; Winer, S.; Furman, D.; Winer, D.A. Chronic inflammation and the hallmarks of aging. Mol. Metab. 2023, 74, 101755. [Google Scholar] [CrossRef]
- Feres, M.; Teles, F.; Teles, R.; Figueiredo, L.C.; Faveri, M. The subgingival periodontal microbiota of the aging mouth. Periodontology 2000 2016, 72, 30–53. [Google Scholar] [CrossRef]
- Kohanski, R.A.; DePinho, R.A.; Collins, J.J. How bacteria cause age-related disease in C. elegans. Nature 2017, 547, 458–461. [Google Scholar] [CrossRef]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette smoking and inflammation: Cellular and molecular mechanisms. J. Dent. Res. 2019, 88, 392–396. [Google Scholar] [CrossRef]
- Guo, X.; Wang, S.; Wang, Y.; Zhang, S. LPS-induced senescence is regulated by the activation of NF-κB and JAK/STAT3 signaling pathways in murine preosteoblasts. J. Cell Physiol. 2021, 236, 4146–4159. [Google Scholar] [CrossRef]
- Chen, J.; Li, K.; Shao, J.; Lai, Z.; Gao, R.; Wang, C.; Song, X.; Guo, W.; Yu, X.; Du, F.; et al. Irisin suppresses nicotine-mediated atherosclerosis by attenuating endothelial cell migration, proliferation, cell cycle arrest, and cell senescence. Front. Cardiovasc. Med. 2022, 9, 851603. [Google Scholar] [CrossRef]
- Centner, A.M.; Bhide, P.G.; Salazar, G. Nicotine in senescence and atherosclerosis. Cells 2020, 9, 1035. [Google Scholar] [CrossRef]
- Jun, N.R.; Jang, J.H.; Lee, J.Y.; Lee, S.I. Autophagy may mediate cellular senescence by nicotine stimulation in gingival fibroblasts. J. Dent. Hyg. Sci. 2022, 22, 164–170. [Google Scholar] [CrossRef]
- Isik Andrikopoulos, G.; Farsalinos, K.; Poulas, K. Electronic nicotine delivery systems (ENDS) and their relevance in oral health. Toxics 2019, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, Z.; Lan, S.; Hao, H.; Baz, A.A.; Yan, X.; Gao, P.; Chen, S.; Chu, Y. The dual roles of activating transcription factor 3 (ATF3) in inflammation, apoptosis, ferroptosis, and pathogen infection responses. Int. J. Mol. Sci. 2024, 25, 824. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, X.; Huang, L.; Guan, Y.; Huang, X.; Tian, X.; Zhang, L.; Tao, W. ATF3 drives senescence by reconstructing accessible chromatin profiles. Aging Cell 2021, 20, e13315. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, J.; Yang, J. ATF3 drives cell senescence through TGFb/Pdcd5 pathway in cardiac myocyte. Int. J. Cardiol. 2021, 348, 118. [Google Scholar] [CrossRef]
- Kim, K.H.; Park, B.; Rhee, D.K.; Pyo, S. Acrylamide induces senescence in macrophages through a process involving ATF3, ROS, p38/JNK, and a telomerase-independent pathway. Chem. Res. Toxicol. 2015, 28, 71–86. [Google Scholar] [CrossRef]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular senescence: A key therapeutic target in aging and diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Zhu, R.; Zhang, L.; Zhang, H.; Hu, Z. BRD4 promotes LPS-induced endothelial cells senescence via activating and cooperating STING-IRF3 pathway. Cell Signal. 2024, 118, 111127. [Google Scholar] [CrossRef]
- Aquino-Martinez, R.; Rowsey, J.L.; Fraser, D.G.; Eckhardt, B.A.; Khosla, S.; Farr, J.N.; Monroe, D.G. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 2020, 132, 115220. [Google Scholar] [CrossRef]
- Feng, G.; Zheng, K.; Cao, T.; Zhang, J.; Lian, M.; Huang, D.; Wei, C.; Gu, Z.; Feng, X. Repeated stimulation by LPS promotes the senescence of DPSCs via TLR4/MyD88-NF-kappaB-p53/p21 signaling. Cytotechnology 2018, 70, 1023–1035. [Google Scholar] [CrossRef]
- Sattari, M.; Masoudnia, M.; Mashayekhi, K.; Hashemi, S.M.; Khannazer, N.; Sattari, S.; Haftcheshmeh, S.M.; Momtazi-Borojeni, A.A. Evaluating the effect of LPS from periodontal pathogenic bacteria on the expression of senescence-related genes in human dental pulp stem cells. J. Cell. Mol. Med. 2022, 26, 5647–5656. [Google Scholar] [CrossRef] [PubMed]
- Junhui, Z.; Xiaojing, H.; Binquan, Z.; Xudong, X.; Junzhu, C.; Guosheng, F. Nicotine-reduced endothelial progenitor cell senescence through augmentation of telomerase activity via the PI3K/Akt pathway. Cytotherapy 2009, 11, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Bodas, M.; Van Westphal, C.; Carpenter-Thompson, R.; Mohanty, D.K.; Vij, N. Nicotine exposure induces bronchial epithelial cell apoptosis and senescence via ROS mediated autophagy-impairment. Free Radic. Biol. Med. 2016, 97, 441–453. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, D.; Liu, O.; Chen, H.; Wang, Y.; Zhou, Y. Cellular senescence and periodontitis: Mechanisms and therapeutics. Biology 2022, 11, 1419. [Google Scholar] [CrossRef]
- Fukasawa, K.; Park, G.; Iezaki, T.; Horie, T.; Kanayama, T.; Ozaki, K.; Onishi, Y.; Takahata, Y.; Yoneda, Y.; Takarada, T.; et al. ATF3 controls proliferation of osteoclast precursor and bone remodeling. Sci. Rep. 2016, 6, 30918. [Google Scholar] [CrossRef]
- Park, Y.; Kang, S.; Kim, D.; Kang, K.; Lee, S.; Lee, H.; Kim, E. Role of resistin in the inflammatory response induced by nicotine plus lipopolysaccharide in human periodontal ligament cells in vitro. J. Periodontal Res. 2015, 50, 602–613. [Google Scholar] [CrossRef]
- Bae, W.J.; Park, J.S.; Kang, S.K.; Kwon, I.K.; Kim, E.C. Effects of melatonin and its underlying mechanism on ethanol-stimulated senescence and osteoclastic differentiation in human periodontal ligament cells and cementoblasts. Int. J. Mol. Sci. 2018, 19, 1742. [Google Scholar] [CrossRef]
- Song, Y.; Chung, J. Aging aggravates periodontal inflammatory responses and alveolar bone resorption by Porphyromonas gingivalis infection. Curr. Issues Mol. Biol. 2023, 45, 6593–6604. [Google Scholar] [CrossRef]
- Jeong, B.C.; Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Kim, N. ATF3 modulates calcium signaling in osteoclast differentiation and activity by associating with c-Fos and NFATc1 proteins. Bone 2017, 95, 33–40. [Google Scholar] [CrossRef]
- Yin, Y.; Chen, G.; Yang, C.; Wang, J.; Peng, J.; Huang, X.; Tang, Q.; Chen, L. Osteocyte ferroptosis induced by ATF3/TFR1 contributes to cortical bone loss during ageing. Cell Prolif. 2024, 57, e13657. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amer, Y. NF-kappaB signaling and bone resorption. Osteoporos. Int. 2013, 24, 2377–2386. [Google Scholar] [CrossRef]
- Li, J.; Yin, Z.; Huang, B.; Xu, K.; Su, J. Stat3 signaling pathway: A future therapeutic target for bone-related diseases. Front. Pharmacol. 2022, 13, 897539. [Google Scholar] [CrossRef]
- Lee, S.I.; Yi, J.K.; Bae, W.J.; Lee, S.; Cha, H.J.; Kim, E.C. Thymosin Beta-4 suppresses osteoclastic differentiation and inflammatory responses in human periodontal ligament cells. PLoS ONE 2016, 11, e0146708. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequences | Tm | Size |
|---|---|---|---|
| qPCR primer | |||
| Human β-actin | Forward 5′-CACCATTGGCAATGAGCGGTTC-3′ Reverse 5′-AGGTCTTTGCGGATGTCCACGT-3′ | 60 | 135 |
| Human IFN-γ | Forward 5′-TGTCGCCAGCAGCTAAAACA-3′ Reverse 5′-TGCAGGCAGGACAACCATTA-3′ | 60 | 91 |
| Human IL6 | Forward 5′-CACCGGGAACGAAAGAGAAGC-3′ Reverse 5′-CGAAGGCGCTTGTGGAGA-3′ | 60 | 75 |
| Human IL8 | Forward 5′-CCACCGGAGCACTCCATAAG-3′ Reverse 5′-GATGGTTCCTTCCGGTGGTT-3′ | 60 | 97 |
| Human ATF3 | Forward 5′-GTTTGAGGATTTTGCTAACCTGAC-3′ Reverse 5′-AGCTGCAATCTTATTTCTTTCTCG-3′ | 55 | 211 |
| Human RANKL | Forward 5′-AAAGCCGGGCTCCAAGTC-3′ Reverse 5′-TTCTTGTCTGCGGCCAACTC-3′ | 60 | 72 |
| Human OPG | Forward 5′-CCTGGCACCAAAGTAAACGC-3′ Reverse 5′-GCACGCTGTTTTCACAGAGG-3′ | 60 | 163 |
| RT-PCR primer | |||
| Human GAPDH | Forward 5′-CTCTTCACCACCATGGAGAAG -3′ Reverse 5′-GTTGTCATGGATGACCTTGGC -3′ | 58 | 201 |
| Human TNF-α | Forward 5′-GGAAGACCCCTCCCAGATAG-3′ Reverse 5′-CCCCAGGGACCTCTCTCTAA-3′ | 52 | 413 |
| Human IL1β | Forward 5′-GGATATGGAGCAACAAGTGG-3′ Reverse 5′-ATGTACCAGTTGGGGAACTG-3′ | 60 | 264 |
| Mouse GAPDH | Forward 5′-GAGAGTGTTTCCTCGTCCCG-3′ Reverse 5′-ACTGTGCCGTTGAATTTGCC-3′ | 60 | 201 |
| Mouse Cathepsin K | Forward 5′-GCCACGCTTCCTATCCGAAA-3′ Reverse 5′-AGCTGAAAGCCCAACAGGAA-3′ | 60 | 481 |
| Mouse MMP-9 | Forward 5′-AACCTCCAACCTCACGGACA-3′ Reverse 5′-CGCGGCAAGTCTTCAGAGTA-3′ | 65 | 294 |
| Mouse NFATc1 | Forward 5′-GGTAACTCTGTCTTTCTAACCTTAAGCTC-3′ Reverse 5′-GTGATGACCCCAGCATGCACCAGTCACAG-3′ | 60 | 240 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, W.-J.; Lee, S.-I. Activating Transcription Factor 3 (ATF3) Regulates Cellular Senescence and Osteoclastogenesis via STAT3/ERK and p65/AP-1 Pathways in Human Periodontal Ligament Cells. Int. J. Mol. Sci. 2025, 26, 4959. https://doi.org/10.3390/ijms26104959
Bae W-J, Lee S-I. Activating Transcription Factor 3 (ATF3) Regulates Cellular Senescence and Osteoclastogenesis via STAT3/ERK and p65/AP-1 Pathways in Human Periodontal Ligament Cells. International Journal of Molecular Sciences. 2025; 26(10):4959. https://doi.org/10.3390/ijms26104959
Chicago/Turabian StyleBae, Won-Jung, and Sang-Im Lee. 2025. "Activating Transcription Factor 3 (ATF3) Regulates Cellular Senescence and Osteoclastogenesis via STAT3/ERK and p65/AP-1 Pathways in Human Periodontal Ligament Cells" International Journal of Molecular Sciences 26, no. 10: 4959. https://doi.org/10.3390/ijms26104959
APA StyleBae, W.-J., & Lee, S.-I. (2025). Activating Transcription Factor 3 (ATF3) Regulates Cellular Senescence and Osteoclastogenesis via STAT3/ERK and p65/AP-1 Pathways in Human Periodontal Ligament Cells. International Journal of Molecular Sciences, 26(10), 4959. https://doi.org/10.3390/ijms26104959