
Genetic Structure, Selective Signatures, and Single Nucleotide Polymorphism Fingerprints of Blue Tilapia (Oreochromis aureus), Nile Tilapia Oreochromis niloticus), and Red Tilapia (Oreochromis spp.), as Determined by Whole-Genome Resequencing

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Whole-Genome Resequencing Data Statistics
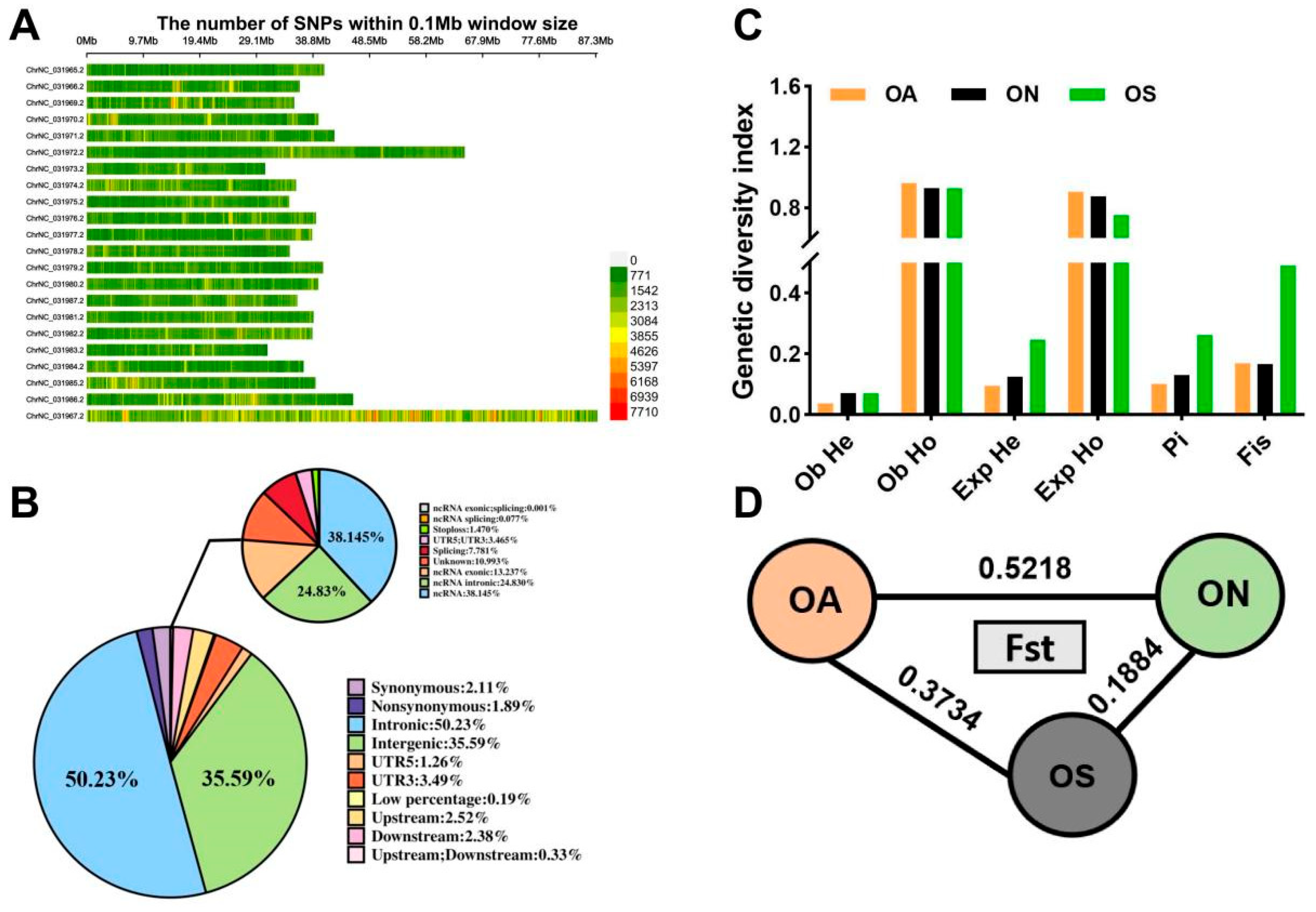
2.2. Whole-Genome SNP Statistics and Genetic Diversity Analysis
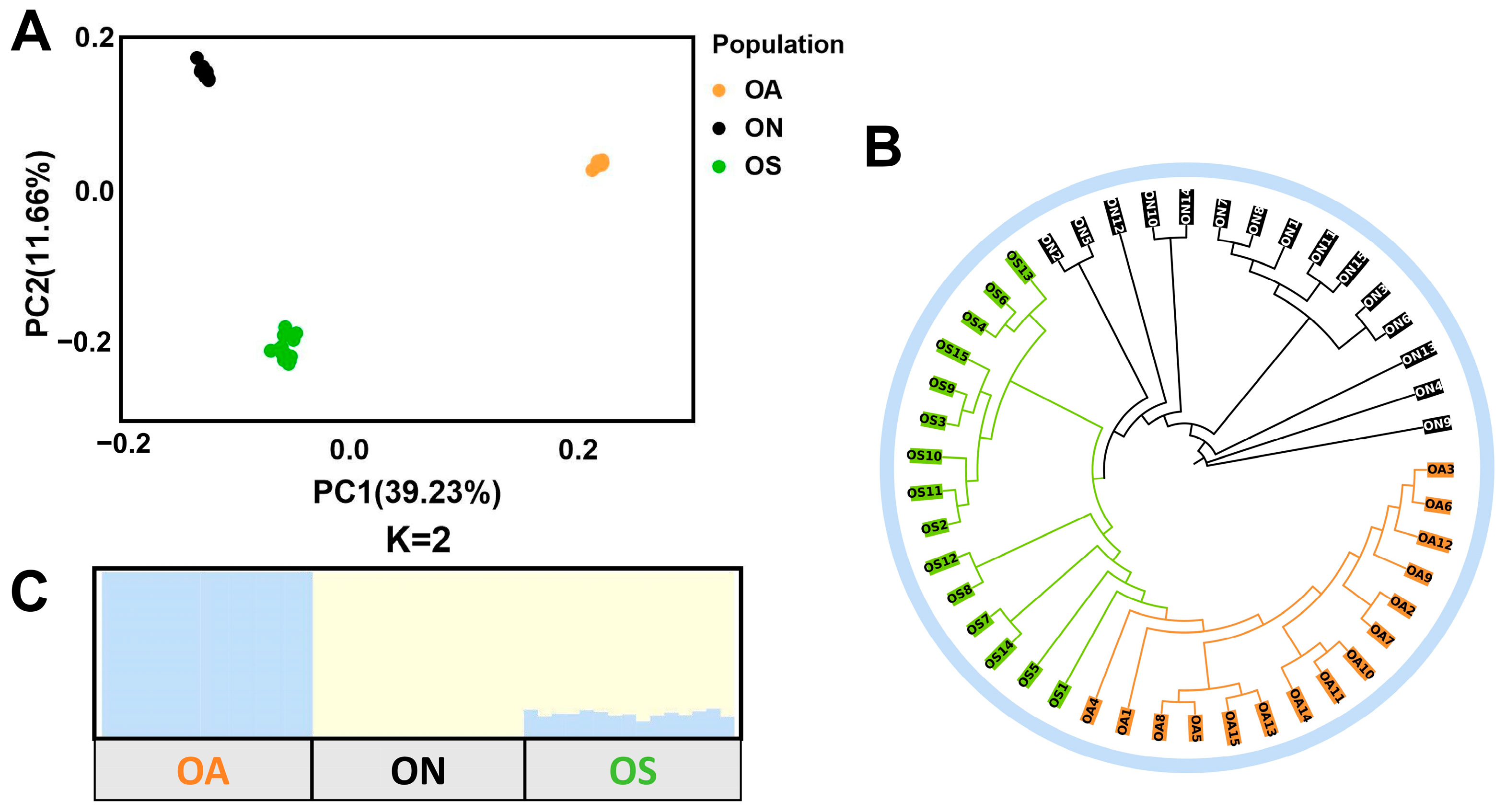
2.3. Population Genetic Structure Analysis
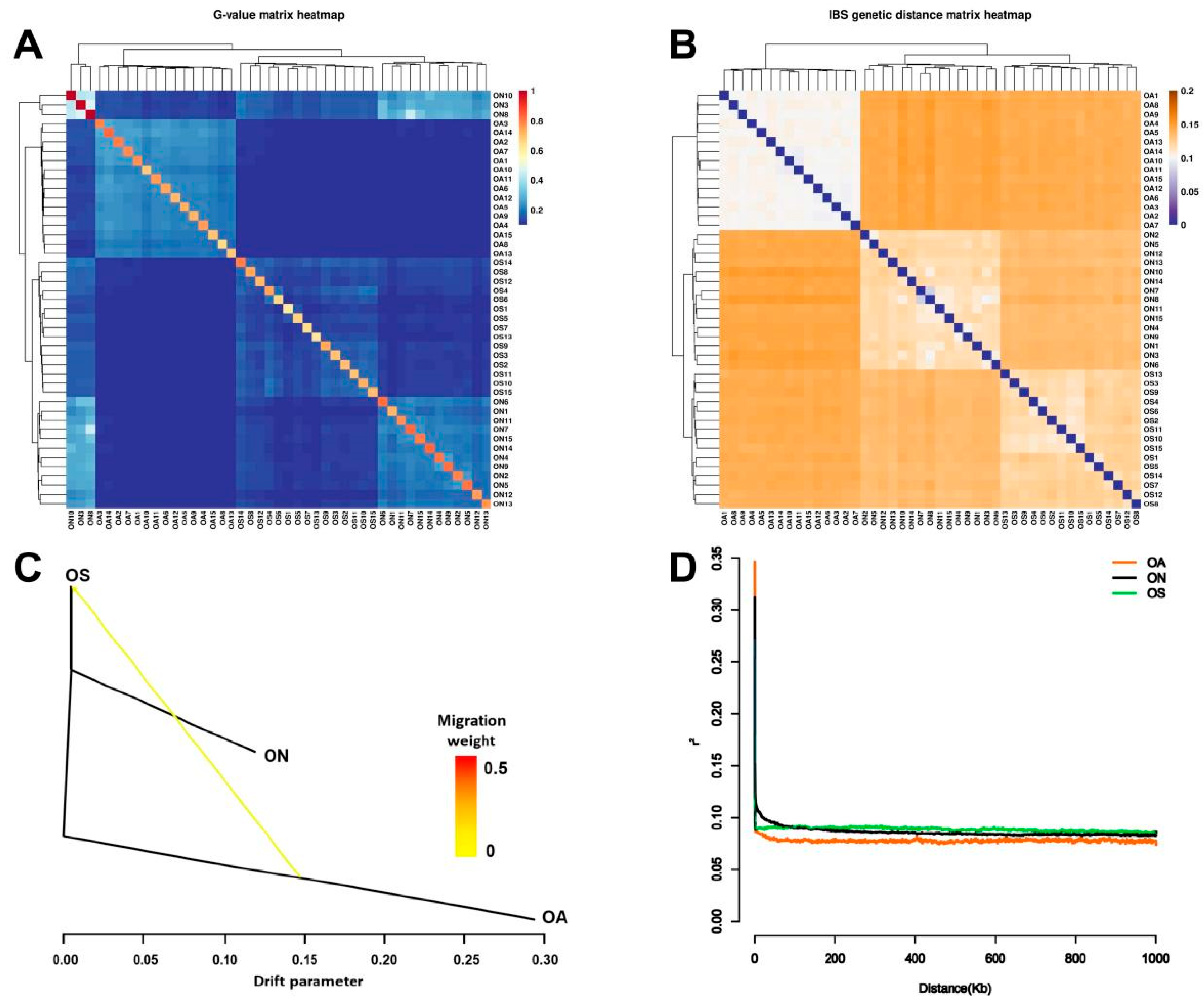
2.4. Population Kinship, Gene Flow, and Linkage Disequilibrium Analysis
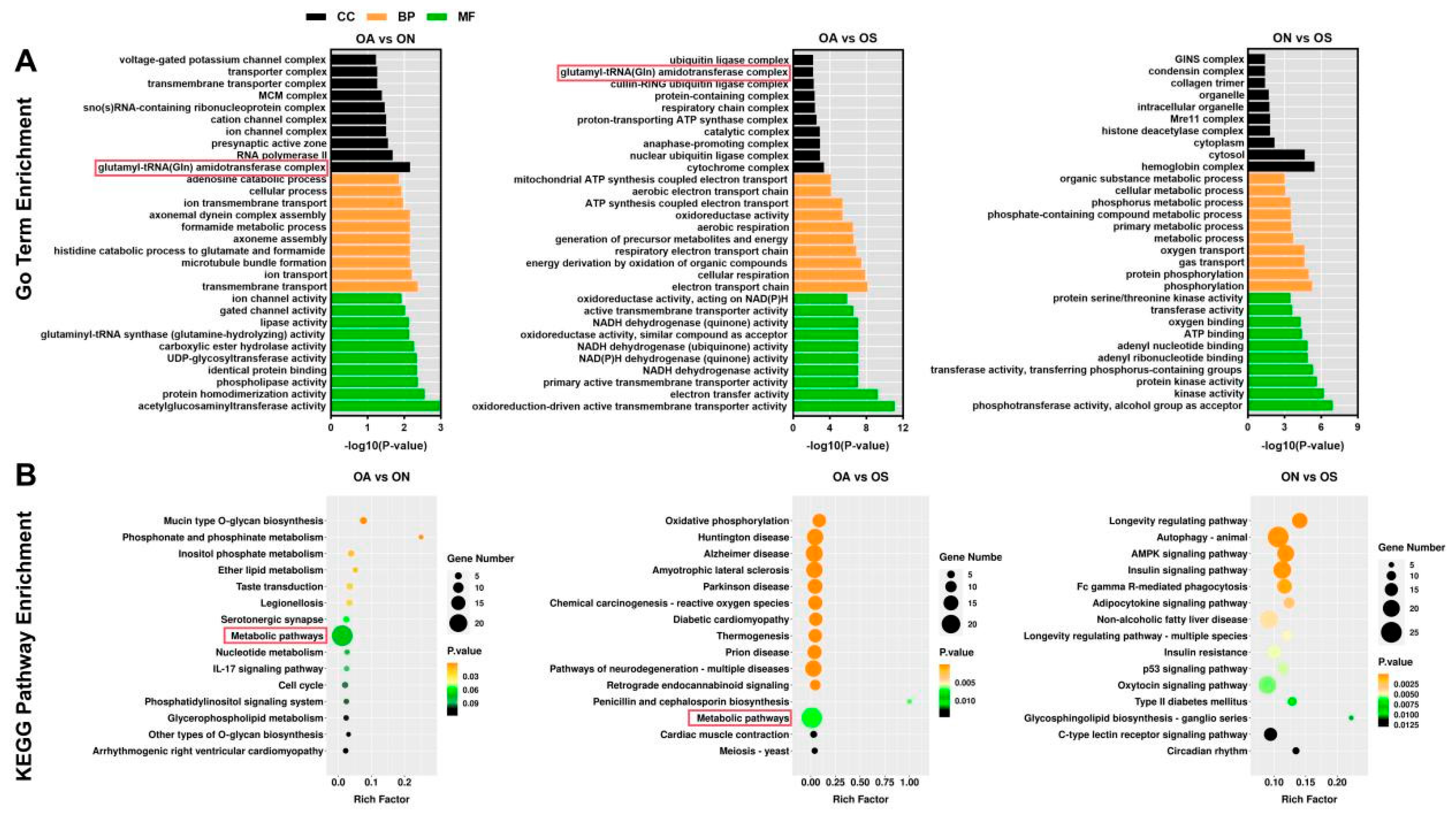
2.5. Identification of Selected Regions in Genomes of the Three Populations and Candidate Gene Analysis
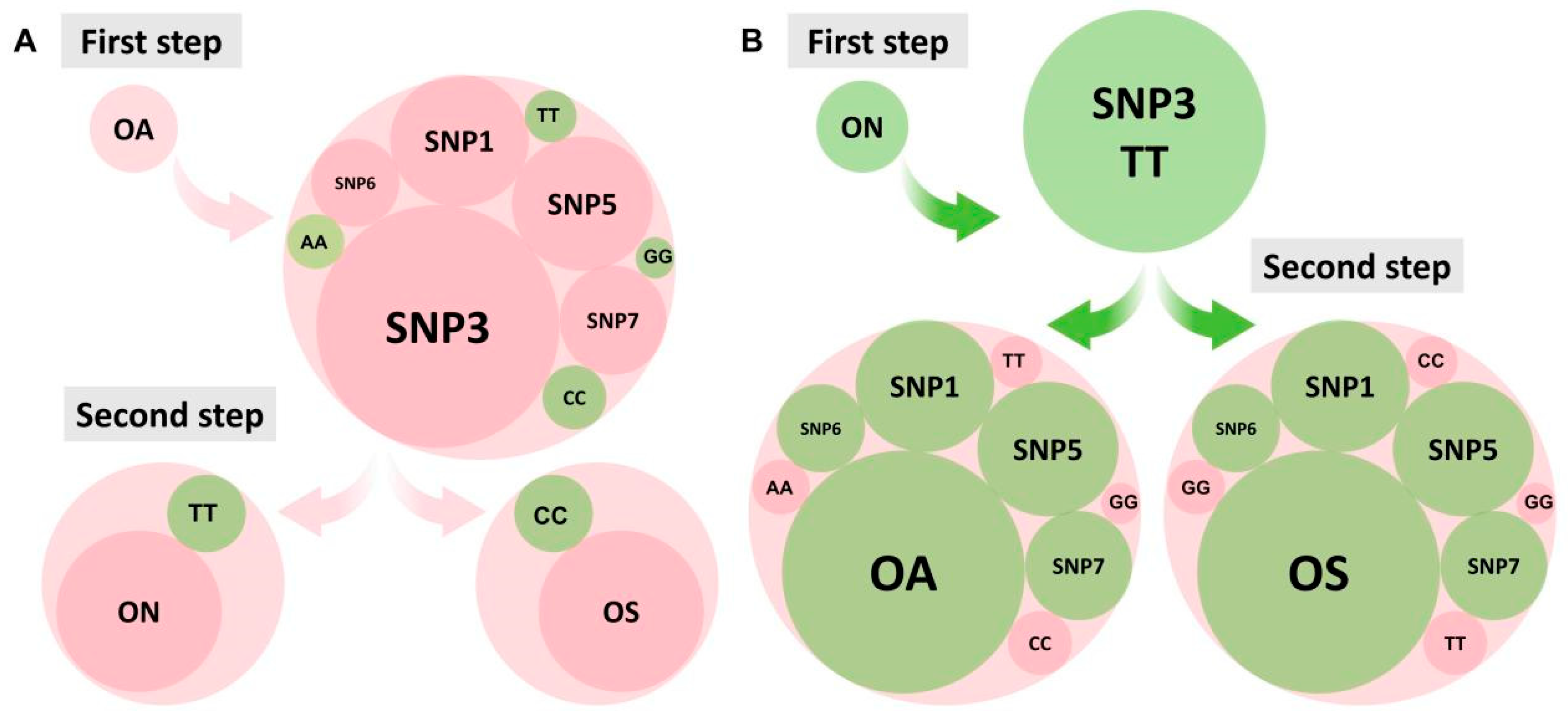
2.6. SNP Fingerprint Construction and Validation
2.7. Combinations of SNP Markers for Population Identification
3. Discussion
3.1. Genetic Diversity and Genetic Structure
3.2. Genes Selected in the Three Populations and Their Pathways and Mechanisms
3.3. SNP Fingerprint Construction
4. Materials and Methods
4.1. Sample Collection
4.2. DNA Extraction, Library Construction, and High-Throughput Sequencing
4.3. Whole Genome SNP Screening
4.4. Population Genetic Diversity Analysis
4.5. Population Genetic Structure and Population Kinship Analysis
4.6. Gene Flow and Linkage Disequilibrium Analyses
4.7. Identification of Selected Genomic Regions and Gene Enrichment Analysis
4.8. Core SNP Loci Screening and Fingerprint Construction
4.9. SNP Fingerprint Validation and Determination of SNP Marker Combinations for Population Identification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Renuhadevi, M.; Jeevagan, I.J.M.A.; Ahilan, B.; Rajagopalsamy, C.B.T.; Prabu, E. Tilapia—An Excellent Candidate Species for World Aquaculture: A Review. Annu. Res. Rev. Biol. 2019, 31, 1–14. [Google Scholar] [CrossRef]
- Zhu, J.; Zou, Z.; Li, D.; Xiao, W.; Yu, J.; Chen, B.; Yang, H. Comparative transcriptomes reveal different tolerance mechanisms to Streptococcus agalactiae in hybrid tilapia, nile tilapia, and blue tilapia. Fish Shellfish. Immunol. 2023, 142, 109121. [Google Scholar] [CrossRef] [PubMed]
- Kariuki, J.; Tibihika, P.D.; Curto, M.; Alemayehu, E.; Winkler, G.; Meimberg, H. Application of microsatellite genotyping by amplicon sequencing for delimitation of African tilapiine species relevant for aquaculture. Aquaculture 2021, 537, 736501. [Google Scholar] [CrossRef]
- Kwikiriza, G.; Vijayan, T.; Tibihika, P.D.; Curto, M.; Winkler, G.; Nattabi, J.K.; Kariuki, J.; Meimberg, H. Introgressive hybridization levels of Tilapiine species in Lake Victoria basin, Kenya inferred from microsatellite and mitochondrial DNA genotyping based on next-generation sequencing. Conserv. Genet. 2023, 25, 305–318. [Google Scholar] [CrossRef]
- Liang, Y.; Tian, P.; Lu, Y.; Qin, Q.; Wang, Z.; Xiong, G.; Wang, X.; Hu, Y. Establishment and population genetic analysis of SNP fingerprinting of Chinese soft-shelled turtle (Pelodiscus sinensis). Aquac. Rep. 2024, 38, 102340. [Google Scholar] [CrossRef]
- Oget, C.; Servin, B.; Palhière, I. Genetic diversity analysis of French goat populations reveals selective sweeps involved in their differentiation. Anim. Genet. 2018, 50, 54–63. [Google Scholar] [CrossRef]
- Agresti, J.J.; Seki, S.; Cnaani, A.; Poompuang, S.; Hallerman, E.M.; Umiel, N.; Hulata, G.; Gall, G.A.E.; May, B. Breeding new strains of tilapia: Development of an artificial center of origin and linkage map based on AFLP and microsatellite loci. Aquaculture 2000, 185, 43–56. [Google Scholar] [CrossRef]
- Romana-Eguia, M.R.R.; Ikeda, M.; Basiao, Z.U.; Taniguchi, N. Genetic diversity in farmed Asian Nile and red hybrid tilapia stocks evaluated from microsatellite and mitochondrial DNA analysis. Aquaculture 2004, 236, 131–150. [Google Scholar] [CrossRef]
- Hua, J.; Zhong, C.; Chen, W.; Fu, J.; Wang, J.; Wang, Q.; Zhu, G.; Li, Y.; Tao, Y.; Zhang, M.; et al. Single nucleotide polymorphism SNP19140160 A > C is a potential breeding locus for fast-growth largemouth bass (Micropterus salmoides). BMC Genom. 2024, 25, 64. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Chu, P.; Shang, R.; Yin, S.; Wang, T. Construction of a high-density genetic linkage map and QTL mapping of growth and cold tolerance traits in Takifugu fasciatus. BMC Genom. 2023, 24, 645. [Google Scholar] [CrossRef]
- Guo, F.; Ye, Y.; Zhu, K.; Lin, S.; Wang, Y.; Dong, Z.; Yao, R.; Li, H.; Wang, W.; Liao, Z.; et al. Genetic Diversity, Population Structure, and Environmental Adaptation Signatures of Chinese Coastal Hard-Shell Mussel Mytilus coruscus Revealed by Whole-Genome Sequencing. Int. J. Mol. Sci. 2023, 24, 13641. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Jian, J.; Niu, B.; Zhang, X.; Yu, J.; Xu, X. Germplasm Resources Evaluation of Cultured Largemouth Bass (Micropterus salmoides) in China Based on Whole Genome Resequencing. Genes 2024, 15, 1307. [Google Scholar] [CrossRef]
- Cui, R.; Huang, J.; Wang, S.; Zhang, X. Whole-genome resequencing reveals genetic diversity and selection signatures in five populations of Mandarin Fish (Siniperca chuatsi). Aquac. Rep. 2024, 39, 102406. [Google Scholar] [CrossRef]
- Li, Z.; Fang, M.; Tang, X.; Zhang, D.; Wang, Z. Disentangling genetic variation for endurance and resistance to visceral white-nodules disease in large yellow croaker (Larimichthys crocea) using genome information. Aquaculture 2023, 564, 739045. [Google Scholar] [CrossRef]
- Hong Xia, J.; Bai, Z.; Meng, Z.; Zhang, Y.; Wang, L.; Liu, F.; Jing, W.; Yi Wan, Z.; Li, J.; Lin, H.; et al. Signatures of selection in tilapia revealed by whole genome resequencing. Sci. Rep. 2015, 5, srep14168. [Google Scholar] [CrossRef]
- Cádiz, M.I.; López, M.E.; Díaz-Domínguez, D.; Cáceres, G.; Yoshida, G.M.; Gomez-Uchida, D.; Yáñez, J.M. Whole genome re-sequencing reveals recent signatures of selection in three strains of farmed Nile tilapia (Oreochromis niloticus). Sci. Rep. 2020, 10, 11514. [Google Scholar] [CrossRef]
- Ciezarek, A.; Ford, A.G.P.; Etherington, G.J.; Kasozi, N.; Malinsky, M.; Mehta, T.K.; Penso-Dolfin, L.; Ngatunga, B.P.; Shechonge, A.; Tamatamah, R.; et al. Whole genome resequencing data enables a targeted SNP panel for conservation and aquaculture of Oreochromis cichlid fishes. Aquaculture 2022, 548, 737637. [Google Scholar] [CrossRef]
- Yáñez, J.M.; Yoshida, G.; Barria, A.; Palma-Véjares, R.; Travisany, D.; Díaz, D.; Cáceres, G.; Cádiz, M.I.; López, M.E.; Lhorente, J.P.; et al. High-Throughput Single Nucleotide Polymorphism (SNP) Discovery and Validation Through Whole-Genome Resequencing in Nile Tilapia (Oreochromis niloticus). Mar. Biotechnol. 2020, 22, 109–117. [Google Scholar] [CrossRef]
- Sun, C.-F.; Zhang, X.-H.; Dong, J.-J.; You, X.-X.; Tian, Y.-Y.; Gao, F.-Y.; Zhang, H.-T.; Shi, Q.; Ye, X. Whole-genome resequencing reveals recent signatures of selection in five populations of largemouth bass (Micropterus salmoides). Zool. Res. 2023, 44, 78–89. [Google Scholar] [CrossRef]
- Zhang, D.-Y.; Liu, X.-M.; Huang, W.-J.; Wang, Y.; Anwarullah, K.; Luo, L.-F.; Gao, Z.-X. Whole-genome resequencing reveals genetic diversity and signatures of selection in mono-female grass carp (Ctenopharyngodon idella). Aquaculture 2023, 575, 739816. [Google Scholar] [CrossRef]
- Sayouh, M.; Ali, M.; Li, Y.; Tao, Y.-F.; Lu, S.-Q.; Qiang, J. Differences in Growth Performance and Meat Quality between Male and Female Juvenile Nile Tilapia (Oreochromis niloticus) during Separate Rearing. Animals 2024, 14, 2954. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, L.; Fan, Z.; Lu, B.; Chen, J.; Tan, D.; Jiang, D.; Tao, W.; Wang, D. Screening and characterization of sex-linked DNA markers and marker-assisted selection in blue tilapia (Oreochromis aureus). Aquaculture 2021, 530, 735934. [Google Scholar] [CrossRef]
- Hassanien, H.A.; Gilbey, J. Genetic diversity and differentiation of Nile tilapia (Oreochromis niloticus) revealed by DNA microsatellites. Aquac. Res. 2005, 36, 1450–1457. [Google Scholar] [CrossRef]
- Nayfa, M.G.; Jones, D.B.; Benzie, J.A.H.; Jerry, D.R.; Zenger, K.R. Comparing Genomic Signatures of Selection Between the Abbassa Strain and Eight Wild Populations of Nile Tilapia (Oreochromis niloticus) in Egypt. Front. Genet. 2020, 11, 567969. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Fang, W.; Li, J.; Cai, W.; Lu, J. Full-length transcriptome reveals alternative splicing regulation pattern of skin color variant in red tilapia (Oreochromis spp.). Aquaculture 2025, 598, 741963. [Google Scholar] [CrossRef]
- Jiang, B.; Fu, J.; Dong, Z.; Fang, M.; Zhu, W.; Wang, L. Maternal ancestry analyses of red tilapia strains based on D-loop sequences of seven tilapia populations. PeerJ 2019, 7, e7007. [Google Scholar] [CrossRef]
- Zhu, W.B.; Yang, H.; Yuan, X.H.; Dong, Z.J.; Fu, J.J.; Wang, L.M.; Su, S.Y.; Liu, N.; Song, F.B.; Chen, X.T. High genetic diversity and differentiation in three red tilapia stocks revealed by microsatellite DNA marker analysis. Aquac. Int. 2017, 25, 1997–2006. [Google Scholar] [CrossRef]
- Tang, H.; Mao, S.; Xu, X.; Li, J.; Shen, Y. Genetic diversity analysis of different geographic populations of black carp (Mylopharyngodon piceus) based on whole genome SNP markers. Aquaculture 2024, 582, 740542. [Google Scholar] [CrossRef]
- Du, J.; Li, S.; Shao, J.; Song, H.; Jiang, P.; Lei, C.; Bai, J.; Han, L. Genetic diversity analysis and development of molecular markers for the identification of largemouth bass (Micropterus salmoides L.) based on whole-genome re-sequencing. Front. Genet. 2022, 13, 936610. [Google Scholar] [CrossRef]
- Wang, S.-L.; Luo, L.-F.; Yu, Y.; Fu, Y.-Y.; Gao, Z.-X. Whole-genome re-sequencing reveals genetic structure and selection signals of different populations in Megalobrama amblycephala. Aquaculture 2025, 595, 741548. [Google Scholar] [CrossRef]
- Yu, C.; Tang, H.; Jiang, Y.; Lu, H.; Chen, Q.; Gui, L.; Qiu, J.; Xu, X.; Li, J.; Shen, Y. Growth performance and selection signatures revealed by whole-genome resequencing in genetically selected grass carp (Ctenopharyngodon idella). Aquaculture 2024, 587, 740885. [Google Scholar] [CrossRef]
- Shen, M.; Wang, J.; Chen, R.; Lv, T.; Chen, J.; Lu, X.; Liu, G.; Xu, X.; Liu, Z.; Deng, Y.; et al. Heterosis, genetic diversity and fertility of reciprocal hybrids between Argopecten irradians concentricus and A. purpuratus. Aquaculture 2024, 586, 740765. [Google Scholar] [CrossRef]
- Pujolar, J.M.; Maes, G.E.; Vancoillie, C.; Volckaert, F.A.M. Growth Rate Correlates To Individual Heterozygosity in the European Eel, Anguilla anguilla L. Evolution 2005, 59, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Heber, S.O.L.; Briskie, J.V. Population Bottlenecks and Increased Hatching Failure in Endangered Birds. Conserv. Biol. 2010, 24, 1674–1678. [Google Scholar] [CrossRef]
- Luo, X.; Li, J.; Xiao, C.; Sun, L.; Xiang, W.; Chen, N.; Lei, C.; Lei, H.; Long, Y.; Long, T.; et al. Whole-Genome Resequencing of Xiangxi Cattle Identifies Genomic Diversity and Selection Signatures. Front. Genet. 2022, 13, 816379. [Google Scholar] [CrossRef]
- Qu, J.; Kachman, S.D.; Garrick, D.; Fernando, R.L.; Cheng, H. Exact Distribution of Linkage Disequilibrium in the Presence of Mutation, Selection, or Minor Allele Frequency Filtering. Front. Genet. 2020, 11, 362. [Google Scholar] [CrossRef]
- Wilson, J.F.; Goldstein, D.B. Consistent Long-Range Linkage Disequilibrium Generated by Admixture in a Bantu-Semitic Hybrid Population. Am. J. Hum. Genet. 2000, 67, 926–935. [Google Scholar] [CrossRef]
- Guo, C.; Li, Y.; Xie, J.; Han, L.; Wang, Y.; Zhang, X.; Wu, Y.; Song, J.; Chang, Y.; Ding, J. Revealing Selection in Breeding and Genetic Characteristics of Economically Important Traits of New Species of Apostichopus Japonicas Based on Genome Resequencing and GWAS Analysis. Front. Mar. Sci. 2022, 9, 948882. [Google Scholar] [CrossRef]
- Baumung, R.; Simianer, H.; Hoffmann, I. Genetic diversity studies in farm animals—A survey. J. Anim. Breed. Genet. 2004, 121, 361–373. [Google Scholar] [CrossRef]
- Lan, D.; Xiong, X.; Mipam, T.-D.; Fu, C.; Li, Q.; Ai, Y.; Hou, D.; Chai, Z.; Zhong, J.; Li, J. Genetic Diversity, Molecular Phylogeny, and Selection Evidence of Jinchuan Yak Revealed by Whole-Genome Resequencing. G3 2018, 8, 945–952. [Google Scholar] [CrossRef]
- Mao, J.; Tian, Y.; Liu, Q.; Li, D.; Ge, X.; Wang, X.; Hao, Z. Revealing genetic diversity, population structure, and selection signatures of the Pacific oyster in Dalian by whole-genome resequencing. Front. Ecol. Evol. 2024, 11. [Google Scholar] [CrossRef]
- Friederich, M.W.; Timal, S.; Powell, C.A.; Dallabona, C.; Kurolap, A.; Palacios-Zambrano, S.; Bratkovic, D.; Derks, T.G.J.; Bick, D.; Bouman, K.; et al. Pathogenic variants in glutamyl-tRNAGln amidotransferase subunits cause a lethal mitochondrial cardiomyopathy disorder. Nat. Commun. 2018, 9, 4065. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, M.; Shen, Z.-G. Molecular biological, physiological, cytological, and epigenetic mechanisms of environmental sex differentiation in teleosts: A systematic review. Ecotoxicol. Environ. Saf. 2023, 267, 115654. [Google Scholar] [CrossRef]
- Peng, K.; Song, L.; Qiao, X.; Lin, Y. Identification and expression analysis of a key LncRNA in the gonads of female Nile tilapia (Oreochromis niloticus). J. Southwest China Norm. Univ. 2019, 44, 40–45. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Wu, B.; Song, Z.; He, S. Transcriptome analysis of the plateau fish (Triplophysa dalaica): Implications for adaptation to hypoxia in fishes. Gene 2015, 565, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Rajanbabu, V.; Pan, C.-Y.; Lee, S.-C.; Lin, W.-J.; Lin, C.-C.; Li, C.-L.; Chen, J.-Y. Tilapia Hepcidin 2-3 Peptide Modulates Lipopolysaccharide-induced Cytokines and Inhibits Tumor Necrosis Factor-α through Cyclooxygenase-2 and Phosphodiesterase 4D. J. Biol. Chem. 2010, 285, 30577–30586. [Google Scholar] [CrossRef]
- Yu, H.; Mo, H.; Gao, J.; Yao, M.; Du, Y.; Liu, K.; Zhang, Q.; Yu, J.; Li, Y.; Wang, L. Fibroblast growth factor 1 (FGF1) improves glucose homeostasis, modulates gut microbial composition, and reduces inflammatory responses in rainbow trout (Oncorhynchus mykiss) fed a high-fat diet. Int. J. Biol. Macromol. 2024, 281, 136226. [Google Scholar] [CrossRef]
- Takahashi, T.; Ogiwara, K. cAMP signaling in ovarian physiology in teleosts: A review. Cell. Signal. 2023, 101, 110499. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.L.C.; Richard, F.J.; Kuo, W.-P.; D’Ercole, A.J.; Conti, M. Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc. Natl. Acad. Sci. USA 1999, 96, 11998–12003. [Google Scholar] [CrossRef]
- Cao, L.; Zhou, J.; Gou, P.; Hu, Y.; Wang, Z.; Hu, Z.; Yu, Y.; Qiu, X.; Liu, Y.; Wang, X.; et al. Identification of growth-associated miRNAs, circRNAs, and their corresponding regulatory networks in fast- and slow- growing families of Takifugu rubripes. Aquaculture 2024, 592, 741206. [Google Scholar] [CrossRef]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Müller, M.; Green, A.R.; Adams, P.D.; et al. Notch Signaling Mediates Secondary Senescence. Cell Rep. 2019, 27, 997–1007.e1005. [Google Scholar] [CrossRef] [PubMed]
- Rotchell, J.M.; Lee, J.-S.; Chipman, J.K.; Ostrander, G.K. Structure, expression and activation of fish ras genes. Aquat. Toxicol. 2001, 55, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Futami, K.; Aoyama, K.; Fukuda, K.; Maita, M.; Katagiri, T. Increased expression of hras induces early, but not full, senescence in the immortal fish cell line, EPC. Gene 2021, 765, 145116. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NF-κB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 2017, 47, 450–465.e455. [Google Scholar] [CrossRef]
- Liu, S.; Yan, L.; Zhang, Y.; Junaid, M.; Wang, J. Toxicological effects of polystyrene nanoplastics and perfluorooctanoic acid to Gambusia affinis. Fish Shellfish Immunol. 2022, 127, 1100–1112. [Google Scholar] [CrossRef]
- Luo, K.; Tian, X.; Wang, B.; Wei, C.; Wang, L.; Zhang, S.; Liu, Y.; Li, T.; Dong, S. Evaluation of paraprobiotic applicability of Clostridium butyricum CBG01 in improving the growth performance, immune responses and disease resistance in Pacific white shrimp, Penaeus vannamei. Aquaculture 2021, 544, 737041. [Google Scholar] [CrossRef]
- Zhuang, Y.; Huang, H.; Liu, X.-L.; Wang, N.-A.; Zhong, G.-F. Effect of bovine lactoferricin on the growth performance, digestive capacity, immune responses and disease resistance in Pacific white shrimp, Penaeus vannamei. Fish Shellfish Immunol. 2022, 123, 282–289. [Google Scholar] [CrossRef]
- Ji, S.; Sun, J.; Bian, C.; Huang, X.; Chang, Z.; Yang, M.; Lu, R.-H.; Ji, H. cAMP-dependent protein kinase A in grass carp Ctenopharyngodon idella: Molecular characterization, gene structure, tissue distribution and mRNA expression in endoplasmic reticulum stress-induced adipocyte lipolysis. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2020, 250, 110479. [Google Scholar] [CrossRef]
- Li, D.; Yang, Y.; Wang, T.; Zhang, W.; Hua, S.; Ruan, Q.; Wang, X.; Zhu, C.; Meng, Z. Liver Transcriptome Shows Differences between Acute Hypoxia-Tolerant and Intolerant Individuals of Greater Amberjack (Seriola dumerili). Animals 2023, 13, 2717. [Google Scholar] [CrossRef]
- Yin, Y.; Zhang, Y.; Hua, Z.; Wu, A.; Pan, X.; Yang, J.; Wang, X. Muscle transcriptome analysis provides new insights into the growth gap between fast- and slow-growing Sinocyclocheilus grahami. Front. Genet. 2023, 14, 1217952. [Google Scholar] [CrossRef]
- Jenneckens, I.; Müller-Belecke, A.; Hörstgen-Schwark, G.; Meyer, J.-N. Proof of the successful development of Nile tilapia (Oreochromis niloticus) clones by DNA fingerprinting. Aquaculture 1999, 173, 377–388. [Google Scholar] [CrossRef]
- Xing, X.; Hu, T.; Wang, Y.; Li, Y.; Wang, W.; Hu, H.; Wei, Q.; Yan, Y.; Gan, D.; Bao, C.; et al. Construction of SNP fingerprints and genetic diversity analysis of radish (Raphanus sativus L.). Front. Plant Sci. 2024, 15, 1329890. [Google Scholar] [CrossRef]
- Xu, C.; Ren, Y.; Jian, Y.; Guo, Z.; Zhang, Y.; Xie, C.; Fu, J.; Wang, H.; Wang, G.; Xu, Y.; et al. Development of a maize 55K SNP array with improved genome coverage for molecular breeding. Mol. Breed. 2017, 37, 20. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.; Zhang, L.; Luo, J.; Zhao, H.; Zhang, J.; Wen, C. A new SNP genotyping technology Target SNP-seq and its application in genetic analysis of cucumber varieties. Sci. Rep. 2020, 10, 5623. [Google Scholar] [CrossRef]
- Cui, X.; Deng, J.; Zhang, Y.; Han, Y.; Ou, M.; Sun, Y. Investigation of genetic diversity in the loach Misgurnus anguillicaudatus revealed by whole-genome resequencing. BMC Genom. 2024, 25, 1126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, B.; Ren, L.; Li, Q.; Liu, S. SNP Fingerprinting for Germplasm Identification of the Fast-Growing Pacific oyster (Crassostrea gigas) “Haida No. 1” Variety. Mar. Biotechnol. 2024, 27, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; He, L.; Li, Y.; Huang, W.; Xi, F.; Lin, L.; Zhi, Q.; Zhang, W.; Tang, Y.T.; Geng, C.; et al. Correction: OTG-snpcaller: An Optimized Pipeline Based on TMAP and GATK for SNP Calling from Ion Torrent Data. PLoS ONE 2015, 10, e0138824. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, P.; Zhu, Y.; Sun, Y.; Dong, W.; Zhou, S. Constructing Cucumber Fingerprints Based on SNP Molecular Markers. Mol. Plant Breed. 2024, 22, 7054–7066. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, J.; Tao, Y.; Lu, S.; Wang, Q.; Sun, H.; Dong, Y.; Qiang, J. Genetic Structure, Selective Signatures, and Single Nucleotide Polymorphism Fingerprints of Blue Tilapia (Oreochromis aureus), Nile Tilapia Oreochromis niloticus), and Red Tilapia (Oreochromis spp.), as Determined by Whole-Genome Resequencing. Int. J. Mol. Sci. 2025, 26, 4910. https://doi.org/10.3390/ijms26104910
Hua J, Tao Y, Lu S, Wang Q, Sun H, Dong Y, Qiang J. Genetic Structure, Selective Signatures, and Single Nucleotide Polymorphism Fingerprints of Blue Tilapia (Oreochromis aureus), Nile Tilapia Oreochromis niloticus), and Red Tilapia (Oreochromis spp.), as Determined by Whole-Genome Resequencing. International Journal of Molecular Sciences. 2025; 26(10):4910. https://doi.org/10.3390/ijms26104910
Chicago/Turabian StyleHua, Jixiang, Yifan Tao, Siqi Lu, Qingchun Wang, Hui Sun, Yalun Dong, and Jun Qiang. 2025. "Genetic Structure, Selective Signatures, and Single Nucleotide Polymorphism Fingerprints of Blue Tilapia (Oreochromis aureus), Nile Tilapia Oreochromis niloticus), and Red Tilapia (Oreochromis spp.), as Determined by Whole-Genome Resequencing" International Journal of Molecular Sciences 26, no. 10: 4910. https://doi.org/10.3390/ijms26104910
APA StyleHua, J., Tao, Y., Lu, S., Wang, Q., Sun, H., Dong, Y., & Qiang, J. (2025). Genetic Structure, Selective Signatures, and Single Nucleotide Polymorphism Fingerprints of Blue Tilapia (Oreochromis aureus), Nile Tilapia Oreochromis niloticus), and Red Tilapia (Oreochromis spp.), as Determined by Whole-Genome Resequencing. International Journal of Molecular Sciences, 26(10), 4910. https://doi.org/10.3390/ijms26104910