

Controlled Exit from the G2/M Checkpoint in RPE-1 Cells Using RO3306: Enrichment of Phase-Specific Cell Populations for In-Depth Analyses of Mitotic Events


Abstract
1. Introduction
2. Results
2.1. Enrichment of Mitotic Cells Using an RO3306 Block-And-Release Method
2.2. Controlled Enrichment of Mitotic Populations
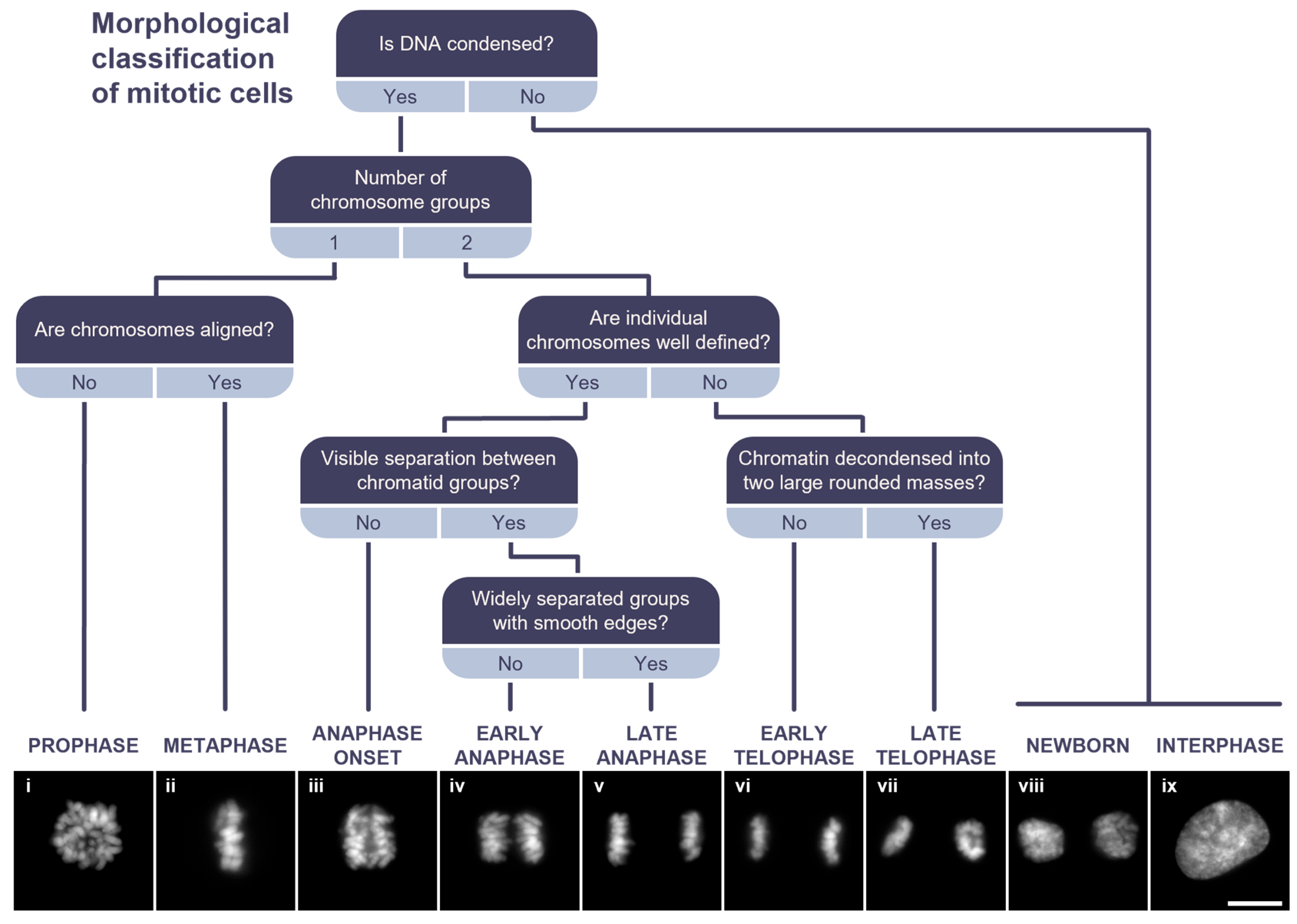
2.2.1. Morphological Classification of Mitotic Cells
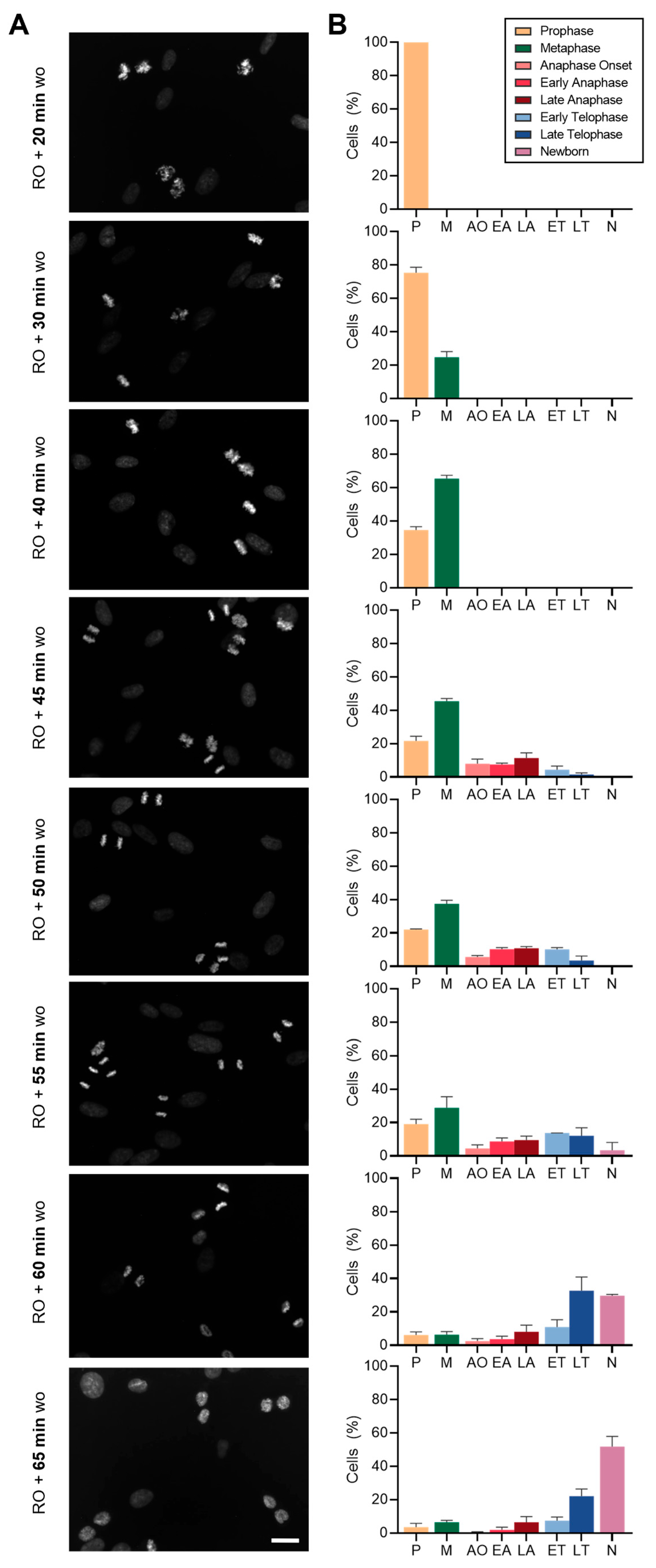
2.2.2. Mitosis on Demand: Precise Timing for the Enrichment of Specific Mitotic Sub-Populations
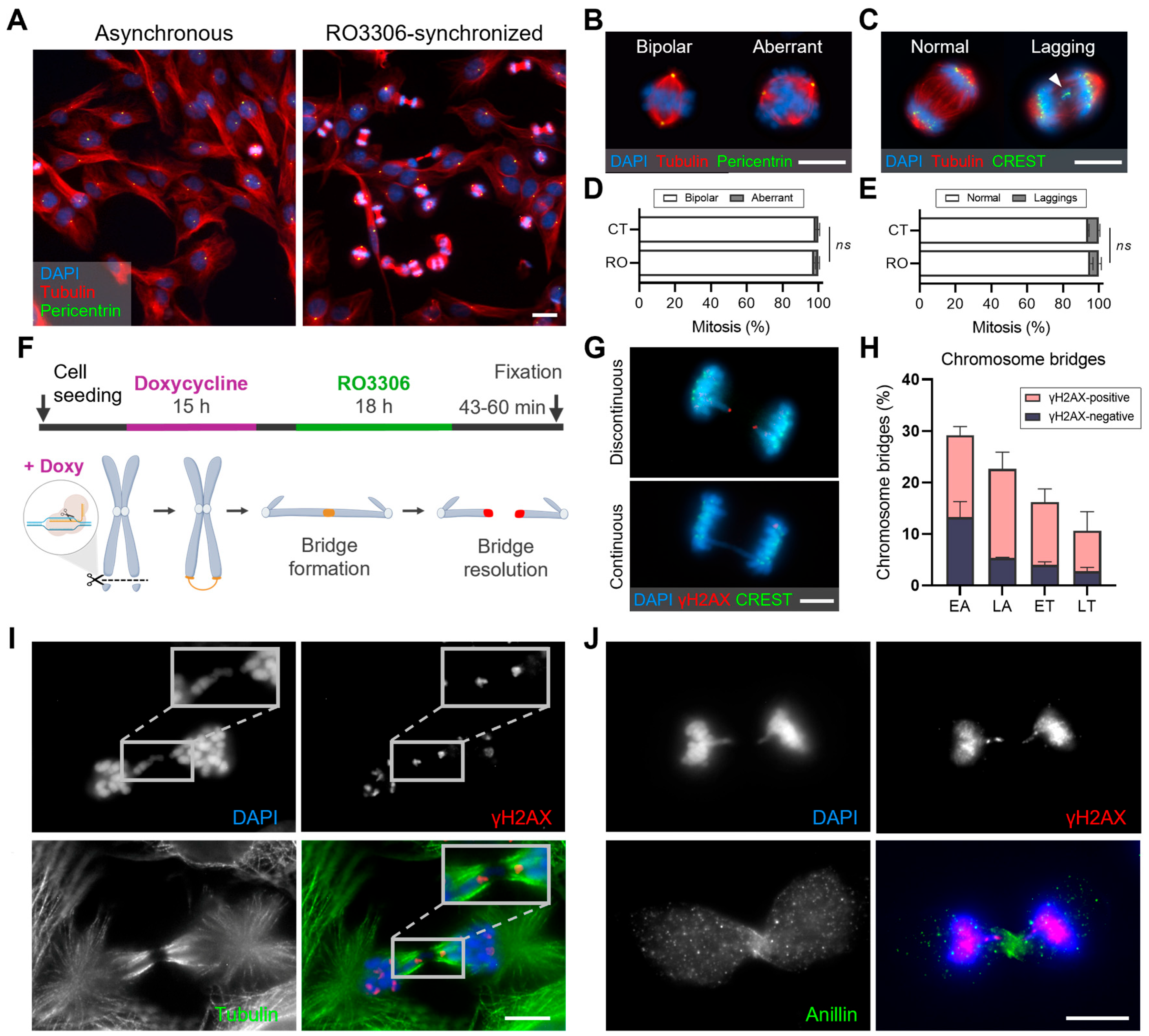
2.3. Applications of RO3306 Synchronization and Precise Washout Protocol
2.3.1. Enrichment of Metaphase Cells for Preparing Chromosome Spreads
2.3.2. Enrichment of Cells at Mitotic Exit for Chromosomal Segregation Studies
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Lentiviral Particle Production and Transduction
4.3. Cell Cycle Synchronization and Cell Treatments
4.4. Cell Cycle
4.5. Live/Dead Assay
4.6. Immunofluorescence
4.7. Fluorescence In Situ Hybridization
4.8. Live-Cell Imaging
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells 2021, 10, 3327. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, A.; Altmeyer, M. The Hammer and the Dance of Cell Cycle Control. Trends Biochem. Sci. 2021, 46, 301–314. [Google Scholar] [CrossRef]
- Bashir, T.; Pagano, M. Cdk1: The dominant sibling of Cdk2. Nat. Cell Biol. 2005, 7, 779–781. [Google Scholar] [CrossRef]
- Ligasová, A.; Koberna, K. Strengths and Weaknesses of Cell Synchronization Protocols Based on Inhibition of DNA Synthesis. Int. J. Mol. Sci. 2021, 22, 10759. [Google Scholar] [CrossRef]
- Davis, P.K.; Ho, A.; Dowdy, S.F. Biological Methods for Cell-Cycle Synchronization of Mammalian Cells. Biotechniques 2001, 30, 1322–1331. [Google Scholar] [CrossRef]
- Chen, G.; Deng, X. Cell Synchronization by Double Thymidine Block. Bio Protoc. 2018, 8, e2994. [Google Scholar] [CrossRef]
- Romsdahl, M.M. Synchronization of human cell lines with colcemid. Exp. Cell Res. 1968, 50, 463–467. [Google Scholar] [CrossRef]
- Zieve, G.W.; Turnbull, D.; Mullins, J.M.; McIntosh, J.R. Production of large numbers of mitotic mammalian cells by use of the reversible microtubule inhibitor Nocodazole. Exp. Cell Res. 1980, 126, 397–405. [Google Scholar] [CrossRef]
- Cavazza, T.; Malgaretti, P.; Vernos, I. The sequential activation of the mitotic microtubule assembly pathways favors bipolar spindle formation. Mol. Biol. Cell 2016, 27, 2935–2945. [Google Scholar] [CrossRef] [PubMed]
- Worrall, J.T.; Tamura, N.; Mazzagatti, A.; Shaikh, N.; van Lingen, T.; Bakker, B.; Spierings, D.C.J.; Vladimirou, E.; Foijer, F.; McClelland, S.E. Non-random Mis-segregation of Human Chromosomes. Cell Rep. 2018, 23, 3366–3380. [Google Scholar] [CrossRef]
- Tipton, A.R.; Gorbsky, G.J. More than two populations of microtubules comprise the dynamic mitotic spindle. J. Cell Sci. 2022, 135, jcs258745. [Google Scholar] [CrossRef]
- Daum, J.R.; Potapova, T.A.; Sivakumar, S.; Daniel, J.J.; Flynn, J.N.; Rankin, S.; Gorbsky, G.J. Cohesion Fatigue Induces Chromatid Separation in Cells Delayed at Metaphase. Curr. Biol. 2011, 21, 1018–1024. [Google Scholar] [CrossRef]
- Matsui, Y.; Nakayama, Y.; Okamoto, M.; Fukumoto, Y.; Yamaguchi, N. Enrichment of cell populations in metaphase, anaphase, and telophase by synchronization using nocodazole and blebbistatin: A novel method suitable for examining dynamic changes in proteins during mitotic progression. Eur. J. Cell Biol. 2012, 91, 413–419. [Google Scholar] [CrossRef]
- Wang, Z. Cell-Cycle Synchronization Methods and Protocols, 7th ed.; Humana Press: New York, NY, USA, 2022. [Google Scholar]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef]
- Flemming, W. Zellsubstanz, Kern und Zelltheilung; F. C. W. Vogel: Leipzig, Germany, 1882; Available online: https://scholar.google.com/scholar_lookup?title=Zellsubstanz,%20kern%20und%20zelltheilung&author=W.%20Flemming&publication_year=1882& (accessed on 18 May 2025).
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Lukow, D.A.; Sausville, E.L.; Suri, P.; Chunduri, N.K.; Wieland, A.; Leu, J.; Smith, J.C.; Girish, V.; Kumar, A.A.; Kendall, J.; et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 2021, 56, 2427–2439.e4. [Google Scholar] [CrossRef]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes. Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Umbreit, N.T.; Zhang, C.-Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, eaba0712. [Google Scholar] [CrossRef] [PubMed]
- Skoufias, D.A.; DeBonis, S.; Saoudi, Y.; Lebeau, L.; Crevel, I.; Cross, R.; Wade, R.H.; Hackney, D.; Kozielski, F. S-Trityl-L-cysteine Is a Reversible, Tight Binding Inhibitor of the Human Kinesin Eg5 That Specifically Blocks Mitotic Progression. J. Biol. Chem. 2006, 281, 17559–17569. [Google Scholar] [CrossRef] [PubMed]
- Gayek, A.S.; Ohi, R. CDK-1 Inhibition in G2 Stabilizes Kinetochore-Microtubules in the following Mitosis. PLoS ONE 2016, 11, e0157491. [Google Scholar] [CrossRef]
- Scott, S.J.; Suvarna, K.S.; D’Avino, P.P. Synchronization of human retinal pigment epithelial-1 cells in mitosis. J. Cell. Sci. 2020, 133, jcs247940. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Reddy, S.; Suzuki, A. Reversible and effective cell cycle synchronization method for studying stage-specific investigations. bioRxiv 2024. [Google Scholar] [CrossRef]
- Voets, E.; Marsman, J.; Demmers, J.; Beijersbergen, R.; Wolthuis, R. The lethal response to Cdk1 inhibition depends on sister chromatid alignment errors generated by KIF4 and isoform 1 of PRC1. Sci. Rep. 2015, 5, 14798. [Google Scholar] [CrossRef]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef]
- Vukušić, K.; Tolić, I.M. Anaphase B: Long-standing models meet new concepts. Semin. Cell Dev. Biol. 2021, 117, 127–139. [Google Scholar] [CrossRef]
- Anglada, T.; Rodriguez-Muñoz, M.; Pulido-Artola, N.; Genesca, A. Engineering Chromosome Bridges through CRISPR/Cas9: Deciphering the Impact of Intercentromeric Distance on Resolution Dynamics. FASEB J. 2025, 39, e70599. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Juan, G.; Bedner, E. Determining Cell Cycle Stages by Flow Cytometry. Curr. Protoc. Cell Biol. 1999, 1, 8.4.1–8.4.18. [Google Scholar] [CrossRef]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid Name | Targeting Sequence + PAM | Antibiotic Resistance | Vector Type | Reference |
|---|---|---|---|---|
| shp21/Rb | - | Blasticidin | Lentiviral | Pellman’s Laboratory |
| H2B-RFP | - | - | Lentiviral | Addgene #26001 |
| Tubulin-GFP | - | - | Lentiviral | Addgene #64060 |
| sgChr1 | TGACGTTGGACAGCCGCTGGAGG | Blasticidin | Lentiviral | Anglada et al., [31] |
| sgChr2 | ATATTAAGGGCTCCCCGTCGGGG | Blasticidin | Lentiviral | |
| sgChr3 | AGGGCTGGTCACCATTCAAGAGG | Blasticidin | Lentiviral | |
| psPAX2 | - | - | Packaging | Addgene #12260 |
| pMD2.G | - | - | Envelope | Addgene #12259 |
| Antibody | Host | Reference | Working Dilution |
|---|---|---|---|
| PRIMARY ANTIBODIES | |||
| Anti- α-Tubulin | Mouse | Sigma-Aldrich (St. Louis, MO, USA), T5168 | 1:1000 |
| Anti-Pericentrin | Rabbit | Abcam (Cambridge, UK), ab4448 | 1:1500 |
| Anti-CREST | Human | Antibodies Incorporated (Davis, CA, USA),15-234 | 1:50 |
| Anti- γH2AX (Ser139) | Mouse | Millipore (Burlington, MA, USA), 05-636, clone JBW301 | 1:1000 |
| Anti- γH2AX (Ser139) | Rabbit | Abcam, ab81299, clone EP854(2)Y | 1:500 |
| Anti-Anillin | Rabbit | Abcam, ab99352 | 1:1000 |
| SECONDARY ANTIBODIES | |||
| Anti-Mouse Cyanine Cy™3 | Goat | Jackson ImmunoResearch Inc. (West Grove, PA, USA), 115-165-146 | 1:500 |
| Anti-Mouse Alexa Fluor®488 | Goat | Jackson ImmunoResearch Inc., 115-545-205 | 1:800 |
| Anti-Rabbit Alexa Fluor®488 | Goat | Thermo Fisher Scientific (Waltham, MA, USA), A-11034 | 1:500 |
| Anti-Rabbit Alexa Fluor®594 | Goat | Thermo Fisher Scientific A-11037 | 1:500 |
| Anti-Human FITC | Goat | Antibodies Incorporated, 52-241-0100 | 1:100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anglada, T.; Pulido-Artola, N.; Rodriguez-Muñoz, M.; Genesca, A. Controlled Exit from the G2/M Checkpoint in RPE-1 Cells Using RO3306: Enrichment of Phase-Specific Cell Populations for In-Depth Analyses of Mitotic Events. Int. J. Mol. Sci. 2025, 26, 4951. https://doi.org/10.3390/ijms26104951
Anglada T, Pulido-Artola N, Rodriguez-Muñoz M, Genesca A. Controlled Exit from the G2/M Checkpoint in RPE-1 Cells Using RO3306: Enrichment of Phase-Specific Cell Populations for In-Depth Analyses of Mitotic Events. International Journal of Molecular Sciences. 2025; 26(10):4951. https://doi.org/10.3390/ijms26104951
Chicago/Turabian StyleAnglada, Teresa, Núria Pulido-Artola, Marina Rodriguez-Muñoz, and Anna Genesca. 2025. "Controlled Exit from the G2/M Checkpoint in RPE-1 Cells Using RO3306: Enrichment of Phase-Specific Cell Populations for In-Depth Analyses of Mitotic Events" International Journal of Molecular Sciences 26, no. 10: 4951. https://doi.org/10.3390/ijms26104951
APA StyleAnglada, T., Pulido-Artola, N., Rodriguez-Muñoz, M., & Genesca, A. (2025). Controlled Exit from the G2/M Checkpoint in RPE-1 Cells Using RO3306: Enrichment of Phase-Specific Cell Populations for In-Depth Analyses of Mitotic Events. International Journal of Molecular Sciences, 26(10), 4951. https://doi.org/10.3390/ijms26104951