


RNA Modifications in Osteoarthritis: Epitranscriptomic Insights into Pathogenesis and Therapeutic Targets
 ,
,  , and
, and 
Abstract

1. Introduction
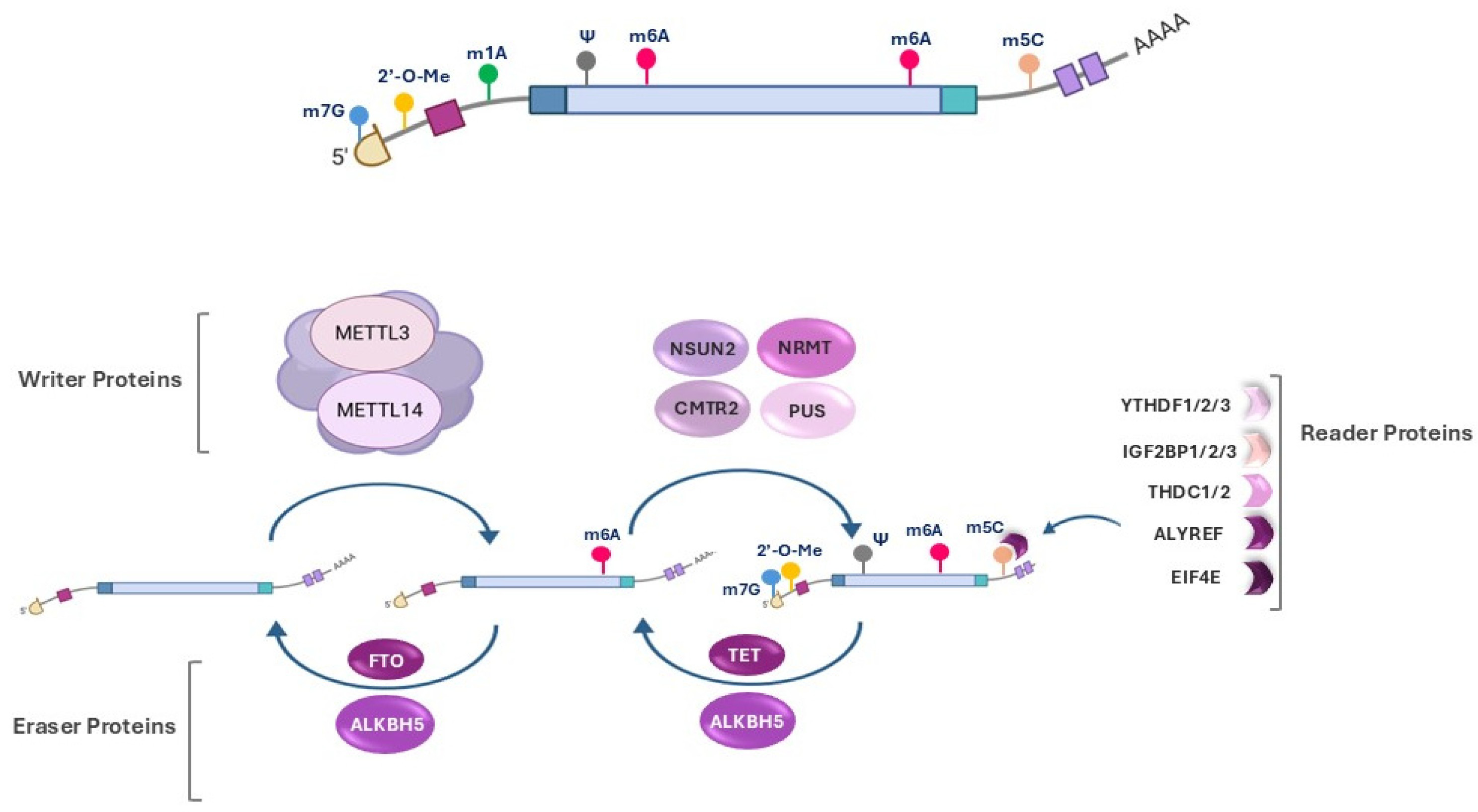
2. Epitranscriptomic Modifications
3. Epitranscriptomic Modifications in OA
4. N6-Methyladenosine (m6A)
4.1. Impact of m6A Modification on OA-Related mRNAs
4.1.1. Inflammation
4.1.2. ECM Degradation
4.1.3. Ferroptosis
4.1.4. Pyroptosis
4.1.5. Cellular Senescence
4.1.6. Autophagy

{kind=link}
{kind=link}
{kind=link}
| Pathogenic Mechanism | Target of Modification | Models | Effect on OA Pathogenesis | References |
|---|---|---|---|---|
| Inflammation | mRNA (IL-6, IL-8, IL-12, and TNF-α) | In vitro: IL-1β-treated mouse chondrocyte cell line ATDC5 In vivo: Collagenase-induced OA mouse model | METTL3 regulates OA progression by modulating pro-inflammatory factors, such as IL-6, IL-8, IL-12, and TNF-α, and NF-κB signaling | [76] |
| mRNA (NLRP3) | In vivo: DMM-induced OA mouse model In vitro: IL-1β-treated human OA primary chondrocytes | Targeting METTL3 via miR-1208 decreases NLRP3 mRNA levels and activity, thereby reducing inflammatory factor levels | [78] | |
| ECM Degradation | mRNA (MMP-1, MMP-3, MMP-13, TIMP-1, TIMP-2) | In vitro: IL-1β-treated human chondrocyte Human OA cartilage explants | METTL3 overexpression modulates ECM degradation by modulating the TIMP/MMP balance | [61] |
| mRNA (MMP-13, and COL2A1) | In vitro: IL-1β-treated mouse chondrocyte cell line ATDC5 In vivo: Collagenase-induced OA mouse model | METTL3 deletion reduces MMP-13 expression and enhances that of COL2A1 | [76] | |
| mRNA (SOX9, COL2A1) | Human OA cartilage explants | METTL3 regulates SOX9 expression and suppresses COL2A1 levels | [77] | |
| mRNA (COL2A1, ADAMTS5 and MMP-13) | In vivo: DMM-induced OA mouse model Human OA cartilage explants | METTL3 inhibition increases COL2A1 and aggrecan levels while decreasing ADAMTS5 and MMP-13 expression | [78] | |
| mRNA (STAT1) | In vitro: IL-1β-treated rat primary chondrocytes In vivo: ALCT induced OA rat model Human OA cartilage explants | METTL3 enhances the expression of STAT1, and upregulates ADAMTS12 | [79] | |
| mRNA (RPL38 and SOCS2) | In vitro: IL-1β-treated human OA primary chondrocytes In vivo: DMM-induced OA mouse model Human OA cartilage explants | Silencing RPL38 upregulates SOCS2 expression via METTL3-mediated methylation | [80] | |
| mRNA ((Ctsk) | In vitro: IL-1β-treated rat primary chondrocytes In vivo: T-2 toxin-induced cartilage injury rat model | T-2 toxin induces cartilage damage via METTL3-m6A methylation of Ctsk | [82] | |
| mRNA (CA12) | In vitro: IL-1β-treated human chondrocyte Human normal and OA cartilage explants | WTAP enhances CA12 mRNA stability and leads to chondrocyte apoptosis | [83] | |
| mRNA (FRZB) | In vivo: DMM-induced OA mouse model Human OA cartilage explants | WTAP-mediated m6A modification reduces FRZB expression, and activates the Wnt/β-catenin pathway | [68] | |
| mRNA (Runx2) | In vitro: LPS-treated mouse primary NP cells In vivo: LPS-induced IVDD mouse model, MIA-induced OA rat model | ALKBH5 facilitates Runx2 mRNA demethylation and upregulates MMPs and ADAMTSs | [86,87] | |
| mRNA (SMAD2) | In vitro: IL-1β-treated mouse primary chondrocytes In vivo: DMM-induced OA mouse model Human OA cartilage explants | Decreased levels of FTO stabilizes SMAD2 mRNA and regulates TGF-β signaling | [88] | |
| Ferroptosis | mRNA (GPX4) | In vitro: IL-1β-treated mouse primary chondrocytes In vivo: MIA-induced OA rat model | Silencing METTL14 suppresses GPX4 mRNA m6A modification and inhibits ferroptosis | [62] |
| mRNA (HMGB1) | In vitro: IL-1β-treated rat primary chondrocytes In vivo: MIA-induced OA rat model | METTL3 promotes m6A methylation of HMGB1 mRNA, and increases ferroptosis | [90] | |
| mRNA (SLC7A11) | In vivo: Acupuncture-induced IVDD rat model | YTHDF1 and HIF-1α overexpression reduces NPC ferroptosis by promoting SLC7A11 translation | [91] | |
| Pyroptosis | mRNA (NeK7) | In vitro LPS-treated human OA primary chondrocytes In vivo: DMM-induced OA mouse model Human OA cartilage explants | METTL3 knockdown reduces NEK7 mRNA levels, and suppresses chondrocyte pyroptosis | [93] |
| Cell senescence | mRNA (SLC1A5) | In vitro: H2O2-treated primary rat osteoblasts In vivo: si-Igf2bp2 and Cpd-564-treated osteoporosis rat model | METTL3 and IGF2BP2 inhibition modulates the METTL3/IGF2BP2-SLC1A5 axis and reduces osteoblast senescence | [97] |
| mRNA (CYP1B1) | In vitro: Human normal cartilage explants In vivo: ALCT induced OA mice model | m6A methylation stabilizes CYP1B1 mRNA and promotes MSC senescence | [95] | |
| Autophagy | mRNA (ATG7) | In vivo: DMM-induced OA mouse model Human normal cartilage explants | METTL3-mediated m6A modification reduces ATG7 mRNA and impairs autophagy | [100] |
| mRNA (PIK3R5) | In vitro: IL-1β-treated rat primary chondrocytes In vivo: MIA-induced OA rat model Human OA cartilage explants | FTO activates the PI3K/AKT/mTOR axis | [101] |
4.2. Impact of m6A Modification on Non-Coding RNAs
4.2.1. MicroRNAs (miRNAs)
4.2.2. Long Non-Coding RNAs (lncRNAs)
4.2.3. Circular RNAs (circRNAs)
| Type of Non-Coding RNAs | Target of Modification | Models | Effect on OA Pathogenesis | References |
|---|---|---|---|---|
| Micro RNAs (miRNAs) | miR-126-5p | In vitro: IL-1β-treated human OA primary chondrocytes Human OA cartilage explants | METTL3 regulates miR-126-5p maturation and inhibits the PI3K/Akt axis | [102] |
| miR-92b-5p | Human OA cartilage explants | WTAP enhances miR-92b-5p maturation, thereby reducing TIMP4 expression | [103] | |
| pri-miR-515-5p | In vitro: LPS-treated human chondrocyte In vivo: MIA-induced OA rat model | FTO modulates pri-miR-515-5p activity and regulates the TLR4/MyD88/NF-κB pathway | [104] | |
| pri-miR-3591 | Human OA cartilage explants | FTO regulates pri-miR-3591 maturation, and suppresses its inhibitory effects on PRKAA2 | [105] | |
| Long non-coding RNAs (lncRNAs) | lncRNA: LINC00680 | In vitro: IL-1β-treated human chondrocytes | METTL3 upregulates LINC00680, which interacts with SIRT1 mRNA | [106] |
| lncRNA: IGFBP7-OT | In vitro: LPS-treated human chondrocytes In vivo: Mouse model of MIA-induced OA Human OA cartilage explants | METTL3-mediated m6A modification of IGFBP7-OT regulates the DNMT1/DNMT3a-IGFBP7 axis | [107] | |
| lncRNA: FAS-AS1 | In vitro: IL-1β-treated human chondrocytes In vivo: ALCT induced OA rat model | METTL14 upregulates FAS-AS1, and activates the JAK/STAT3 signaling pathway | [108] | |
| lncRNA: NORAD | Human normal and DDD NP tissues | WTAP-mediated m6A modification of NORAD regulates the PUMILIO/E2F3 pathway, promoting senescence | [109] | |
| lncRNA: AC008 | In vitro: IL-1β-treated human normal primary chondrocytes In vivo: MIA-induced OA rat model Human normal and OA cartilage explants | FTO suppresses AC008 transcription, thereby elevating miR-328-3p levels and downregulating AQP1 and ANKH | [110] | |
| lncRNA: HS3ST3B1-IT1 | In vivo: MIA-induced OA mouse model Human normal and OA cartilage explants | ALKBH5-mediated demethylation stabilizes HS3ST3B1-IT1 RNA | [111] | |
| lncRNA: AK311120 | In vitro: Human adipose-derived stem cells (hASCs) In vivo: Mouse model of mandibular defect | ALKBH5 demethylates lnc-AK311120, regulates MAP2K7/JNK signaling, and promotes osteogenesis | [112] | |
| lncRNA: LOC102555094 | In vivo: Acupuncture-induced IVDD rat model | Demethylation of LOC102555094 by ZFP217 and FTO activates the miR-431/GSK-3β/Wnt pathway, which promotes IVDD | [113] | |
| Circular RNAs (circRNAs) | circRNA: circMYO1C | Human normal and OA cartilage explants | An m6A-modified circMYO1C enhances HMGB1 mRNA stability, promoting chondrocyte apoptosis | [115] |
| circular RNA: circRERE | In vitro: IL-1β-treated human chondrocytes In vivo: DMM-induced OA mouse model | m6A modification promotes circRERE degradation and contributes to OA pathogenesis | [116] |
5. 5-Methylcytosine (m5C)
6. N7-Methylguanosine (m7G)
7. Pseudouridylation (Ψ)
8. 2′-O-Ribose Methylation (2′-O-Me)
9. Polyadenylation
10. Therapeutic Potential of Targeting RNA Modifications in OA
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2′-O-Me | 2′-O-ribose methylation |
| ADAMTS | A Disintegrin and Metalloproteinase with Thrombospondin Motifs |
| ALKBH5 | AlkB Homolog 5 |
| ECM | Extracellular Matrix |
| EGFR | Epidermal Growth Factor Receptor |
| EIF4E | Eukaryotic Translation Initiation Factor 4E |
| EIF4E2 | Eukaryotic Translation Initiation Factor 4E2 |
| hm5C | Cytosine-5-Hydroxymethylation |
| IGF2BP1 | Insulin-like Growth Factor 2 Binding Protein 1 |
| IL-12 | Interleukin 12 |
| IL1 α | Interleukin 1 Alpha |
| IL1β | Interleukin 1 Beta |
| IL-6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| IVDD | Intervertebral Disc Degeneration |
| KOA | Knee Osteoarthritis |
| lncRNA | Long Non-Coding RNA |
| m1A | Methylation of Adenosine at Position N1 |
| m5C | 5-methylcytosine |
| m6A | N6-methyladenosine |
| m7G | N7-methylguanosine |
| MeRIP-Seq | Methylated RNA Immunoprecipitation Sequencing |
| METTL | Methyltransferase-Like |
| miCLIP | Methylation-induced Crosslinking and Immunoprecipitation |
| miRNA | MicroRNA |
| mRNA | Messenger RNA |
| ROS | Reactive Oxygen Species |
| rRNA | Ribosomal RNA |
| SONFH | Steroid-Associated Osteonecrosis of the Femoral Head |
| SUMOylation | Small Ubiquitin-like Modifier Modification |
| TET | Ten-Eleven Translocation |
| TGFβ1 | Transforming Growth Factor Beta 1 |
| tRNA | Transfer RNA |
| VDR | Vitamin D Receptor |
| Ψ | Pseudouridylation |
| OA | Osteoarthritis |
| PI3K/AKT/mTOR | Phosphoinositide 3-Kinase/Protein Kinase B/Mechanistic Target of Rapamycin |
| WTAP | Wilms Tumor 1-Associated Protein |
| YTH | YTH Domain-Containing Proteins |
| ZFP217 | Zinc Finger Protein 217 |
References
- Tang, S.; Zhang, C.; Oo, W.M.; Fu, K.; Risberg, M.A.; Bierma-Zeinstra, S.M.; Neogi, T.; Atukorala, I.; Malfait, A.-M.; Ding, C.; et al. Osteoarthritis. Nat. Rev. Dis. Primers 2025, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Courties, A.; Kouki, I.; Soliman, N.; Mathieu, S.; Sellam, J. Osteoarthritis year in review 2024: Epidemiology and therapy. Osteoarthr. Cartil. 2024, 32, 1397–1404. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.-P. Osteoarthritis. Nat. Rev. Dis. Primers 2016, 2, 16072. [Google Scholar] [CrossRef]
- Tong, L.; Yu, H.; Huang, X.; Shen, J.; Xiao, G.; Chen, L.; Wang, H.; Xing, L.; Chen, D. Current understanding of osteoarthritis pathogenesis and relevant new approaches. Bone Res. 2022, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- De Roover, A.; Escribano-Núñez, A.; Monteagudo, S.; Lories, R. Fundamentals of osteoarthritis: Inflammatory mediators in osteoarthritis. Osteoarthr. Cartil. 2023, 31, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Wu, X.; Tao, C.; Gong, W.; Chen, M.; Qu, M.; Zhong, Y.; He, T.; Chen, S.; Xiao, G. Osteoarthritis: Pathogenic signaling pathways and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 56. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Lajeunesse, D.; Fahmi, H.; Tardif, G.; Pelletier, J.P. New thoughts on the pathophysiology of osteoarthritis: One more step toward new therapeutic targets. Curr. Rheumatol. Rep. 2006, 8, 30–36. [Google Scholar] [CrossRef]
- Reginato, A.M.; Olsen, B.R. The role of structural genes in the pathogenesis of osteoarthritic disorders. Arthritis. Res. 2002, 4, 337–345. [Google Scholar] [CrossRef]
- Zhai, G.; Huang, J. Genetics of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2024, 38, 101972. [Google Scholar] [CrossRef]
- Loughlin, J. Osteoarthritis Linkage Scan: More Loci for the Geneticists to Investigate. Ann. Rheum. Dis. 2006, 65, 1265–1266. [Google Scholar] [CrossRef]
- Vikkula, M.; Madman, E.C.; Lui, V.C.; Zhidkova, N.I.; Tiller, G.E.; Goldring, M.B.; van Beersum, S.E.; de Waal Malefijt, M.C.; van den Hoogen, F.H.; Ropers, H.-H. Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 1995, 80, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Zhai, G.; Rivadeneira, F.; Houwing-Duistermaat, J.; Meulenbelt, I.; Bijkerk, C.; Hofman, A.; van Meurs, J.; Uitterlinden, A.; Pols, H.; Slagboom, P. Insulin-like growth factor I gene promoter polymorphism, collagen type II α1 (COL2A1) gene, and the prevalence of radiographic osteoarthritis: The Rotterdam Study. Ann. Rheum. Dis. 2004, 63, 544–548. [Google Scholar] [CrossRef]
- Vincent, T.L. IL-1 in osteoarthritis: Time for a critical review of the literature. F1000Research 2019, 8, 934. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.J.; Beier, F.; Young, D.A.; Loughlin, J. Interplay between genetics and epigenetics in osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Izda, V.; Martin, J.; Sturdy, C.; Jeffries, M.A. DNA methylation and noncoding RNA in OA: Recent findings and methodological advances. Osteoarthr. Cartil. Open 2021, 3, 100208. [Google Scholar] [CrossRef]
- Alsabri, S.G.; Guedi, G.G.; Najar, M.; Merimi, M.; Lavoie, F.; Grabs, D.; Fernandes, J.; Pelletier, J.-P.; Martel-Pelletier, J.; Benderdour, M. Epigenetic regulation of 15-Lipoxygenase-1 expression in human chondrocytes by promoter methylation. Inflamm. Res. 2023, 72, 2145–2153. [Google Scholar] [CrossRef]
- Najar, M.; Alsabri, S.G.; Guedi, G.G.; Merimi, M.; Lavoie, F.; Grabs, D.; Pelletier, J.-P.; Martel-Pelletier, J.; Benderdour, M.; Fahmi, H. Role of epigenetics and the transcription factor Sp1 in the expression of the D prostanoid receptor 1 in human cartilage. Front. Cell Dev. Biol. 2023, 11, 1256998. [Google Scholar] [CrossRef]
- Núñez-Carro, C.; Blanco-Blanco, M.; Villagrán-Andrade, K.M.; Blanco, F.J.; de Andrés, M.C. Epigenetics as a therapeutic target in osteoarthritis. Pharmaceuticals 2023, 16, 156. [Google Scholar] [CrossRef]
- Farrajota, K.; Cheng, S.; Martel-Pelletier, J.; Afif, H.; Pelletier, J.P.; Li, X.; Ranger, P.; Fahmi, H. Inhibition of Interleukin-1beta-induced cyclooxygenase 2 expression in human synovial fibroblasts by 15-Deoxy-Delta12,14-prostaglandin J2 through a histone deacetylase-independent mechanism. Arthritis Rheum. 2005, 52, 94–104. [Google Scholar] [CrossRef]
- El Mansouri, F.E.; Nebbaki, S.S.; Kapoor, M.; Afif, H.; Martel-Pelletier, J.; Pelletier, J.P.; Benderdour, M.; Fahmi, H. Lysine-specific demethylase 1-mediated demethylation of histone H3 lysine 9 contributes to interleukin 1β-induced microsomal prostaglandin E synthase 1 expression in human osteoarthritic chondrocytes. Arthritis Res. Ther. 2014, 16, R113. [Google Scholar] [CrossRef]
- Panagopoulos, P.K.; Lambrou, G.I. The Involvement of MicroRNAs in Osteoarthritis and Recent Developments: A Narrative Review. Mediterr. J. Rheumatol. 2018, 29, 67–79. [Google Scholar] [CrossRef] [PubMed]
- He, C. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Boer, C.G. Osteoarthritis year in review 2024: Genetics, genomics, and epigenetics. Osteoarthr. Cartil. 2025, 33, 50–57. [Google Scholar] [CrossRef]
- Cai, Z.; Long, T.; Zhao, Y.; Lin, R.; Wang, Y. Epigenetic regulation in knee osteoarthritis. Front. Genet. 2022, 13, 942982. [Google Scholar] [CrossRef]
- Fathollahi, A.; Aslani, S.; Jamshidi, A.; Mahmoudi, M. Epigenetics in osteoarthritis: Novel spotlight. J. Cell. Physiol. 2019, 234, 12309–12324. [Google Scholar] [CrossRef]
- Simon, T.C.; Jeffries, M.A. The epigenomic landscape in osteoarthritis. Curr. Rheumatol. Rep. 2017, 19, 30. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, Y.-L.; Wang, X.-Q. The emerging roles of N6-methyladenosine in osteoarthritis. Front. Mol. Neurosci. 2022, 15, 1040699. [Google Scholar] [CrossRef]
- Arcidiacono, O.A.; Krejčí, J.; Legartová, S.; Stixova, L.; Bártová, E. Chapter 1—Epigenetic processes and DNA repair in embryonic stem cells☆. In Stem Cell Epigenetics; Meshorer, E., Testa, G., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 17, pp. 1–23. [Google Scholar]
- Cappannini, A.; Ray, A.; Purta, E.; Mukherjee, S.; Boccaletto, P.; Moafinejad, S.N.; Lechner, A.; Barchet, C.; Klaholz, B.P.; Stefaniak, F.; et al. MODOMICS: A database of RNA modifications and related information. 2023 update. Nucleic Acids Res. 2023, 52, D239–D244. [Google Scholar] [CrossRef]
- Zheng, H.; Aihaiti, Y.; Cai, Y.; Yuan, Q.; Yang, M.; Li, Z.; Xu, K.; Xu, P. The m6A/m1A/m5C-Related Methylation Modification Patterns and Immune Landscapes in Rheumatoid Arthritis and Osteoarthritis Revealed by Microarray and Single-Cell Transcriptome. J. Inflamm. Res. 2023, 16, 5001–5025. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, L.; Chen, S.; Liu, H. A brief review of RNA modification related database resources. Methods 2022, 203, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Chen, L.; Chen, Z.; Pang, J.; Huang, J.; Lin, J.; Zheng, L.; Li, B.; Qu, L.; Yang, J. RMBase v3.0: Decode the landscape, mechanisms and functions of RNA modifications. Nucleic Acids Res. 2023, 52, D273–D284. [Google Scholar] [CrossRef] [PubMed]
- Ofusa, K.; Chijimatsu, R.; Ishii, H. Detection techniques for epitranscriptomic marks. Am. J. Physiol. Cell Physiol. 2022, 322, C787–C793. [Google Scholar] [CrossRef] [PubMed]
- Flamand, M.N.; Tegowski, M.; Meyer, K.D. The Proteins of mRNA Modification: Writers, Readers, and Erasers. Annu. Rev. Biochem. 2023, 92, 145–173. [Google Scholar] [CrossRef]
- Nie, F.; Feng, P.; Song, X.; Wu, M.; Tang, Q.; Chen, W. RNAWRE: A resource of writers, readers and erasers of RNA modifications. Database 2020, 2020, baaa049. [Google Scholar] [CrossRef]
- Schaefer, M.R. The Regulation of RNA Modification Systems: The Next Frontier in Epitranscriptomics? Genes 2021, 12, 345. [Google Scholar] [CrossRef]
- Yang, Y.; Hsu, P.J.; Chen, Y.-S.; Yang, Y.-G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef]
- Liu, W.-W.; Zheng, S.-Q.; Li, T.; Fei, Y.-F.; Wang, C.; Zhang, S.; Wang, F.; Jiang, G.-M.; Wang, H. RNA modifications in cellular metabolism: Implications for metabolism-targeted therapy and immunotherapy. Signal Transduct. Target. Ther. 2024, 9, 70. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, J. RNA 5-Methylcytosine modification and its emerging role as an epitranscriptomic mark. RNA Biol. 2021, 18 (Suppl. S1), 117–127. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, W.-Y.; Shen, S.-Y.; Shen, J.-H.; Chen, X.-D. Biological roles of RNA m7G modification and its implications in cancer. Biol. Direct 2023, 18, 58. [Google Scholar] [CrossRef]
- Zhou, K.I.; Pecot, C.V.; Holley, C.L. 2′-O-methylation (Nm) in RNA: Progress, challenges, and future directions. RNA 2024, 30, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Imbriano, C.; Moresi, V.; Belluti, S.; Renzini, A.; Cavioli, G.; Maretti, E.; Molinari, S. Epitranscriptomics as a New Layer of Regulation of Gene Expression in Skeletal Muscle: Known Functions and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 15161. [Google Scholar] [CrossRef] [PubMed]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Zhai, G.; Xiao, L.; Jiang, C.; Yue, S.; Zhang, M.; Zheng, J.; Liu, Z.; Dong, Y. Regulatory Role of N6-Methyladenosine (m6A) Modification in Osteoarthritis. Front. Cell Dev. Biol. 2022, 10, 946219. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, D.; Yang, X.; Li, Q.; Zhang, R.; Xiong, Y. Expression of m6A Methylation Regulator in Osteoarthritis and Its Prognostic Markers. Cartilage 2023, 14, 321–328. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, Y.; Yu, J.; Jiang, F.; Wu, C. Significance of m6A regulatory factor in gene expression and immune function of osteoarthritis. Front. Physiol. 2022, 13, 918270. [Google Scholar] [CrossRef]
- Ouyang, Y.; Tu, Y.; Chen, S.; Min, H.; Wen, Z.; Zheng, G.; Wan, T.; Fan, H.; Yang, W.; Sun, G. Characterization of immune microenvironment infiltration and m6A regulator-mediated RNA methylation modification patterns in osteoarthritis. Front. Immunol. 2022, 13, 1018701. [Google Scholar] [CrossRef]
- Mi, H.; Wang, M.; Chang, Y. The potential impact of polymorphisms in METTL3 gene on knee osteoarthritis susceptibility. Heliyon 2024, 10, e28035. [Google Scholar] [CrossRef]
- Bian, A.; Wang, C.; Zhang, H.; Yan, Y.; Zhang, L.; Cheng, W. Diagnostic value and immune infiltration characterization of YTHDF2 as a critical m6A regulator in osteoarthritic synovitis. J. Orthop. Surg. Res. 2023, 18, 535. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, H.; Li, D.; Ma, L.; Lu, T.; Sun, H.; Zhang, Y.; Yang, H. Comprehensive analysis of m6A RNA methylation modification patterns and the immune microenvironment in osteoarthritis. Front. Immunol. 2023, 14, 1128459. [Google Scholar] [CrossRef]
- Zhang, W.; He, L.; Liu, Z.; Ren, X.; Qi, L.; Wan, L.; Wang, W.; Tu, C.; Li, Z. Multifaceted functions and novel insight into the regulatory role of RNA N6-methyladenosine modification in musculoskeletal disorders. Front. Cell Dev. Biol. 2020, 8, 870. [Google Scholar] [CrossRef]
- Chen, X.; Hua, W.; Huang, X.; Chen, Y.; Zhang, J.; Li, G. Regulatory role of RNA N6-methyladenosine modification in bone biology and osteoporosis. Front. Endocrinol. 2020, 10, 911. [Google Scholar] [CrossRef]
- Luo, J.; Xu, T.; Sun, K. N6-methyladenosine RNA modification in inflammation: Roles, mechanisms, and applications. Front. Cell Dev. Biol. 2021, 9, 670711. [Google Scholar] [CrossRef]
- Ignatova, V.V.; Stolz, P.; Kaiser, S.; Gustafsson, T.H.; Lastres, P.R.; Sanz-Moreno, A.; Cho, Y.-L.; Amarie, O.V.; Aguilar-Pimentel, A.; Klein-Rodewald, T. The rRNA m6A methyltransferase METTL5 is involved in pluripotency and developmental programs. Genes Dev. 2020, 34, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 2017, 169, 824–835.E14. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Moindrot, B.; Cerase, A.; Coker, H.; Masui, O.; Grijzenhout, A.; Pintacuda, G.; Schermelleh, L.; Nesterova, T.B.; Brockdorff, N. A pooled shRNA screen identifies Rbm15, Spen, and Wtap as factors required for Xist RNA-mediated silencing. Cell Rep. 2015, 12, 562–572. [Google Scholar] [CrossRef]
- Liao, S.; Sun, H.; Xu, C. YTH domain: A family of N6-methyladenosine (m6A) readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef]
- Delaunay, S.; Helm, M.; Frye, M. RNA modifications in physiology and disease: Towards clinical applications. Nat. Rev. Genet. 2024, 25, 104–122. [Google Scholar] [CrossRef]
- Sang, W.; Xue, S.; Jiang, Y.; Lu, H.; Zhu, L.; Wang, C.; Ma, J. METTL3 involves the progression of osteoarthritis probably by affecting ECM degradation and regulating the inflammatory response. Life Sci. 2021, 278, 119528. [Google Scholar] [CrossRef]
- Liu, D.; Ren, L.; Liu, J. METTL14 promotes chondrocyte ferroptosis in osteoarthritis via m6A modification of GPX4. Int. J. Rheum. Dis. 2024, 27, e15297. [Google Scholar] [CrossRef] [PubMed]
- Knights, A.J.; Redding, S.J.; Maerz, T. Inflammation in osteoarthritis: The latest progress and ongoing challenges. Curr. Opin. Rheumatol. 2023, 35, 128–134. [Google Scholar] [CrossRef]
- Cai, L.; Li, D.; Feng, Z.; Gu, X.; Xu, Q.; Li, Q. YTHDF2 Regulates Macrophage Polarization through NF-κB and MAPK Signaling Pathway Inhibition or p53 Degradation. Dis. Markers 2022, 2022, 3153362. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, H.; Chaby, R. Selective refractoriness of macrophages to endotoxin-induced production of tumor necrosis factor, elicited by an autocrine mechanism. J. Leukoc. Biol. 1993, 53, 45–52. [Google Scholar] [CrossRef]
- Fahmi, H.; Charon, D.; Mondange, M.; Chaby, R. Endotoxin-induced desensitization of mouse macrophages is mediated in part by nitric oxide production. Infect. Immun. 1995, 63, 1863–1869. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, H.; Pan, H.; Zhang, Z.; Zeng, H.; Xie, H.; Yin, J.; Tang, W.; Lin, R.; Zeng, C.; et al. Expression pattern analysis of m6A regulators reveals IGF2BP3 as a key modulator in osteoarthritis synovial macrophages. J. Transl. Med. 2023, 21, 339. [Google Scholar] [CrossRef]
- An, X.; Wang, R.; Lv, Z.; Wu, W.; Sun, Z.; Wu, R.; Yan, W.; Jiang, Q.; Xu, X. WTAP-mediated m6A modification of FRZB triggers the inflammatory response via the Wnt signaling pathway in osteoarthritis. Exp. Mol. Med. 2024, 56, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhong, Y.; Wang, F.; Guo, Y.; Mao, R.; Xie, H.; Zhang, Y.; Li, J. WTAP mediated N6-methyladenosine RNA modification of ELF3 drives cellular senescence by upregulating IRF8. Int. J. Biol. Sci. 2024, 20, 1763. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, W.; Hua, Y. Expression patterns of eight RNA-modified regulators correlating with immune infiltrates during the progression of osteoarthritis. Front. Immunol. 2023, 14, 1019445. [Google Scholar] [CrossRef]
- Duan, Y.; Yu, C.; Yan, M.; Ouyang, Y.; Ni, S. m6A regulator-mediated RNA methylation modification patterns regulate the immune microenvironment in osteoarthritis. Front. Genet. 2022, 13, 921256. [Google Scholar] [CrossRef]
- Hu, S.; Shen, C.; Yao, X.; Zou, Y.; Wang, T.; Sun, X.; Nie, M. m6A regulator-mediated methylation modification patterns and immune microenvironment infiltration characterization in osteoarthritis. BMC Med. Genom. 2022, 15, 273. [Google Scholar] [CrossRef] [PubMed]
- Zayed, N.; Afif, H.; Chabane, N.; Mfuna-Endam, L.; Benderdour, M.; Martel-Pelletier, J.; Pelletier, J.P.; Motiani, R.K.; Trebak, M.; Duval, N. Inhibition of Interleukin-1β–induced matrix metalloproteinases 1 and 13 production in human osteoarthritic chondrocytes by prostaglandin D2. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2008, 58, 3530–3540. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, M.D.; Malfait, A.M.; Deccico, C.; Arner, E. The role of ADAM-TS4 (aggrecanase-1) and ADAM-TS5 (aggrecanase-2) in a model of cartilage degradation. Osteoarthr. Cartil. 2001, 9, 539–552. [Google Scholar] [CrossRef]
- Mukherjee, A.; Das, B. The role of inflammatory mediators and matrix metalloproteinases (MMPs) in the progression of osteoarthritis. Biomater. Biosyst. 2024, 13, 100090. [Google Scholar] [CrossRef]
- Liu, Q.; Li, M.; Jiang, L.; Jiang, R.; Fu, B. METTL3 promotes experimental osteoarthritis development by regulating inflammatory response and apoptosis in chondrocyte. Biochem. Biophys. Res. Commun. 2019, 516, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Hu, B.; Ding, B.; Zhao, Q.; Liu, C.; Öner, F.; Xu, H. N(6)-methyladenosine RNA methyltransferase like 3 inhibits extracellular matrix synthesis of endplate chondrocytes by downregulating sex-determining region Y-Box transcription factor 9 expression under tension. Osteoarthr. Cartil. 2022, 30, 613–625. [Google Scholar] [CrossRef]
- Zhou, H.; Shen, X.; Yan, C.; Xiong, W.; Ma, Z.; Tan, Z.; Wang, J.; Li, Y.; Liu, J.; Duan, A. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate osteoarthritis of the knee in mice model by interacting with METTL3 to reduce m6A of NLRP3 in macrophage. Stem Cell Res. Ther. 2022, 13, 322. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, X.; Jia, Z.; Zhang, M.; Yuan, M.; Zhou, Y.; Wang, J.; Xia, D. Epigenetic regulatory mechanism of ADAMTS12 expression in osteoarthritis. Mol. Med. 2023, 29, 86. [Google Scholar] [CrossRef]
- Shi, L.; Hu, H.; Sun, P.; Li, Z.; Ji, L.; Liu, S.; Zhang, J. RPL38 knockdown inhibits the inflammation and apoptosis in chondrocytes through regulating METTL3-mediated SOCS2 m6A modification in osteoarthritis. Inflamm. Res. 2022, 71, 977–989. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, X.; Deng, X.; Niu, H.; Zhao, Y.; Wen, J.; Wang, S.; Liu, H.; Guo, X.; Wu, C. Transcriptome-wide RNA m6A methylation profiles in an endemic osteoarthropathy, Kashin-Beck disease. J. Cell. Mol. Med. 2024, 28, e70047. [Google Scholar] [CrossRef]
- Zhang, B.; Li, H.; Qi, F.; Yu, Q.; Jiang, H.; Lin, B.; Dong, H.; Li, H.; Yu, J. T-2 toxin induces chondrocyte extracellular matrix degradation by regulating the METTL3-mediated Ctsk m6A modification. Int. Immunopharmacol. 2024, 143, 113390. [Google Scholar] [CrossRef]
- Deng, G.; Xu, Y.; Li, Z.; Zeng, G. WTAP mediates IL-1β-induced chondrocyte injury by enhancing CA12 mRNA stability depending on m6A modification. J. Orthop. Surg. Res. 2024, 19, 826. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liao, W. Hydroxysafflor yellow A attenuates oxidative stress injury-induced apoptosis in the nucleus pulposus cell line and regulates extracellular matrix balance via CA XII. Exp. Ther. Med. 2022, 23, 182. [Google Scholar] [CrossRef]
- Bentz, M.; Zaouter, C.; Shi, Q.; Fahmi, H.; Moldovan, F.; Fernandes, J.C.; Benderdour, M. Inhibition of inducible nitric oxide synthase prevents lipid peroxidation in osteoarthritic chondrocytes. J. Cell. Biochem. 2012, 113, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhan, E.; Chen, C.; Hu, Y.; Lv, Z.; He, Q.; Wang, X.; Li, X.; Zhang, F. ALKBH5-mediated m6A demethylation of Runx2 mRNA promotes extracellular matrix degradation and intervertebral disc degeneration. Cell Biosci. 2024, 14, 79. [Google Scholar]
- Nie, G.; Li, Y.; Zhao, H.; Liu, C.; Zhang, Y.; Yang, X.; Tian, F.; Wen, X. Inflammatory microenvironment promotes extracellular matrix degradation of chondrocytes through ALKBH5-dependent Runx2 m6A modification in the pathogenesis of osteoarthritis. Int. Immunopharmacol. 2025, 144, 113638. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xie, Z.; Qian, Y.; Ni, W.; Cui, L.; Fang, X.; Wan, S.; Zhao, X.; Qin, A.; Fan, S. FTO-mediated SMAD2 m6A modification protects cartilage against Osteoarthritis. Exp. Mol. Med. 2024, 56, 2283–2295. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, J.; Si, H.; Wu, Y.; Zhou, S.; Shen, B. The Role Played by Ferroptosis in Osteoarthritis: Evidence Based on Iron Dyshomeostasis and Lipid Peroxidation. Antioxidants 2022, 11, 1668. [Google Scholar] [CrossRef]
- Bao, T.; Liao, T.; Cai, X.; Lu, B.; Dai, G.; Pei, S.; Zhang, Y.; Li, Y.; Xu, B. METTL3 mediated ferroptosis in chondrocytes and promoted pain in KOA via HMGB1 m6A modification. Cell Biol. Int. 2024, 48, 1755–1765. [Google Scholar] [CrossRef]
- Lu, X.; Li, D.; Lin, Z.; Gao, T.; Gong, Z.; Zhang, Y.; Wang, H.; Xia, X.; Lu, F.; Song, J.; et al. HIF-1α-induced expression of the m6A reader YTHDF1 inhibits the ferroptosis of nucleus pulposus cells by promoting SLC7A11 translation. Aging Cell 2024, 23, e14210. [Google Scholar] [CrossRef]
- Chang, X.; Kang, Y.; Yang, Y.; Chen, Y.; Shen, Y.; Jiang, C.; Shen, Y. Pyroptosis: A Novel Intervention Target in the Progression of Osteoarthritis. J. Inflamm. Res. 2022, 15, 3859–3871. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Xiong, H.; Peng, J.; Liu, Y.; Zong, Y. METTL3 Regulates the m6A Modification of NEK7 to Inhibit the Formation of Osteoarthritis. Cartilage 2023, 16, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Du, Y.; Tan, Z.; Li, D.; Xie, J. METTL14-mediated HOXA5 m6A modification alleviates osteoporosis via promoting WNK1 transcription to suppress NLRP3-dependent macrophage pyroptosis. J. Orthop. Transl. 2024, 48, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Li, J.; Yu, W.; Xie, Z.; Zheng, G.; Liu, W.; Wang, S.; Cao, Q.; Lin, J.; Su, Z.; et al. ALKBH5 facilitates CYP1B1 mRNA degradation via m6A demethylation to alleviate MSC senescence and osteoarthritis progression. Exp. Mol. Med. 2023, 55, 1743–1756. [Google Scholar] [CrossRef]
- Benderdour, M.; Martel-Pelletier, J.; Pelletier, J.P.; Kapoor, M.; Zunzunegui, M.V.; Fahmi, H. Cellular Aging, Senescence and Autophagy Processes in Osteoarthritis. Curr. Aging Sci. 2015, 8, 147–157. [Google Scholar] [CrossRef]
- Liu, X.-W.; Xu, H.-W.; Yi, Y.-Y.; Zhang, S.-B.; Chang, S.-J.; Pan, W.; Wang, S.-J. Inhibition of Mettl3 ameliorates osteoblastic senescence by mitigating m6A modifications on Slc1a5 via Igf2bp2-dependent mechanisms. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2024, 1870, 167273. [Google Scholar] [CrossRef]
- Duan, R.; Xie, H.; Liu, Z.Z. The Role of Autophagy in Osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 608388. [Google Scholar] [CrossRef]
- Wen, X.; Wang, J.; Wang, Q.; Liu, P.; Zhao, H. Interaction between N6-methyladenosine and autophagy in the regulation of bone and tissue degeneration. Front. Bioeng. Biotechnol. 2022, 10, 978283. [Google Scholar] [CrossRef]
- Chen, X.; Gong, W.; Shao, X.; Shi, T.; Zhang, L.; Dong, J.; Shi, Y.; Shen, S.; Qin, J.; Jiang, Q. METTL3-mediated m6A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann. Rheum. Dis. 2022, 81, 85–97. [Google Scholar] [CrossRef]
- Lv, G.; Wang, B.; Li, L.; Li, Y.; Li, X.; He, H.; Kuang, L. Exosomes from dysfunctional chondrocytes affect osteoarthritis in Sprague-Dawley rats through FTO-dependent regulation of PIK3R5 mRNA stability. Bone Jt. Res. 2022, 11, 652–668. [Google Scholar] [CrossRef]
- Xiao, L.; Zhao, Q.; Hu, B.; Wang, J.; Liu, C.; Xu, H. METTL3 promotes IL-1β–induced degeneration of endplate chondrocytes by driving m6A-dependent maturation of miR-126-5p. J. Cell. Mol. Med. 2020, 24, 14013–14025. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Jiang, T.; Zheng, W.; Zhang, J.; Li, A.; Lu, C.; Liu, W. N6-methyladenosine (m6A) methyltransferase WTAP-mediated miR-92b-5p accelerates osteoarthritis progression. Cell Commun. Signal. 2023, 21, 199. [Google Scholar] [CrossRef]
- Cai, D.; Zhang, J.; Yang, J.; Lv, Q.; Zhong, C. Overexpression of FTO alleviates osteoarthritis by regulating the processing of miR-515-5p and the TLR4/MyD88/NF-κB axis. Int. Immunopharmacol. 2023, 114, 109524. [Google Scholar] [CrossRef]
- Liu, W.; Jiang, T.; Zheng, W.; Zhang, J.; Li, A.; Lu, C.; Lin, Z. FTO-mediated m6A demethylation of pri-miR-3591 alleviates osteoarthritis progression. Arthritis Res. Ther. 2023, 25, 53. [Google Scholar] [CrossRef]
- Ren, J.; Li, Y.; Wuermanbieke, S.; Hu, S.; Huang, G. N6-methyladenosine (m6A) methyltransferase METTL3-mediated LINC00680 accelerates osteoarthritis through m6A/SIRT1 manner. Cell Death Discov. 2022, 8, 240. [Google Scholar] [CrossRef]
- Tang, Y.; Hong, F.; Ding, S.; Yang, J.; Zhang, M.; Ma, Y.; Zheng, Q.; Yang, D.; Jin, Y.; Ma, C. METTL3-mediated m6A modification of IGFBP7-OT promotes osteoarthritis progression by regulating the DNMT1/DNMT3a-IGFBP7 axis. Cell Rep. 2023, 42, 112589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Mao, H.; Li, F.; Wang, D.; Liu, Y. METTL14-mediated lncRNA-FAS-AS1 promotes osteoarthritis progression by up-regulating ADAM8. Int. J. Rheum. Dis. 2024, 27, e15323. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ma, L.; He, S.; Luo, R.; Wang, B.; Zhang, W.; Song, Y.; Liao, Z.; Ke, W.; Xiang, Q. WTAP-mediated m6A modification of lncRNA NORAD promotes intervertebral disc degeneration. Nat. Commun. 2022, 13, 1469. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, M.; Yang, D.; Ma, Y.; Tang, Y.; Xing, M.; Li, L.; Chen, L.; Jin, Y.; Ma, C. m6A-mediated Upregulation of AC008 Promotes Osteoarthritis Progression through the miR-328-3p-AQP1/ANKH axis. Exp. Mol. Med. 2021, 53, 1723–1734. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, Y.; Zhu, X.; Chen, Y.; Jiang, X.; Ding, S.; Zheng, Q.; Zhang, M.; Yang, J.; Ma, Y. ALKBH5-mediated m6A demethylation of HS3ST3B1-IT1 prevents osteoarthritis progression. Iscience 2023, 26, 107838. [Google Scholar] [CrossRef]
- Song, Y.; Gao, H.; Pan, Y.; Gu, Y.; Sun, W.; Wang, Y.; Liu, J. ALKBH5 Regulates Osteogenic Differentiation via the lncRNA/mRNA Complex. J. Dent. Res. 2024, 103, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, N.; Du, Z.; Ling, Z.; Zhang, P.; Yang, J.; Khaleel, M.; Khoury, A.N.; Li, J.; Li, S. Bioinformatics analysis integrating metabolomics of m6A RNA microarray in intervertebral disc degeneration. Epigenomics 2020, 12, 1419–1441. [Google Scholar] [CrossRef]
- Song, M.; Yao, H.; Sun, Z.; Chen, D.; Xu, X.; Long, G.; Wu, L.; Hu, W. METTL3/YTHDC1-medicated m6A modification of circRNA3634 regulates the proliferation and differentiation of antler chondrocytes by miR-124486-5-MAPK1 axis. Cell. Mol. Biol. Lett. 2023, 28, 101. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Chu, X.; Qian, W.; Yin, H. CircMYO1C silencing alleviates chondrocytes inflammation and apoptosis through m6A/HMGB1 axis in osteoarthritis. Biotechnol. Appl. Biochem. 2024, 71, 1360–1369. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Lin, Y.; Wei, B.; Hu, X.; Xu, L.; Zhang, W.; Lu, J. N6-methyladenosine-modified circRNA RERE modulates osteoarthritis by regulating β-catenin ubiquitination and degradation. Cell Prolif. 2023, 56, e13297. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, W.; Liu, J.; Gu, N.; Zhang, R. Genome-wide identification of mRNA 5-Methylcytosine in mammals. Nat. Struct. Mol. Biol. 2019, 26, 380–388. [Google Scholar] [CrossRef]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-Methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Y.; Sun, B.-F.; Chen, Y.-S.; Xu, J.-W.; Lai, W.-Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W. 5-Methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef]
- Arguello, A.E.; Li, A.; Sun, X.; Eggert, T.W.; Mairhofer, E.; Kleiner, R.E. Reactivity-dependent profiling of RNA 5-Methylcytidine dioxygenases. Nat. Commun. 2022, 13, 4176. [Google Scholar] [CrossRef]
- Yu, Y.; Lu, S.; Liu, X.; Li, Y.; Xu, J. Identification and analysis of RNA-5-methylcytosine-related key genes in osteoarthritis. BMC Genom. 2023, 24, 539. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, S.; Xu, C.; Ni, Z.; Wang, X.; Wang, F. The role of m5C RNA methylation regulators in the diagnosis and immune microenvironment of osteoarthritis. Comput. Methods Biomech. Biomed. Eng. 2024, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Tan, S.; Ku, J.; Widowati, T.A.; Ku, D.; Lee, K.; You, K.; Kim, Y. RNA 5-Methylcytosine marks mitochondrial double-stranded RNAs for degradation and cytosolic release. Mol. Cell 2024, 84, 2935–2948.E7. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ren, Z.; Yan, S.; Zhao, L.; Liu, J.; Zhao, L.; Li, Z.; Ye, S.; Liu, A.; Li, X. Nsun4 and Mettl3 mediated translational reprogramming of Sox9 promotes BMSC chondrogenic differentiation. Commun. Biol. 2022, 5, 495. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Zheng, J.C. Internal m7G methylation: A novel epitranscriptomic contributor in brain development and diseases. Mol. Ther. Nucleic Acids 2023, 31, 295–308. [Google Scholar] [CrossRef]
- Long, D.; Deng, Z.; Zhao, X.; Xu, Y.; Li, W.; Mo, X.; Zhong, Y.; Li, M.; He, A.; Zhang, Z.; et al. m7G-modified mt-tRF3b-LeuTAA regulates mitophagy and metabolic reprogramming via SUMOylation of SIRT3 in chondrocytes. Biomaterials 2025, 314, 122903. [Google Scholar] [CrossRef]
- Hao, L.; Shang, X.; Wu, Y.; Chen, J.; Chen, S. Construction of a Diagnostic m7G Regulator-Mediated Scoring Model for Identifying the Characteristics and Immune Landscapes of Osteoarthritis. Biomolecules 2023, 13, 539. [Google Scholar] [CrossRef]
- Chen, Z.; Hua, Y. Identification of m7G-related hub biomarkers and m7G regulator expression pattern in immune landscape during the progression of osteoarthritis. Cytokine 2023, 170, 156313. [Google Scholar] [CrossRef]
- Huo, Z.; Hao, K.; Wang, X.; Fan, C.; Niu, Y.; Wang, F. Identification of m7G RNA Methylation Regulators in Osteoarthritis and Its Prognostic Markers. Fortune J. Rheumatol. 2024, 6, 46–56. [Google Scholar] [CrossRef]
- Ge, J.; Yu, Y.T. RNA pseudouridylation: New insights into an old modification. Trends Biochem. Sci. 2013, 38, 210–218. [Google Scholar] [CrossRef]
- Davis, D.R. Stabilization of RNA stacking by pseudouridine. Nucleic. Acids Res. 1995, 23, 5020–5026. [Google Scholar] [CrossRef] [PubMed]
- Chabronova, A.; van den Akker, G.; Housmans, B.A.C.; Caron, M.M.J.; Cremers, A.; Surtel, D.A.M.; Peffers, M.J.; van Rhijn, L.W.; Marchand, V.; Motorin, Y.; et al. Depletion of SNORA33 Abolishes ψ of 28S-U4966 and Affects the Ribosome Translational Apparatus. Int. J. Mol. Sci. 2023, 24, 12578. [Google Scholar] [CrossRef] [PubMed]
- Höfler, S.; Carlomagno, T. Structural and functional roles of 2′-O-ribose methylations and their enzymatic machinery across multiple classes of RNAs. Curr. Opin. Struct. Biol. 2020, 65, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Chabronova, A.; van den Akker, G.G.H.; Housmans, B.A.C.; Caron, M.M.J.; Cremers, A.; Surtel, D.A.M.; Wichapong, K.; Peffers, M.M.J.; van Rhijn, L.W.; Marchand, V.; et al. Ribosomal RNA-based epitranscriptomic regulation of chondrocyte translation and proteome in osteoarthritis. Osteoarthr. Cartil. 2023, 31, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Farrell, R.E. Chapter 5—Isolation of Polyadenylated RNA. In RNA Methodologies, 4th ed.; Farrell, R.E., Ed.; Academic Press: San Diego, CA, USA, 2010; pp. 121–137. [Google Scholar] [CrossRef]
- Winstanley-Zarach, P.; Rot, G.; Kuba, S.; Smagul, A.; Peffers, M.J.; Tew, S.R. Analysis of RNA Polyadenylation in Healthy and Osteoarthritic Human Articular Cartilage. Int. J. Mol. Sci. 2023, 24, 6611. [Google Scholar] [CrossRef]
- Winstanley-Zarach, P.; Kuba, S.; Smagul, A.; Peffers, M.; Rot, G.; Tew, S. Use of A 3′RNA Sequencing Approach to Determine Differential Transcript Expression and Alternative Polyadenylation Site Usage Between Age Matched, Healthy and Osteoarthritic Knee Cartilage. Osteoarthr. Cartil. 2022, 30, S351–S352. [Google Scholar] [CrossRef]
- Ashraf, S.; Radhi, M.; Gowler, P.; Burston, J.J.; Gandhi, R.D.; Thorn, G.J.; Piccinini, A.M.; Walsh, D.A.; Chapman, V.; De Moor, C.H. The polyadenylation inhibitor cordycepin reduces pain, inflammation and joint pathology in rodent models of osteoarthritis. Sci. Rep. 2019, 9, 4696. [Google Scholar] [CrossRef]
- Fahmi, H.; He, Y.; Zhang, M.; Martel-Pelletier, J.; Pelletier, J.-P.; Di Battista, J. Nimesulide reduces Interleukin-1β-induced Cyclooxygenase-2 gene expression in human synovial fibroblasts. Osteoarthr. Cartil. 2001, 9, 332–340. [Google Scholar] [CrossRef]
- Deyle, G.D.; Allen, C.S.; Allison, S.C.; Gill, N.W.; Hando, B.R.; Petersen, E.J.; Dusenberry, D.I.; Rhon, D.I. Physical therapy versus glucocorticoid injection for osteoarthritis of the knee. N. Engl. J. Med. 2020, 382, 1420–1429. [Google Scholar] [CrossRef]
- Kalajdzic, T.; Faour, W.H.; He, Q.W.; Fahmi, H.; Martel-Pelletier, J.; Pelletier, J.P.; Di Battista, J.A. Nimesulide, a preferential cyclooxygenase 2 inhibitor, suppresses peroxisome proliferator-activated receptor induction of cyclooxygenase 2 gene expression in human synovial fibroblasts: Evidence for receptor antagonism. Arthritis Rheum. 2002, 46, 494–506. [Google Scholar] [CrossRef]
- Di Battista, J.A.; Fahmi, H.; He, Y.; Zhang, M.; Martel-Pelletier, J.; Pelletier, J.P. Differential regulation of Interleukin-1 beta-induced Cyclooxygenase-2 gene expression by nimesulide in human synovial fibroblasts. Clin. Exp. Rheumatol. 2001, 19, S3–S5. [Google Scholar] [PubMed]
- Li, X.; Shen, L.; Deng, Z.; Huang, Z. New treatment for osteoarthritis: Gene therapy. Precis. Clin. Med. 2023, 6, pbad014. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Martel-Pelletier, J.; Pelletier, J.P.; Fahmi, H. Mesenchymal Stromal Cell Immunology for Efficient and Safe Treatment of Osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 567813. [Google Scholar] [CrossRef]
- Najar, M.; Fayyad-Kazan, H.; Faour, W.H.; Merimi, M.; Sokal, E.M.; Lombard, C.A.; Fahmi, H. Immunological modulation following bone marrow-derived mesenchymal stromal cells and Th17 lymphocyte co-cultures. Inflamm. Res. 2019, 68, 203–213. [Google Scholar] [CrossRef]
- Zelinka, A.; Roelofs, A.J.; Kandel, R.A.; De Bari, C. Cellular therapy and tissue engineering for cartilage repair. Osteoarthr. Cartil. 2022, 30, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yu, J.; Zhou, C.; Luo, Z.; Lu, X.; Zhu, L. Bushen Huoxue decotion-containing serum prevents chondrocyte pyroptosis in a m6A-dependent manner in facet joint osteoarthritis. Transpl. Immunol. 2024, 86, 102083. [Google Scholar] [CrossRef]
- Fan, D.; Liu, B.; Gu, X.; Zhang, Q.; Ye, Q.; Xi, X.; Xia, Y.; Wang, Q.; Wang, Z.; Wang, B.; et al. Potential Target Analysis of Triptolide Based on Transcriptome-Wide m6A Methylome in Rheumatoid Arthritis. Front. Pharmacol. 2022, 13, 843358. [Google Scholar] [CrossRef]
- Cui, L.; Shen, G.; Yu, Y.; Yan, Z.; Zeng, H.; Ye, X.; Xu, K.; Zhu, C.; Li, Y.; Shen, Z. Gubi decoction mitigates knee osteoarthritis via promoting chondrocyte autophagy through METTL3-mediated ATG7 m6A methylation. J. Cell. Mol. Med. 2024, 28, e70019. [Google Scholar] [CrossRef]
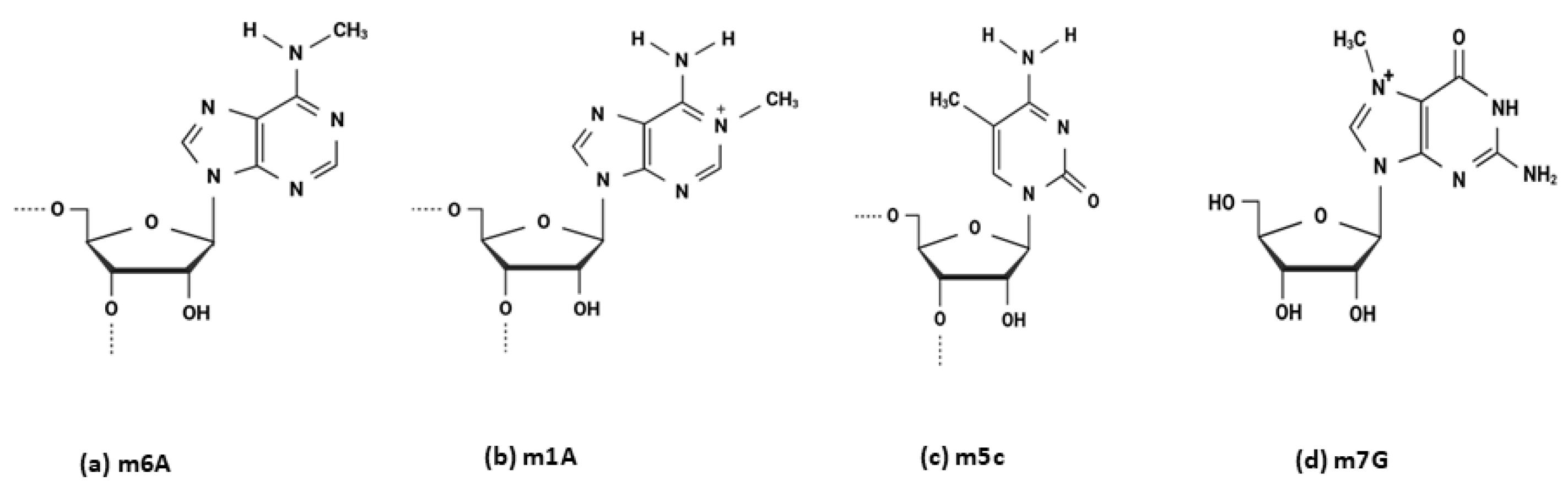
| Type of Modifications | Target of Modification | Models | Effect on OA Pathogenesis | References |
|---|---|---|---|---|
| m5C Methylation | mRNA (Sox9) | In vivo: Surgically induced full-thickness osteochondral defect model in rat | NSUN4 methylates the 3′ UTR of Sox9 mRNA, regulating chondrogenic differentiation | [125] |
| m7G Methylation | Mitochondrial transfer RNA (mt-tRF3b-LeuTAA) | In vitro: IL-1β-treated human OA primary chondrocytes In vivo: DMM-induced OA mouse model | METTL1-mediated m7G modification promotes mt-tRF3b-LeuTAA, SIRT3 SUMOylation, and cartilage degeneration | [127] |
| Pseudouridylation | rRNA (at sites 28S- 4966) | In vitro: IL-1β-treated human normal primary chondrocytes | OA microenvironment alters site-specific changes in rRNA pseudouridylation | [133] |
| 2′-O-ribose methylation (2′-O-Me) | rRNA (U14in 5.8S) | In vitro: IL-1β-stimulated human chondrocytes | Reduced 2′-O-Me of U14 in 5.8S rRNA impairs ribosome function and contributes to OA pathogenesis | [135] |
| Polyadenylation | mRNA (OSMR) | Human OA cartilage explants | Disruption of polyadenylation alters 3′UTR length, stabilizes OSMR mRNA, and promotes ECM degradation | [138] |
| mRNA (Runx2, MMPS-3/-13, ADAMTS-4) | In vivo: DMM-induced OA mouse model Human OA cartilage explants | Cordycepin inhibits polyadenylation, and reduces Runx2 and MMP-3/-13 levels | [139] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radbakhsh, S.; Najar, M.; Merimi, M.; Benderdour, M.; Fernandes, J.C.; Martel-Pelletier, J.; Pelletier, J.-P.; Fahmi, H. RNA Modifications in Osteoarthritis: Epitranscriptomic Insights into Pathogenesis and Therapeutic Targets. Int. J. Mol. Sci. 2025, 26, 4955. https://doi.org/10.3390/ijms26104955
Radbakhsh S, Najar M, Merimi M, Benderdour M, Fernandes JC, Martel-Pelletier J, Pelletier J-P, Fahmi H. RNA Modifications in Osteoarthritis: Epitranscriptomic Insights into Pathogenesis and Therapeutic Targets. International Journal of Molecular Sciences. 2025; 26(10):4955. https://doi.org/10.3390/ijms26104955
Chicago/Turabian StyleRadbakhsh, Shabnam, Mehdi Najar, Makram Merimi, Mohamed Benderdour, Julio C. Fernandes, Johanne Martel-Pelletier, Jean-Pierre Pelletier, and Hassan Fahmi. 2025. "RNA Modifications in Osteoarthritis: Epitranscriptomic Insights into Pathogenesis and Therapeutic Targets" International Journal of Molecular Sciences 26, no. 10: 4955. https://doi.org/10.3390/ijms26104955
APA StyleRadbakhsh, S., Najar, M., Merimi, M., Benderdour, M., Fernandes, J. C., Martel-Pelletier, J., Pelletier, J.-P., & Fahmi, H. (2025). RNA Modifications in Osteoarthritis: Epitranscriptomic Insights into Pathogenesis and Therapeutic Targets. International Journal of Molecular Sciences, 26(10), 4955. https://doi.org/10.3390/ijms26104955