
Genetic Basis of Motor Neuron Diseases: Insights, Clinical Management, and Future Directions

 , ,
, ,  ,
,  , ,
, , 
 and
and 
Abstract
1. Introduction
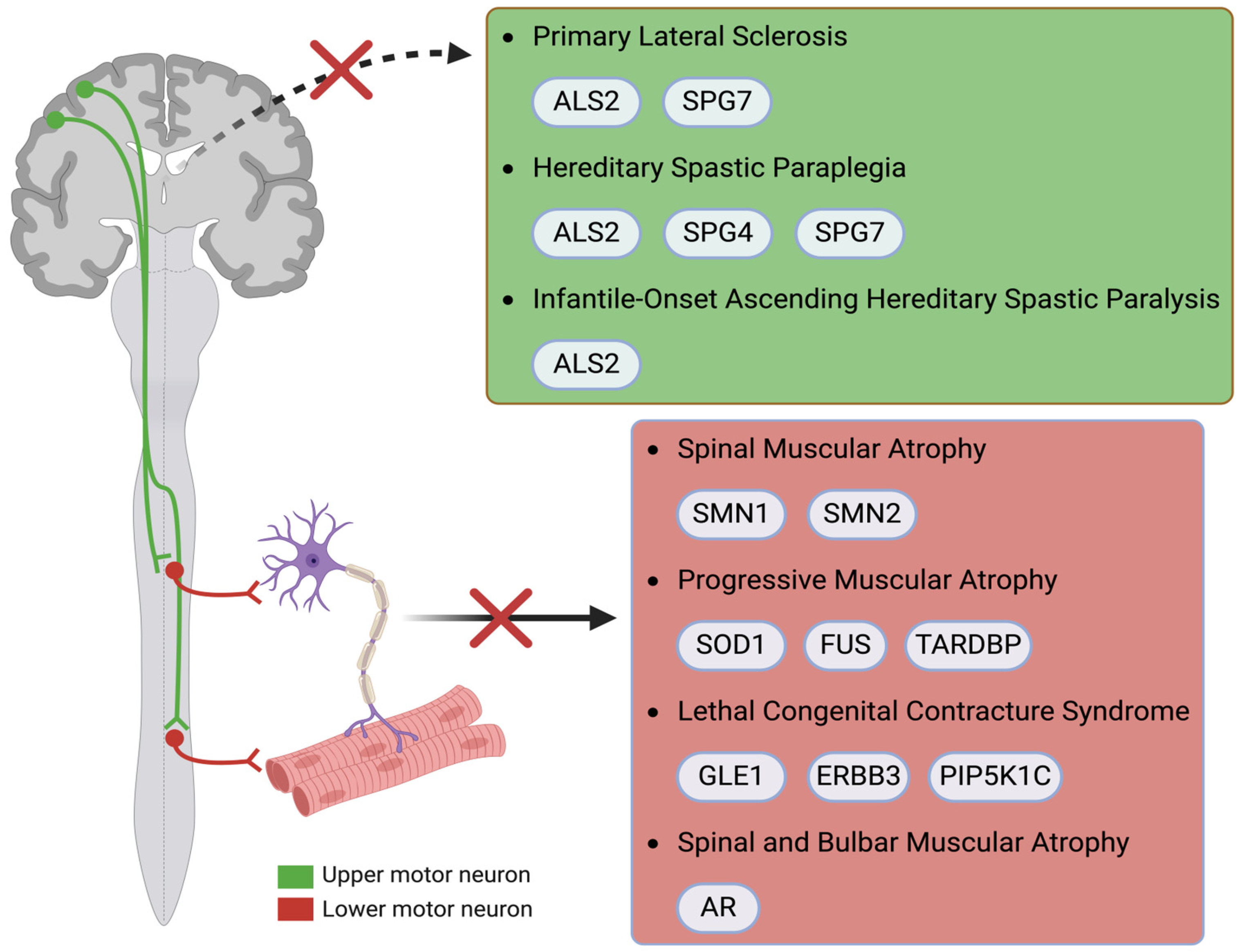
2. Diseases and Associated Genes
2.1. ALS
2.1.1. SOD
2.1.2. TARDBP
2.1.3. FUS
2.1.4. C9orf72
2.1.5. ALS2
2.1.6. ATAXIN-2
2.2. SMA
SMN1/SMN2
2.3. PLS
2.4. IAHSP
2.5. HSP
2.6. PMA
2.7. SBMA
2.8. LCCS
3. Epigenetics
3.1. Epigenetic Mechanisms in ALS
3.2. Epigenetic Mechanisms in SMA
4. Current Treatment Approaches of MNDs
4.1. Current Treatment for ALS
4.2. Current Treatment for SMA
4.3. Current Treatment for Other MNDs
4.4. Potential Treatment for IAHSP
5. From Molecular Landscape to Therapy: Future Perspectives in MNDs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic Lateral Sclerosis |
| ALS-FTD | Amyotrophic Lateral Sclerosis–Frontotemporal Dementia |
| ALS2 | Amyotrophic Lateral Sclerosis 2 |
| ANG | Angiogenin |
| AR | Androgen Receptor |
| ASO | Antisense Oligonucleotide |
| ATL1 | Atlastin GTPase 1 |
| ATP | Adenosine Triphosphate |
| ATXN2 | Ataxin-2 |
| BBB | Blood–Brain Barrier |
| C9orf72 | Chromosome 9 Open Reading Frame 72 |
| CAG | Cytosine–Adenine–Guanine |
| CCND1 | Cyclin D1 |
| CHIP | Carboxyl Terminus of Hsc70-Interacting Protein |
| CHMP2B | Charged Multivesicular Body Protein 2B |
| CNS | Central Nervous System |
| DAPI | 4′,6-Diamidino-2-Phenylindole |
| DOAJ | Directory of Open Access Journals |
| DPR | Dipeptide Repeat Proteins |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ERBB3 | Erb-B2 Receptor Tyrosine Kinase 3 |
| FBS | Fetal Bovine Serum |
| FIG4 | FIG4 Phosphoinositide 5-Phosphatase |
| FTD | Frontotemporal Dementia |
| FUS | Fused in Sarcoma |
| GFAP | Glial Fibrillary Acidic Protein |
| GLE1 | GLE1 RNA Export Mediator |
| GRN | Progranulin |
| HDAC | Histone Deacetylase |
| HSP | Hereditary Spastic Paraplegia |
| HSP70 | Heat Shock Protein 70 |
| IAHSP | Infantile-Onset Ascending Hereditary Spastic Paraplegia |
| KIF5A | Kinesin Family Member 5A |
| LCCS | Lethal Congenital Contracture Syndrome |
| LD | Linear Dichroism |
| MAPT | Microtubule-Associated Protein Tau |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MND | Motor Neuron Disease |
| mRNA | Messenger Ribonucleic Acid |
| NES | Nuclear Export Signal |
| NMJ | Neuromuscular Junction |
| Nrf2/ARE | Nuclear Factor Erythroid 2-Related Factor 2/Antioxidant Response Element |
| OPTN | Optineurin |
| PBS | Phosphate-Buffered Saline |
| PIP5K1C | Phosphatidylinositol-4-Phosphate 5-Kinase Type I Gamma |
| PLS | Primary Lateral Sclerosis |
| PMA | Progressive Muscular Atrophy |
| PolyQ | Polyglutamine |
| REEP1 | Receptor Expression-Enhancing Protein 1 |
| RNA | Ribonucleic Acid |
| RNP | Ribonucleoprotein |
| ROS | Reactive Oxygen Species |
| RT-PCR | Reverse Transcription Polymerase Chain Reaction |
| Rab5 | Ras-related Protein Rab-5 |
| SBMA | Spinal and Bulbar Muscular Atrophy |
| siRNA | Small Interfering RNA |
| SMA | Spinal Muscular Atrophy |
| SMN | Survival Motor Neuron |
| SOD1 | Superoxide Dismutase 1 |
| SPAST | Spastin |
| SPG4 | Spastin |
| SPG7 | Paraplegin |
| TARDBP | TAR DNA-Binding Protein Gene |
| TDP-43 | TAR DNA-Binding Protein 43 |
| TFEB | Transcription Factor EB |
| TUBA4A | Tubulin Alpha 4A |
| UBQLN2 | Ubiquilin-2 |
| UPR | Unfolded Protein Response |
| UPS | Ubiquitin–Proteasome System |
| VCP | Valosin-Containing Protein |
| 5hmC | 5-hydroxymethylcytosine |
References
- Foster, L.A.; Salajegheh, M.K. Motor Neuron Disease: Pathophysiology, Diagnosis, and Management. Am. J. Med. 2019, 132, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Radakovic, R.; Radakovic, C.; Abrahams, S.; Simmons, Z.; Carroll, A. Quality of Life, Cognitive and Behavioural Impairment in People with Motor Neuron Disease: A Systematic Review. Qual. Life Res. 2024, 33, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.; Elman, L. Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases. Continuum Lifelong Learn. Neurol. 2020, 26, 1323–1347. [Google Scholar] [CrossRef]
- Cookson, M.R.; Shaw, P.J. Oxidative Stress and Motor Neurone Disease. Brain Pathol. 1999, 9, 165–186. [Google Scholar] [CrossRef]
- Xu, J.; Su, X.; Burley, S.K.; Zheng, X.F.S. Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer. Antioxidants 2022, 11, 427. [Google Scholar] [CrossRef]
- Brotman, R.G.; Moreno-Escobar, M.C.; Joseph, J.; Munakomi, S.; Pawar, G. Amyotrophic Lateral Sclerosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Masrori, P.; Van Damme, P. Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Verma, A. Clinical Manifestation and Management of Amyotrophic Lateral Sclerosis. In Amyotrophic Lateral Sclerosis; Araki, T., Ed.; Exon Publications: Brisbane, AU, USA, 2021; ISBN 978-0-6450017-7-8. [Google Scholar]
- Zakharova, M.N.; Abramova, A.A. Lower and Upper Motor Neuron Involvement and Their Impact on Disease Prognosis in Amyotrophic Lateral Sclerosis. Neural Regen. Res. 2022, 17, 65–73. [Google Scholar] [CrossRef]
- Tena, A.; Clarià, F.; Solsona, F.; Povedano, M. Detecting Bulbar Involvement in Patients with Amyotrophic Lateral Sclerosis Based on Phonatory and Time-Frequency Features. Sensors 2022, 22, 1137. [Google Scholar] [CrossRef]
- Corcia, P.; Pradat, P.-F.; Salachas, F.; Bruneteau, G.; Forestier, N.L.; Seilhean, D.; Hauw, J.-J.; Meininger, V. Causes of Death in a Post-Mortem Series of ALS Patients. Amyotroph. Lateral Scler. 2008, 9, 59–62. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Masson, G.L. Genetics of Amyotrophic Lateral Sclerosis: A Review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Ticozzi, N.; Tiloca, C.; Morelli, C.; Colombrita, C.; Poletti, B.; Doretti, A.; Maderna, L.; Messina, S.; Ratti, A.; Silani, V. Genetics of Familial Amyotrophic Lateral Sclerosis. Arch. Ital. Biol. 2011, 149, 65–82. [Google Scholar] [CrossRef]
- Gibson, S.B.; Downie, J.M.; Tsetsou, S.; Feusier, J.E.; Figueroa, K.P.; Bromberg, M.B.; Jorde, L.B.; Pulst, S.M. The Evolving Genetic Risk for Sporadic ALS. Neurology 2017, 89, 226–233. [Google Scholar] [CrossRef]
- Hemerková, P.; Vališ, M. Role of Oxidative Stress in the Pathogenesis of Amyotrophic Lateral Sclerosis: Antioxidant Metalloenzymes and Therapeutic Strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative Stress in ALS: A Mechanism of Neurodegeneration and a Therapeutic Target. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2006, 1762, 1051–1067. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Chaudhary, P.; Janmeda, P.; Docea, A.O.; Yeskaliyeva, B.; Abdull Razis, A.F.; Modu, B.; Calina, D.; Sharifi-Rad, J. Oxidative Stress, Free Radicals and Antioxidants: Potential Crosstalk in the Pathophysiology of Human Diseases. Front. Chem. 2023, 11, 1158198. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide Dismutase Multigene Family: A Comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) Gene Structures, Evolution, and Expression. Free. Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F.S. Superoxide Dismutase 1 Acts as a Nuclear Transcription Factor to Regulate Oxidative Stress Resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef]
- Li, Y.; Wang, T.; Li, X.; Li, W.; Lei, Y.; Shang, Q.; Zheng, Z.; Fang, J.; Cao, L.; Yu, D.; et al. SOD2 Promotes the Immunosuppressive Function of Mesenchymal Stem Cells at the Expense of Adipocyte Differentiation. Mol. Ther. 2024, 32, 1144–1157. [Google Scholar] [CrossRef]
- Parascandolo, A.; Laukkanen, M.O. SOD3 Is a Non-Mutagenic Growth Regulator Affecting Cell Migration and Proliferation Signal Transduction. Antioxidants 2021, 10, 635. [Google Scholar] [CrossRef]
- Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592. [Google Scholar] [CrossRef]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dong, H.; Song, E.; Xu, X.; Liu, L.; Song, Y. Nrf2/ARE Pathway Activation, HO-1 and NQO1 Induction by Polychlorinated Biphenyl Quinone Is Associated with Reactive Oxygen Species and PI3K/AKT Signaling. Chem. Biol. Interact. 2014, 209, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-Regulated Glutathione Recycling Independent of Biosynthesis Is Critical for Cell Survival during Oxidative Stress. Free. Radic. Biol. Med. 2008, 46, 443. [Google Scholar] [CrossRef] [PubMed]
- Canella, R.; Benedusi, M.; Martini, M.; Cervellati, F.; Cavicchio, C.; Valacchi, G. Role of Nrf2 in Preventing Oxidative Stress Induced Chloride Current Alteration in Human Lung Cells. J. Cell Physiol. 2018, 233, 6018–6027. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Goodfellow, M.J.; Borcar, A.; Proctor, J.L.; Greco, T.; Rosenthal, R.E.; Fiskum, G. Transcriptional Activation of Antioxidant Gene Expression by Nrf2 Protects against Mitochondrial Dysfunction and Neuronal Death Associated with Acute and Chronic Neurodegeneration. Exp. Neurol. 2020, 328, 113247. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef]
- Bono, S.; Feligioni, M.; Corbo, M. Impaired Antioxidant KEAP1-NRF2 System in Amyotrophic Lateral Sclerosis: NRF2 Activation as a Potential Therapeutic Strategy. Mol. Neurodegener. 2021, 16, 71. [Google Scholar] [CrossRef]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8+ T Lymphocytes Expressing ALS-Causing SOD1 Mutant Selectively Trigger Death of Spinal Motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef]
- Mead, R.J.; Bennett, E.J.; Kennerley, A.J.; Sharp, P.; Sunyach, C.; Kasher, P.; Berwick, J.; Pettmann, B.; Battaglia, G.; Azzouz, M.; et al. Optimised and Rapid Pre-Clinical Screening in the SOD1(G93A) Transgenic Mouse Model of Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 2011, 6, e23244. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Yang, Y.; Deng, H.-X.; Hung, W.-Y.; Siddique, N.; Dellefave, L.; Gellera, C.; Andersen, P.M.; Siddique, T. Age and Founder Effect of SOD1 A4V Mutation Causing ALS. Neurology 2009, 72, 1634–1639. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Arslanbaeva, L.; Bisaglia, M. Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment. Molecules 2022, 27, 1471. [Google Scholar] [CrossRef]
- Soo, K.Y.; Atkin, J.D.; Horne, M.K.; Nagley, P. Recruitment of Mitochondria into Apoptotic Signaling Correlates with the Presence of Inclusions Formed by Amyotrophic Lateral Sclerosis-Associated SOD1 Mutations. J. Neurochem. 2009, 108, 578–590. [Google Scholar] [CrossRef]
- Cozzolino, M.; Pesaresi, M.G.; Amori, I.; Crosio, C.; Ferri, A.; Nencini, M.; Carrì, M.T. Oligomerization of Mutant SOD1 in Mitochondria of Motoneuronal Cells Drives Mitochondrial Damage and Cell Toxicity. Antioxid. Redox Signal 2009, 11, 1547–1558. [Google Scholar] [CrossRef]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 Misplacing and Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis Pathogenesis. Front. Cell Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
- Li, Q.; Spencer, N.Y.; Pantazis, N.J.; Engelhardt, J.F. Alsin and SOD1(G93A) Proteins Regulate Endosomal Reactive Oxygen Species Production by Glial Cells and Proinflammatory Pathways Responsible for Neurotoxicity. J. Biol. Chem. 2011, 286, 40151–40162. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant Superoxide Dismutase 1-Induced IL-1β Accelerates ALS Pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef]
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Tremblay, M.-È.; Petratos, S.; Zoupi, L.; Boziki, M.; Kesidou, E.; Simeonidou, C.; Theotokis, P. Origin and Emergence of Microglia in the CNS—An Interesting (Hi)Story of an Eccentric Cell. CIMB 2023, 45, 2609–2628. [Google Scholar] [CrossRef]
- Ferri, A.; Nencini, M.; Casciati, A.; Cozzolino, M.; Angelini, D.F.; Longone, P.; Spalloni, A.; Rotilio, G.; Carrì, M.T. Cell Death in Amyotrophic Lateral Sclerosis: Interplay between Neuronal and Glial Cells. FASEB J. 2004, 18, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes Expressing ALS-Linked Mutated SOD1 Release Factors Selectively Toxic to Motor Neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Scalcon, V.; Massimino, M.L.; Nies, K.; Lopreiato, R.; Rigobello, M.P.; Bertoli, A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants 2022, 11, 614. [Google Scholar] [CrossRef]
- Colombrita, C.; Onesto, E.; Tiloca, C.; Ticozzi, N.; Silani, V.; Ratti, A. RNA-Binding Proteins and RNA Metabolism: A New Scenario in the Pathogenesis of Amyotrophic Lateral Sclerosis. Arch. Ital. Biol. 2011, 149, 83–99. [Google Scholar] [CrossRef]
- Cohen, T.J.; Lee, V.M.Y.; Trojanowski, J.Q. TDP-43 Functions and Pathogenic Mechanisms Implicated in TDP-43 Proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 Control in Neurodegeneration: Autoregulation, Localization and Aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic Functions of TDP-43 and FUS and Their Role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef]
- Nagano, S.; Jinno, J.; Abdelhamid, R.F.; Jin, Y.; Shibata, M.; Watanabe, S.; Hirokawa, S.; Nishizawa, M.; Sakimura, K.; Onodera, O.; et al. TDP-43 Transports Ribosomal Protein mRNA to Regulate Axonal Local Translation in Neuronal Axons. Acta Neuropathol. 2020, 140, 695–713. [Google Scholar] [CrossRef]
- Wong, C.-E.; Jin, L.-W.; Chu, Y.-P.; Wei, W.-Y.; Ho, P.-C.; Tsai, K.-J. TDP-43 Proteinopathy Impairs mRNP Granule Mediated Postsynaptic Translation and mRNA Metabolism. Theranostics 2021, 11, 330–345. [Google Scholar] [CrossRef]
- Mori, F.; Yasui, H.; Miki, Y.; Kon, T.; Arai, A.; Kurotaki, H.; Tomiyama, M.; Wakabayashi, K. Colocalization of TDP-43 and Stress Granules at the Early Stage of TDP-43 Aggregation in Amyotrophic Lateral Sclerosis. Brain Pathol. 2024, 34, e13215. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 Aggregation in Neurodegeneration: Are Stress Granules the Key? Brain Res. 2012, 1462, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Zhang, Y.-J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.-F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.P.; et al. Novel Mutations in TARDBP (TDP-43) in Patients with Familial Amyotrophic Lateral Sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668. [Google Scholar] [CrossRef]
- Janssens, J.; Wils, H.; Kleinberger, G.; Joris, G.; Cuijt, I.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. Overexpression of ALS-Associated p.M337V Human TDP-43 in Mice Worsens Disease Features Compared to Wild-Type Human TDP-43 Mice. Mol. Neurobiol. 2013, 48, 22–35. [Google Scholar] [CrossRef]
- Zeng, J.; Tang, Y.; Dong, X.; Li, F.; Wei, G. Influence of ALS-Linked M337V Mutation on the Conformational Ensembles of TDP-43321-340 Peptide Monomer and Dimer. Proteins 2024, 92, 1059–1069. [Google Scholar] [CrossRef]
- Takeda, T.; Iijima, M.; Shimizu, Y.; Yoshizawa, H.; Miyashiro, M.; Onizuka, H.; Yamamoto, T.; Nishiyama, A.; Suzuki, N.; Aoki, M.; et al. P.N345K Mutation in TARDBP in a Patient with Familial Amyotrophic Lateral Sclerosis: An Autopsy Case. Neuropathology 2019, 39, 286–293. [Google Scholar] [CrossRef]
- Gendron, T.F.; Rademakers, R.; Petrucelli, L. TARDBP Mutation Analysis in TDP-43 Proteinopathies and Deciphering the Toxicity of Mutant TDP-43. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S35–S45. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The Role of TDP-43 Mislocalization in Amyotrophic Lateral Sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- De Marco, G.; Lupino, E.; Calvo, A.; Moglia, C.; Buccinnà, B.; Grifoni, S.; Ramondetti, C.; Lomartire, A.; Rinaudo, M.T.; Piccinini, M.; et al. Cytoplasmic Accumulation of TDP-43 in Circulating Lymphomonocytes of ALS Patients with and without TARDBP Mutations. Acta Neuropathol. 2011, 121, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J.; Skibinski, G.; Korb, E.; Rao, E.J.; Wu, J.Y.; Finkbeiner, S. Cytoplasmic Mislocalization of TDP-43 Is Toxic to Neurons and Enhanced by a Mutation Associated with Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2010, 30, 639–649. [Google Scholar] [CrossRef]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef] [PubMed]
- Pakravan, D.; Michiels, E.; Bratek-Skicki, A.; De Decker, M.; Van Lindt, J.; Alsteens, D.; Derclaye, S.; Van Damme, P.; Schymkowitz, J.; Rousseau, F.; et al. Liquid-Liquid Phase Separation Enhances TDP-43 LCD Aggregation but Delays Seeded Aggregation. Biomolecules 2021, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.L.; Guo, L. Liquid-Liquid Phase Separation of TDP-43 and FUS in Physiology and Pathology of Neurodegenerative Diseases. Front. Mol. Biosci. 2022, 9, 826719. [Google Scholar] [CrossRef]
- Xiao, X.; Li, M.; Ye, Z.; He, X.; Wei, J.; Zha, Y. FUS Gene Mutation in Amyotrophic Lateral Sclerosis: A New Case Report and Systematic Review. Amyotroph. Lateral Scler. Front. Degener. 2024, 25, 1–15. [Google Scholar] [CrossRef]
- Gijselinck, I.; Cruts, M.; Van Broeckhoven, C. The Genetics of C9orf72 Expansions. Cold Spring Harb. Perspect. Med. 2018, 8, a026757. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.-H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC Repeat Expansion in C9orf72 Compromises Nucleocytoplasmic Transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Orrell, R.W. ALS2-Related Disorder. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 Intermediate-Length Polyglutamine Expansions Are Associated with Increased Risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Honda, D.; Ishigaki, S.; Iguchi, Y.; Fujioka, Y.; Udagawa, T.; Masuda, A.; Ohno, K.; Katsuno, M.; Sobue, G. The ALS/FTLD-Related RNA-Binding Proteins TDP-43 and FUS Have Common Downstream RNA Targets in Cortical Neurons. FEBS Open Bio 2014, 4, 1–10. [Google Scholar] [CrossRef]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-Associated ALS and FTD: Insights from Rodent Models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Assoni, A.F.; Foijer, F.; Zatz, M. Amyotrophic Lateral Sclerosis, FUS and Protein Synthesis Defects. Stem Cell Rev. Rep. 2023, 19, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Bowden, H.A.; Dormann, D. Altered mRNP Granule Dynamics in FTLD Pathogenesis. J. Neurochem. 2016, 138, 112–133. [Google Scholar] [CrossRef]
- López-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-Linked Mutation in FUS Suppresses Intra-Axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 2018, 100, 816–830.e7. [Google Scholar] [CrossRef]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.A.; Capell, A.; Schmid, B.; et al. ALS-Associated Fused in Sarcoma (FUS) Mutations Disrupt Transportin-Mediated Nuclear Import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Robinson, H.K.; Connor-Robson, N.; Buchman, V.L. Recruitment into Stress Granules Prevents Irreversible Aggregation of FUS Protein Mislocalized to the Cytoplasm. Cell Cycle 2013, 12, 3383–3391. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- An, H.; Litscher, G.; Watanabe, N.; Wei, W.; Hashimoto, T.; Iwatsubo, T.; Buchman, V.L.; Shelkovnikova, T.A. ALS-Linked Cytoplasmic FUS Assemblies Are Compositionally Different from Physiological Stress Granules and Sequester hnRNPA3, a Novel Modifier of FUS Toxicity. Neurobiol. Dis. 2022, 162, 105585. [Google Scholar] [CrossRef]
- Barmada, S.J. Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 340–351. [Google Scholar] [CrossRef]
- Scekic-Zahirovic, J.; Sendscheid, O.; El Oussini, H.; Jambeau, M.; Sun, Y.; Mersmann, S.; Wagner, M.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Toxic Gain of Function from Mutant FUS Protein Is Crucial to Trigger Cell Autonomous Motor Neuron Loss. EMBO J. 2016, 35, 1077–1097. [Google Scholar] [CrossRef]
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA Metabolism in Neurodegenerative Disease. Dis. Model. Mech. 2017, 10, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Daigle, J.G.; Lanson, N.A.; Smith, R.B.; Casci, I.; Maltare, A.; Monaghan, J.; Nichols, C.D.; Kryndushkin, D.; Shewmaker, F.; Pandey, U.B. RNA-Binding Ability of FUS Regulates Neurodegeneration, Cytoplasmic Mislocalization and Incorporation into Stress Granules Associated with FUS Carrying ALS-Linked Mutations. Hum. Mol. Genet. 2013, 22, 1193–1205. [Google Scholar] [CrossRef]
- Scotter, E.L.; Chen, H.-J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef]
- Lee, J.W.; Kang, S.-W.; Choi, W.A. Clinical Course of Amyotrophic Lateral Sclerosis According to Initial Symptoms: An Analysis of 500 Cases. Yonsei Med. J. 2021, 62, 338–343. [Google Scholar] [CrossRef]
- Hayes, L.R.; Kalab, P. Emerging Therapies and Novel Targets for TDP-43 Proteinopathy in ALS/FTD. Neurotherapeutics 2022, 19, 1061–1084. [Google Scholar] [CrossRef]
- Yan, Y.-P.; Xu, C.-Y.; Gu, L.-Y.; Zhang, B.; Shen, T.; Gao, T.; Tian, J.; Pu, J.-L.; Yin, X.-Z.; Zhang, B.-R.; et al. Genetic Testing of FUS, HTRA2, and TENM4 Genes in Chinese Patients with Essential Tremor. CNS Neurosci. Ther. 2020, 26, 837–841. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 Is Required for Proper Macrophage and Microglial Function in Mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 Regulates Energy Homeostasis by Stabilizing Mitochondrial Complex I Assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef]
- Beckers, J.; Tharkeshwar, A.K.; Van Damme, P. C9orf72 ALS-FTD: Recent Evidence for Dysregulation of the Autophagy-Lysosome Pathway at Multiple Levels. Autophagy 2021, 17, 3306–3322. [Google Scholar] [CrossRef]
- Marchi, P.M.; Marrone, L.; Brasseur, L.; Coens, A.; Webster, C.P.; Bousset, L.; Destro, M.; Smith, E.F.; Walther, C.G.; Alfred, V.; et al. C9ORF72-Derived Poly-GA DPRs Undergo Endocytic Uptake in iAstrocytes and Spread to Motor Neurons. Life Sci. Alliance 2022, 5, e202101276. [Google Scholar] [CrossRef]
- Pang, W.; Hu, F. Cellular and Physiological Functions of C9ORF72 and Implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic Stress Induces Formation of Stress Granules and Pathological TDP-43 Aggregates in Human ALS Fibroblasts and iPSC-Motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Hasan, G.M.; Hassan, M.I. Unraveling the Role of RNA Mediated Toxicity of C9orf72 Repeats in C9-FTD/ALS. Front. Neurosci. 2017, 11, 711. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, N.; Künzli, C.; Buthey, K.; Saxena, S. C9ORF72 Regulates Stress Granule Formation and Its Deficiency Impairs Stress Granule Assembly, Hypersensitizing Cells to Stress. Mol. Neurobiol. 2017, 54, 3062–3077. [Google Scholar] [CrossRef]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct Brain Transcriptome Profiles in C9orf72-Associated and Sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense Transcripts of the Expanded C9ORF72 Hexanucleotide Repeat Form Nuclear RNA Foci and Undergo Repeat-Associated Non-ATG Translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef]
- Boivin, M.; Pfister, V.; Gaucherot, A.; Ruffenach, F.; Negroni, L.; Sellier, C.; Charlet-Berguerand, N. Reduced Autophagy upon C9ORF72 Loss Synergizes with Dipeptide Repeat Protein Toxicity in G4C2 Repeat Expansion Disorders. EMBO J. 2020, 39, e100574. [Google Scholar] [CrossRef]
- Geng, Y.; Cai, Q. Role of C9orf72 Hexanucleotide Repeat Expansions in ALS/FTD Pathogenesis. Front. Mol. Neurosci. 2024, 17, 1322720. [Google Scholar] [CrossRef]
- Nonaka, T.; Masuda-Suzukake, M.; Hosokawa, M.; Shimozawa, A.; Hirai, S.; Okado, H.; Hasegawa, M. C9ORF72 Dipeptide Repeat Poly-GA Inclusions Promote Intracellular Aggregation of Phosphorylated TDP-43. Hum. Mol. Genet. 2018, 27, 2658–2670. [Google Scholar] [CrossRef]
- Hadano, S.; Kunita, R.; Otomo, A.; Suzuki-Utsunomiya, K.; Ikeda, J.-E. Molecular and Cellular Function of ALS2/Alsin: Implication of Membrane Dynamics in Neuronal Development and Degeneration. Neurochem. Int. 2007, 51, 74–84. [Google Scholar] [CrossRef]
- ALS2—An Overview | ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/als2 (accessed on 14 September 2024).
- Hsu, F.; Spannl, S.; Ferguson, C.; Hyman, A.A.; Parton, R.G.; Zerial, M. Rab5 and Alsin Regulate Stress-Activated Cytoprotective Signaling on Mitochondria. Elife 2018, 7, e32282. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Ganesan, D. Regulation of Neuronal Autophagy and the Implications in Neurodegenerative Diseases. Neurobiol. Dis. 2022, 162, 105582. [Google Scholar] [CrossRef] [PubMed]
- Chandran, J.; Ding, J.; Cai, H. Alsin and the Molecular Pathways of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2007, 36, 224–231. [Google Scholar] [CrossRef]
- Lai, C.; Xie, C.; Shim, H.; Chandran, J.; Howell, B.W.; Cai, H. Regulation of Endosomal Motility and Degradation by Amyotrophic Lateral Sclerosis 2/Alsin. Mol. Brain 2009, 2, 23. [Google Scholar] [CrossRef]
- Otomo, A.; Hadano, S.; Okada, T.; Mizumura, H.; Kunita, R.; Nishijima, H.; Showguchi-Miyata, J.; Yanagisawa, Y.; Kohiki, E.; Suga, E.; et al. ALS2, a Novel Guanine Nucleotide Exchange Factor for the Small GTPase Rab5, Is Implicated in Endosomal Dynamics. Hum. Mol. Genet. 2003, 12, 1671–1687. [Google Scholar] [CrossRef]
- Morishita, S.; Wada, N.; Fukuda, M.; Nakamura, T. Rab5 Activation on Macropinosomes Requires ALS2, and Subsequent Rab5 Inactivation through ALS2 Detachment Requires Active Rab7. FEBS Lett. 2019, 593, 230–241. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox signaling 2014, 20, 460–473. [Google Scholar] [CrossRef]
- The Interplay Between Oxidative Stress and Autophagy: Focus on the Development of Neurological Diseases-PMC. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8799983/ (accessed on 14 September 2024).
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Cai, H.; Lin, X.; Xie, C.; Laird, F.M.; Lai, C.; Wen, H.; Chiang, H.-C.; Shim, H.; Farah, M.H.; Hoke, A.; et al. Loss of ALS2 Function Is Insufficient to Trigger Motor Neuron Degeneration in Knock-Out Mice But Predisposes Neurons to Oxidative Stress. J. Neurosci. 2005, 25, 7567–7574. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef] [PubMed]
- Cannariato, M.; Miceli, M.; Cavaglià, M.; Deriu, M.A. Prediction of Protein-Protein Interactions Between Alsin DH/PH and Rac1 and Resulting Protein Dynamics. Front. Mol. Neurosci. 2021, 14, 772122. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Jara, J.H.; Sekerkova, G.; Yasvoina, M.V.; Martina, M.; Özdinler, P.H. Absence of Alsin Function Leads to Corticospinal Motor Neuron Vulnerability via Novel Disease Mechanisms. Hum. Mol. Genet. 2016, 25, 1074–1087. [Google Scholar] [CrossRef]
- Yoganathan, S.; Kumar, M.; Aaron, R.; Rangan, S.R.; Umakant, B.S.; Thomas, M.; Oommen, S.P.; Danda, S. Phenotype and Genotype of Children with ALS2 Gene-Related Disorder. Neuropediatrics 2024. [Google Scholar] [CrossRef]
- Sprute, R.; Jergas, H.; Ölmez, A.; Alawbathani, S.; Karasoy, H.; Dafsari, H.S.; Becker, K.; Daimagüler, H.-S.; Nürnberg, P.; Muntoni, F.; et al. Genotype-Phenotype Correlation in Seven Motor Neuron Disease Families with Novel ALS2 Mutations. Am. J. Med. Genet. A 2021, 185, 344–354. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; Mullen, B.; Heckman, M.G.; Baker, M.C.; DeJesus-Hernandez, M.; Brown, P.H.; Murray, M.E.; Hsiung, G.-Y.R.; Stewart, H.; Karydas, A.M.; et al. Ataxin-2 as Potential Disease Modifier in C9ORF72 Expansion Carriers. Neurobiol. Aging 2014, 35, 2421.e13–2421.e17. [Google Scholar] [CrossRef]
- Wijegunawardana, D.; Nayak, A.; Vishal, S.S.; Venkatesh, N.; Gopal, P.P. Ataxin-2 Polyglutamine Expansions Aberrantly Sequester TDP-43 Ribonucleoprotein Condensates Disrupting mRNA Transport and Local Translation in Neurons. Dev. Cell 2024, 60, 253–269. [Google Scholar] [CrossRef]
- Laffita-Mesa, J.M.; Paucar, M.; Svenningsson, P. Ataxin-2 Gene: A Powerful Modulator of Neurological Disorders. Curr. Opin. Neurol. 2021, 34, 578–588. [Google Scholar] [CrossRef]
- Mercuri, E.; Sumner, C.J.; Muntoni, F.; Darras, B.T.; Finkel, R.S. Spinal Muscular Atrophy. Nat. Rev. Dis. Primers 2022, 8, 52. [Google Scholar] [CrossRef]
- Spinal Muscular Atrophy|National Institute of Neurological Disorders and Stroke. Available online: https://www.ninds.nih.gov/health-information/disorders/spinal-muscular-atrophy (accessed on 23 November 2024).
- Rizvi, S.B.; Ahmed, H.; Zaman, A.; Ali, A.M.N.; Shah, H.H.; Rauf, S.A.; Dave, T. Spinal Muscular Atrophy Type 1: A Fatal Case in a 1-Year-Old Girl with Delayed Diagnosis. Clin. Case Rep. 2024, 12, e8513. [Google Scholar] [CrossRef]
- Cances, C.; Vlodavets, D.; Comi, G.P.; Masson, R.; Mazurkiewicz-Bełdzińska, M.; Saito, K.; Zanoteli, E.; Dodman, A.; El-Khairi, M.; Gorni, K.; et al. Natural History of Type 1 Spinal Muscular Atrophy: A Retrospective, Global, Multicenter Study. Orphanet J. Rare Dis. 2022, 17, 300. [Google Scholar] [CrossRef] [PubMed]
- Cancès, C.; Richelme, C.; Barnerias, C.; Espil, C. Clinical Features of Spinal Muscular Atrophy (SMA) Type 2. Arch. Pediatr. 2020, 27, 7S18–7S22. [Google Scholar] [CrossRef]
- Salort-Campana, E.; Quijano-Roy, S. Clinical Features of Spinal Muscular Atrophy (SMA) Type 3 (Kugelberg-Welander Disease). Arch. Pediatr. 2020, 27, 7S23–7S28. [Google Scholar] [CrossRef]
- Souza, P.V.S.; Pinto, W.B.V.R.; Ricarte, A.; Badia, B.M.L.; Seneor, D.D.; Teixeira, D.T.; Caetano, L.; Gonçalves, E.A.; Chieia, M.a.T.; Farias, I.B.; et al. Clinical and Radiological Profile of Patients with Spinal Muscular Atrophy Type 4. Eur. J. Neurol. 2021, 28, 609–619. [Google Scholar] [CrossRef]
- Germain-Desprez, D.; Brun, T.; Rochette, C.; Semionov, A.; Rouget, R.; Simard, L.R. The SMN Genes Are Subject to Transcriptional Regulation during Cellular Differentiation. Gene 2001, 279, 109–117. [Google Scholar] [CrossRef]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Burghes, A.H.M.; Kissel, J.T. A Positive Modifier of Spinal Muscular Atrophy in the SMN2 Gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef]
- SMN1 Survival of Motor Neuron 1, Telomeric [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/6606 (accessed on 23 November 2024).
- Workman, E.; Kolb, S.J.; Battle, D.J. Spliceosomal Small Nuclear Ribonucleoprotein Biogenesis Defects and Motor Neuron Selectivity in Spinal Muscular Atrophy. Brain Res. 2012, 1462, 93–99. [Google Scholar] [CrossRef]
- Fallini, C.; Bassell, G.J.; Rossoll, W. Spinal Muscular Atrophy: The Role of SMN in Axonal mRNA Regulation. Brain Res. 2012, 1462, 81–92. [Google Scholar] [CrossRef]
- Donlin-Asp, P.G.; Fallini, C.; Campos, J.; Chou, C.-C.; Merritt, M.E.; Phan, H.C.; Bassell, G.J.; Rossoll, W. The Survival of Motor Neuron Protein Acts as a Molecular Chaperone for mRNP Assembly. Cell Rep. 2017, 18, 1660–1673. [Google Scholar] [CrossRef]
- Karafoulidou, E.; Kesidou, E.; Theotokis, P.; Konstantinou, C.; Nella, M.-K.; Michailidou, I.; Touloumi, O.; Polyzoidou, E.; Salamotas, I.; Einstein, O.; et al. Systemic LPS Administration Stimulates the Activation of Non-Neuronal Cells in an Experimental Model of Spinal Muscular Atrophy. Cells 2024, 13, 785. [Google Scholar] [CrossRef]
- Chaytow, H.; Huang, Y.-T.; Gillingwater, T.H.; Faller, K.M.E. The Role of Survival Motor Neuron Protein (SMN) in Protein Homeostasis. Cell Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef] [PubMed]
- Jedrzejowska, M.; Madej-Pilarczyk, A.; Zimowski, J.; Hausmanowa-Petrusewicz, I. [Pseudodominant inheritance of spinal muscular atrophy—Father and son suffering from SMA]. Neurol. Neurochir. Pol. 2006, 40, 446–449. [Google Scholar]
- Wirth, B. An Update of the Mutation Spectrum of the Survival Motor Neuron Gene (SMN1) in Autosomal Recessive Spinal Muscular Atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Burghes, A.H.M.; Beattie, C.E. Spinal Muscular Atrophy: Why Do Low Levels of SMN Make Motor Neurons Sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef]
- Chemello, F.; Pozzobon, M.; Tsansizi, L.I.; Varanita, T.; Quintana-Cabrera, R.; Bonesso, D.; Piccoli, M.; Lanfranchi, G.; Giacomello, M.; Scorrano, L.; et al. Dysfunctional Mitochondria Accumulate in a Skeletal Muscle Knockout Model of Smn1, the Causal Gene of Spinal Muscular Atrophy. Cell Death Dis. 2023, 14, 162. [Google Scholar] [CrossRef]
- Young, P.J.; Day, P.M.; Zhou, J.; Androphy, E.J.; Morris, G.E.; Lorson, C.L. A Direct Interaction between the Survival Motor Neuron Protein and P53 and Its Relationship to Spinal Muscular Atrophy. J. Biol. Chem. 2002, 277, 2852–2859. [Google Scholar] [CrossRef]
- Beattie, C.E.; Kolb, S.J. Spinal Muscular Atrophy: Selective Motor Neuron Loss and Global Defect in the Assembly of Ribonucleoproteins. Brain Res. 2018, 1693, 92–97. [Google Scholar] [CrossRef]
- SMN Deficiency Causes Tissue-Specific Perturbations in the Repertoire of snRNAs and Widespread Defects in Splicing–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/18485868/ (accessed on 25 November 2024).
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Dosi, C.; Masson, R. The Impact of Three SMN2 Gene Copies on Clinical Characteristics and Effect of Disease-Modifying Treatment in Patients with Spinal Muscular Atrophy: A Systematic Literature Review. Front. Neurol. 2024, 15, 1308296. [Google Scholar] [CrossRef]
- Vacchiano, V.; Bonan, L.; Liguori, R.; Rizzo, G. Primary Lateral Sclerosis: An Overview. J. Clin. Med. 2024, 13, 578. [Google Scholar] [CrossRef]
- Turner, M.R.; Talbot, K. Primary Lateral Sclerosis: Diagnosis and Management. Pract. Neurol. 2020, 20, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.C.; Rowe, A.; Findlater, K.; Orange, J.B.; Grace, G.; Strong, M.J. Differentiation between Primary Lateral Sclerosis and Amyotrophic Lateral Sclerosis: Examination of Symptoms and Signs at Disease Onset and during Follow-Up. Arch. Neurol. 2007, 64, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, L.; Lynch, D.R.; Lukas, T.; Ahmeti, K.; Sleiman, P.M.A.; Ryan, E.; Schadt, K.A.; Newman, J.H.; Deng, H.-X.; et al. Compound Heterozygote Mutations in SPG7 in a Family with Adult-Onset Primary Lateral Sclerosis. Neurol. Genet. 2016, 2, e60. [Google Scholar] [CrossRef]
- Panzeri, C.; De Palma, C.; Martinuzzi, A.; Daga, A.; De Polo, G.; Bresolin, N.; Miller, C.C.; Tudor, E.L.; Clementi, E.; Bassi, M.T. The First ALS2 Missense Mutation Associated with JPLS Reveals New Aspects of Alsin Biological Function. Brain 2006, 129, 1710–1719. [Google Scholar] [CrossRef]
- Agarwal, S.; Highton-Williamson, E.; Caga, J.; Matamala, J.M.; Dharmadasa, T.; Howells, J.; Zoing, M.C.; Shibuya, K.; Geevasinga, N.; Vucic, S.; et al. Primary Lateral Sclerosis and the Amyotrophic Lateral Sclerosis-Frontotemporal Dementia Spectrum. J. Neurol. 2018, 265, 1819–1828. [Google Scholar] [CrossRef]
- Miceli, M.; Exertier, C.; Cavaglià, M.; Gugole, E.; Boccardo, M.; Casaluci, R.R.; Ceccarelli, N.; De Maio, A.; Vallone, B.; Deriu, M.A. ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules. Biology 2022, 11, 77. [Google Scholar] [CrossRef]
- Lesca, G.; Eymard-Pierre, E.; Santorelli, F.M.; Cusmai, R.; Di Capua, M.; Valente, E.M.; Attia-Sobol, J.; Plauchu, H.; Leuzzi, V.; Ponzone, A.; et al. Infantile Ascending Hereditary Spastic Paralysis (IAHSP): Clinical Features in 11 Families. Neurology 2003, 60, 674–682. [Google Scholar] [CrossRef]
- Cai, H.; Shim, H.; Lai, C.; Xie, C.; Lin, X.; Yang, W.J.; Chandran, J. ALS2/Alsin Knockout Mice and Motor Neuron Diseases. Neurodegener. Dis. 2008, 5, 359–366. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, Q.; Luo, J.; Zhou, X.; Yi, S.; Tan, S.; Qin, Z. Clinical Features and Molecular Genetic Investigation of Infantile-Onset Ascending Hereditary Spastic Paralysis (IAHSP) in Two Chinese Siblings Caused by a Novel Splice Site ALS2 Variation. BMC Med. Genomics 2024, 17, 44. [Google Scholar] [CrossRef]
- Eymard-Pierre, E.; Lesca, G.; Dollet, S.; Santorelli, F.M.; di Capua, M.; Bertini, E.; Boespflug-Tanguy, O. Infantile-Onset Ascending Hereditary Spastic Paralysis Is Associated with Mutations in the Alsin Gene. Am. J. Hum. Genet. 2002, 71, 518–527. [Google Scholar] [CrossRef]
- Murala, S.; Nagarajan, E.; Bollu, P.C. Hereditary Spastic Paraplegia. Neurol. Sci. 2021, 42, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Meyyazhagan, A.; Orlacchio, A. Hereditary Spastic Paraplegia: An Update. Int. J. Mol. Sci. 2022, 23, 1697. [Google Scholar] [CrossRef] [PubMed]
- Varghaei, P.; Estiar, M.A.; Ashtiani, S.; Veyron, S.; Mufti, K.; Leveille, E.; Yu, E.; Spiegelman, D.; Rioux, M.-F.; Yoon, G.; et al. Genetic, Structural and Clinical Analysis of Spastic Paraplegia 4. Parkinsonism Relat. Disord. 2022, 98, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Parodi, L.; Rydning, S.L.; Tallaksen, C.; Durr, A. Spastic Paraplegia 4. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X.-J. Loss of Spastin Function Results in Disease-Specific Axonal Defects in Human Pluripotent Stem Cell-Based Models of Hereditary Spastic Paraplegia. Stem Cells 2014, 32, 414–423. [Google Scholar] [CrossRef]
- Wali, G.; Liyanage, E.; Blair, N.F.; Sutharsan, R.; Park, J.-S.; Mackay-Sim, A.; Sue, C.M. Oxidative Stress-Induced Axon Fragmentation Is a Consequence of Reduced Axonal Transport in Hereditary Spastic Paraplegia SPAST Patient Neurons. Front. Neurosci. 2020, 14, 401. [Google Scholar] [CrossRef]
- Solowska, J.M.; Baas, P.W. Hereditary Spastic Paraplegia SPG4: What Is Known and Not Known about the Disease. Brain 2015, 138, 2471–2484. [Google Scholar] [CrossRef]
- Sauter, S.; Miterski, B.; Klimpe, S.; Bönsch, D.; Schöls, L.; Visbeck, A.; Papke, T.; Hopf, H.C.; Engel, W.; Deufel, T.; et al. Mutation Analysis of the Spastin Gene (SPG4) in Patients in Germany with Autosomal Dominant Hereditary Spastic Paraplegia. Hum. Mutat. 2002, 20, 127–132. [Google Scholar] [CrossRef]
- Casari, G.; Marconi, R. Spastic Paraplegia 7. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Wilkinson, P.A.; Crosby, A.H.; Turner, C.; Bradley, L.J.; Ginsberg, L.; Wood, N.W.; Schapira, A.H.; Warner, T.T. A Clinical, Genetic and Biochemical Study of SPG7 Mutations in Hereditary Spastic Paraplegia. Brain 2004, 127, 973–980. [Google Scholar] [CrossRef]
- Elleuch, N.; Depienne, C.; Benomar, A.; Hernandez, A.M.O.; Ferrer, X.; Fontaine, B.; Grid, D.; Tallaksen, C.M.E.; Zemmouri, R.; Stevanin, G.; et al. Mutation Analysis of the Paraplegin Gene (SPG7) in Patients with Hereditary Spastic Paraplegia. Neurology 2006, 66, 654–659. [Google Scholar] [CrossRef]
- Osmanovic, A.; Widjaja, M.; Förster, A.; Weder, J.; Wattjes, M.P.; Lange, I.; Sarikidi, A.; Auber, B.; Raab, P.; Christians, A.; et al. SPG7 Mutations in Amyotrophic Lateral Sclerosis: A Genetic Link to Hereditary Spastic Paraplegia. J. Neurol. 2020, 267, 2732–2743. [Google Scholar] [CrossRef]
- Liewluck, T.; Saperstein, D.S. Progressive Muscular Atrophy. Neurol. Clin. 2015, 33, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Cervenakova, L.; Protas, I.I.; Hirano, A.; Votiakov, V.I.; Nedzved, M.K.; Kolomiets, N.D.; Taller, I.; Park, K.Y.; Sambuughin, N.; Gajdusek, D.C.; et al. Progressive Muscular Atrophy Variant of Familial Amyotrophic Lateral Sclerosis (PMA/ALS). J. Neurol. Sci. 2000, 177, 124–130. [Google Scholar] [CrossRef] [PubMed]
- van Blitterswijk, M.; Vlam, L.; van Es, M.A.; van der Pol, W.-L.; Hennekam, E.A.M.; Dooijes, D.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; et al. Genetic Overlap between Apparently Sporadic Motor Neuron Diseases. PLoS ONE 2012, 7, e48983. [Google Scholar] [CrossRef]
- Cruz, S.D.; Cleveland, D.W. Understanding the Role of TDP-43 and FUS/TLS in ALS and Beyond. Curr. Opin. Neurobiol. 2011, 21, 904. [Google Scholar] [CrossRef]
- Fischbeck, K.H. Spinal and Bulbar Muscular Atrophy Overview. J. Mol. Neurosci. 2016, 58, 317–320. [Google Scholar] [CrossRef]
- Katsuno, M.; Tanaka, F.; Adachi, H.; Banno, H.; Suzuki, K.; Watanabe, H.; Sobue, G. Pathogenesis and Therapy of Spinal and Bulbar Muscular Atrophy (SBMA). Prog. Neurobiol. 2012, 99, 246–256. [Google Scholar] [CrossRef]
- Katsuno, M.; Banno, H.; Suzuki, K.; Adachi, H.; Tanaka, F.; Sobue, G. Clinical Features and Molecular Mechanisms of Spinal and Bulbar Muscular Atrophy (SBMA). Adv. Exp. Med. Biol. 2010, 685, 64–74. [Google Scholar] [CrossRef]
- Spada, A.L. Spinal and Bulbar Muscular Atrophy. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Cortes, C.J.; La Spada, A.R. X-Linked Spinal and Bulbar Muscular Atrophy: From Clinical Genetic Features and Molecular Pathology to Mechanisms Underlying Disease Toxicity. Adv. Exp. Med. Biol. 2018, 1049, 103–133. [Google Scholar] [CrossRef]
- Katsuno, M.; Adachi, H.; Tanaka, F.; Sobue, G. Spinal and Bulbar Muscular Atrophy: Ligand-Dependent Pathogenesis and Therapeutic Perspectives. J. Mol. Med. 2004, 82, 298–307. [Google Scholar] [CrossRef]
- Ranganathan, S.; Harmison, G.G.; Meyertholen, K.; Pennuto, M.; Burnett, B.G.; Fischbeck, K.H. Mitochondrial Abnormalities in Spinal and Bulbar Muscular Atrophy. Hum. Mol. Genet. 2009, 18, 27–42. [Google Scholar] [CrossRef]
- Feng, X.; Cheng, X.-T.; Zheng, P.; Li, Y.; Hakim, J.; Zhang, S.Q.; Anderson, S.M.; Linask, K.; Prestil, R.; Zou, J.; et al. Ligand-Free Mitochondria-Localized Mutant AR-Induced Cytotoxicity in Spinal Bulbar Muscular Atrophy. Brain 2023, 146, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Stiene, D.; Song, T.; Sadayappan, S. Distal Arthrogryposis and Lethal Congenital Contracture Syndrome—An Overview. Front. Physiol. 2020, 11, 689. [Google Scholar] [CrossRef] [PubMed]
- Pakkasjärvi, N.; Ritvanen, A.; Herva, R.; Peltonen, L.; Kestilä, M.; Ignatius, J. Lethal Congenital Contracture Syndrome (LCCS) and Other Lethal Arthrogryposes in Finland—An Epidemiological Study. Am. J. Med. Genet. A 2006, 140A, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Lethal Congenital Contracture Syndrome 1-NIH Genetic Testing Registry (GTR)-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gtr/conditions/C1854664/ (accessed on 25 November 2024).
- Vuopala, K.; Herva, R. Lethal Congenital Contracture Syndrome: Further Delineation and Genetic Aspects. J. Med. Genet. 1994, 31, 521–527. [Google Scholar] [CrossRef]
- Nousiainen, H.O.; Kestilä, M.; Pakkasjärvi, N.; Honkala, H.; Kuure, S.; Tallila, J.; Vuopala, K.; Ignatius, J.; Herva, R.; Peltonen, L. Mutations in mRNA Export Mediator GLE1 Result in a Fetal Motoneuron Disease. Nat. Genet. 2008, 40, 155–157. [Google Scholar] [CrossRef]
- Narkis, G.; Ofir, R.; Landau, D.; Manor, E.; Volokita, M.; Hershkowitz, R.; Elbedour, K.; Birk, O.S. Lethal Contractural Syndrome Type 3 (LCCS3) Is Caused by a Mutation in PIP5K1C, Which Encodes PIPKI Gamma of the Phophatidylinsitol Pathway. Am. J. Hum. Genet. 2007, 81, 530–539. [Google Scholar] [CrossRef]
- Noches, V.; Campos-Melo, D.; Droppelmann, C.A.; Strong, M.J. Epigenetics in the Formation of Pathological Aggregates in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2024, 17, 14179611417961. [Google Scholar] [CrossRef]
- Martin, L.J.; Wong, M. Aberrant Regulation of DNA Methylation in Amyotrophic Lateral Sclerosis: A New Target of Disease Mechanisms. Neurotherapeutics 2013, 10, 722–733. [Google Scholar] [CrossRef]
- Tsekrekou, M.; Giannakou, M.; Papanikolopoulou, K.; Skretas, G. Protein Aggregation and Therapeutic Strategies in SOD1- and TDP-43- Linked ALS. Front. Mol. Biosci. 2024, 11, 1383453. [Google Scholar] [CrossRef]
- Al-Mahdawi, S.; Virmouni, S.A.; Pook, M.A. The Emerging Role of 5-Hydroxymethylcytosine in Neurodegenerative Diseases. Front. Neurosci. 2014, 8, 397. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 Inhibition Reverses Axonal Transport Defects in Motor Neurons Derived from FUS-ALS Patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; Boudreau, M.; Kriz, J.; Couillard-Després, S.; Kaplan, D.R.; Julien, J.-P. Cell Cycle Regulators in the Neuronal Death Pathway of Amyotrophic Lateral Sclerosis Caused by Mutant Superoxide Dismutase 1. J. Neurosci. 2003, 23, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in Amyotrophic Lateral Sclerosis: A Role for Histone Post Translational Modifications in Neurodegenerative Disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef]
- Laneve, P.; Tollis, P.; Caffarelli, E. RNA Deregulation in Amyotrophic Lateral Sclerosis: The Noncoding Perspective. Int. J. Mol. Sci. 2021, 22, 10285. [Google Scholar] [CrossRef]
- Yashooa, R.K.; Duranti, E.; Conconi, D.; Lavitrano, M.; Mustafa, S.A.; Villa, C. Mitochondrial microRNAs: Key Drivers in Unraveling Neurodegenerative Diseases. Int. J. Mol. Sci. 2025, 26, 626. [Google Scholar] [CrossRef]
- Zheleznyakova, G.Y.; Voisin, S.; Kiselev, A.V.; Sällman Almén, M.; Xavier, M.J.; Maretina, M.A.; Tishchenko, L.I.; Fredriksson, R.; Baranov, V.S.; Schiöth, H.B. Genome-Wide Analysis Shows Association of Epigenetic Changes in Regulators of Rab and Rho GTPases with Spinal Muscular Atrophy Severity. Eur. J. Hum. Genet. 2013, 21, 988–993. [Google Scholar] [CrossRef]
- Lunke, S.; El-Osta, A. The Emerging Role of Epigenetic Modifications and Chromatin Remodeling in Spinal Muscular Atrophy. J. Neurochem. 2009, 109, 1557–1569. [Google Scholar] [CrossRef]
- Marasco, L.E.; Dujardin, G.; Sousa-Luís, R.; Liu, Y.H.; Stigliano, J.; Nomakuchi, T.; Proudfoot, N.J.; Krainer, A.R.; Kornblihtt, A.R. Counteracting Chromatin Effects of a Splicing-Correcting Antisense Oligonucleotide Improves Its Therapeutic Efficacy in Spinal Muscular Atrophy. Cell 2022, 185, 2057–2070.e15. [Google Scholar] [CrossRef]
- Coppedè, F. Epigenetics of Neuromuscular Disorders. Epigenomics 2020, 12, 2125–2139. [Google Scholar] [CrossRef]
- Riluzole: Package Insert/Prescribing Information. Available online: https://www.drugs.com/pro/riluzole.html (accessed on 25 February 2025).
- Wang, S.-J.; Wang, K.-Y.; Wang, W.-C. Mechanisms Underlying the Riluzole Inhibition of Glutamate Release from Rat Cerebral Cortex Nerve Terminals (Synaptosomes). Neuroscience 2004, 125, 191–201. [Google Scholar] [CrossRef]
- Saitoh, Y.; Takahashi, Y. Riluzole for the Treatment of Amyotrophic Lateral Sclerosis. Neurodegener. Dis. Manag. 2020, 10, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Roch-Torreilles, I.; Camu, W.; Hillaire-Buys, D. Adverse efects of riluzole (Rilutek) in the treatment of amyotrophic lateral sclerosis. Therapie 2000, 55, 303–312. [Google Scholar] [PubMed]
- Cantara, S.; Simoncelli, G.; Ricci, C. Antisense Oligonucleotides (ASOs) in Motor Neuron Diseases: A Road to Cure in Light and Shade. Int. J. Mol. Sci. 2024, 25, 4809. [Google Scholar] [CrossRef]
- Cerillo, J.L.; Parmar, M. Tofersen. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef]
- Ogbonmide, T.; Rathore, R.; Rangrej, S.B.; Hutchinson, S.; Lewis, M.; Ojilere, S.; Carvalho, V.; Kelly, I. Gene Therapy for Spinal Muscular Atrophy (SMA): A Review of Current Challenges and Safety Considerations for Onasemnogene Abeparvovec (Zolgensma). Cureus 2023, 15, e36197. [Google Scholar] [CrossRef]
- Paik, J. Risdiplam: A Review in Spinal Muscular Atrophy. CNS Drugs 2022, 36, 401–410. [Google Scholar] [CrossRef]
- Finkel, R.S.; Hughes, S.H.; Parker, J.; Civitello, M.; Lavado, A.; Mefford, H.C.; Mueller, L.; Kletzl, H. Risdiplam for Prenatal Therapy of Spinal Muscular Atrophy. N. Engl. J. Med. 2025, 392, 1138–1140. [Google Scholar] [CrossRef]
- Kaneko, K.; Hoskin, J.; Hodis, B. Primary Lateral Sclerosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- McDermott, C.J.; Taylor, R.W.; Hayes, C.; Johnson, M.; Bushby, K.M.D.; Turnbull, D.M.; Shaw, P.J. Investigation of Mitochondrial Function in Hereditary Spastic Paraparesis. Neuroreport 2003, 14, 485–488. [Google Scholar] [CrossRef]
- Bellofatto, M.; De Michele, G.; Iovino, A.; Filla, A.; Santorelli, F.M. Management of Hereditary Spastic Paraplegia: A Systematic Review of the Literature. Front. Neurol. 2019, 10, 3. [Google Scholar] [CrossRef]
- Lim, W.F.; Forouhan, M.; Roberts, T.C.; Dabney, J.; Ellerington, R.; Speciale, A.A.; Manzano, R.; Lieto, M.; Sangha, G.; Banerjee, S.; et al. Gene Therapy with AR Isoform 2 Rescues Spinal and Bulbar Muscular Atrophy Phenotype by Modulating AR Transcriptional Activity. Sci. Adv. 2021, 7, eabi6896. [Google Scholar] [CrossRef]
- Rossi Sebastiano, M.; Ermondi, G.; Sato, K.; Otomo, A.; Hadano, S.; Caron, G. Personalized Treatment for Infantile Ascending Hereditary Spastic Paralysis Based on In Silico Strategies. Molecules 2022, 27, 7063. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Liu, C.-Y.; Che, C.-H.; Huang, H.-P. Toward Precision Medicine in Amyotrophic Lateral Sclerosis. Ann. Transl. Med. 2016, 4, 27. [Google Scholar] [CrossRef]
- Mishra, J.; Bhatti, G.K.; Sehrawat, A.; Singh, C.; Singh, A.; Reddy, A.P.; Reddy, P.H.; Bhatti, J.S. Modulating Autophagy and Mitophagy as a Promising Therapeutic Approach in Neurodegenerative Disorders. Life Sci. 2022, 311, 121153. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; Manai, F.; Davinelli, S.; Tucci, P.; Saso, L.; Amadio, M. Novel Potential Pharmacological Applications of Dimethyl Fumarate—an Overview and Update. Front. Pharmacol. 2023, 14, 1264842. [Google Scholar] [CrossRef]
- Mayer, C.; Riera-Ponsati, L.; Kauppinen, S.; Klitgaard, H.; Erler, J.T.; Hansen, S.N. Targeting the NRF2 Pathway for Disease Modification in Neurodegenerative Diseases: Mechanisms and Therapeutic Implications. Front. Pharmacol. 2024, 15, 1437939. [Google Scholar] [CrossRef]
- Boros, B.D.; Schoch, K.M.; Kreple, C.J.; Miller, T.M. Antisense Oligonucleotides for the Study and Treatment of ALS. Neurotherapeutics 2022, 19, 1145–1158. [Google Scholar] [CrossRef]

{kind=link}
| Gene Name | Encoded Protein | Chromosomal Locus | Pathogenic Mechanism | Ref. |
|---|---|---|---|---|
| SOD1 | Superoxide dismutase 1 | 21q22.11 | Mutant SOD1 misfolds, aggregates, and induces oxidative stress, mitochondrial dysfunction, and motor neuron death. | [39,46] |
| TARDBP | TAR DNA-binding protein 43 | 1p36.22 | TDP-43 mislocalizes, forming toxic aggregates that disrupt RNA processing, autophagy, and neuronal homeostasis. | [62] |
| FUS | Fused in sarcoma | 16p11.2 | Cytoplasmic mislocalization causes toxic aggregation, impaired RNA metabolism, and stress granule dysfunction. | [68] |
| C9orf72 | C9orf72 protein | 9p21.2 | G4C2 repeat expansion forms RNA foci, toxic dipeptides, disrupts autophagy, and induces neurotoxicity. | [69,70] |
| ALS2 | Alsin | 2q33.1 | Loss of function disrupts endosomal trafficking, autophagy, and intracellular transport, causing motor neuron degeneration. | [71] |
| ATXN2 | Ataxin-2 | 12q24.12 | PolyQ expansions enhance TDP-43 aggregation, impair RNA metabolism, and increase ALS risk. | [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonakoudis, A.; Kyriakoudi, S.A.; Chatzi, D.; Dermitzakis, I.; Gargani, S.; Meditskou, S.; Manthou, M.E.; Theotokis, P. Genetic Basis of Motor Neuron Diseases: Insights, Clinical Management, and Future Directions. Int. J. Mol. Sci. 2025, 26, 4904. https://doi.org/10.3390/ijms26104904
Antonakoudis A, Kyriakoudi SA, Chatzi D, Dermitzakis I, Gargani S, Meditskou S, Manthou ME, Theotokis P. Genetic Basis of Motor Neuron Diseases: Insights, Clinical Management, and Future Directions. International Journal of Molecular Sciences. 2025; 26(10):4904. https://doi.org/10.3390/ijms26104904
Chicago/Turabian StyleAntonakoudis, Apostolos, Stella Aikaterini Kyriakoudi, Despoina Chatzi, Iasonas Dermitzakis, Sofia Gargani, Soultana Meditskou, Maria Eleni Manthou, and Paschalis Theotokis. 2025. "Genetic Basis of Motor Neuron Diseases: Insights, Clinical Management, and Future Directions" International Journal of Molecular Sciences 26, no. 10: 4904. https://doi.org/10.3390/ijms26104904
APA StyleAntonakoudis, A., Kyriakoudi, S. A., Chatzi, D., Dermitzakis, I., Gargani, S., Meditskou, S., Manthou, M. E., & Theotokis, P. (2025). Genetic Basis of Motor Neuron Diseases: Insights, Clinical Management, and Future Directions. International Journal of Molecular Sciences, 26(10), 4904. https://doi.org/10.3390/ijms26104904