
Harnessing Genomics for Breeding Lantana camara L.: Genotyping and Ploidy Testing of Clonal Lines Through ddRADseq Applications



Abstract
1. Introduction
2. Results
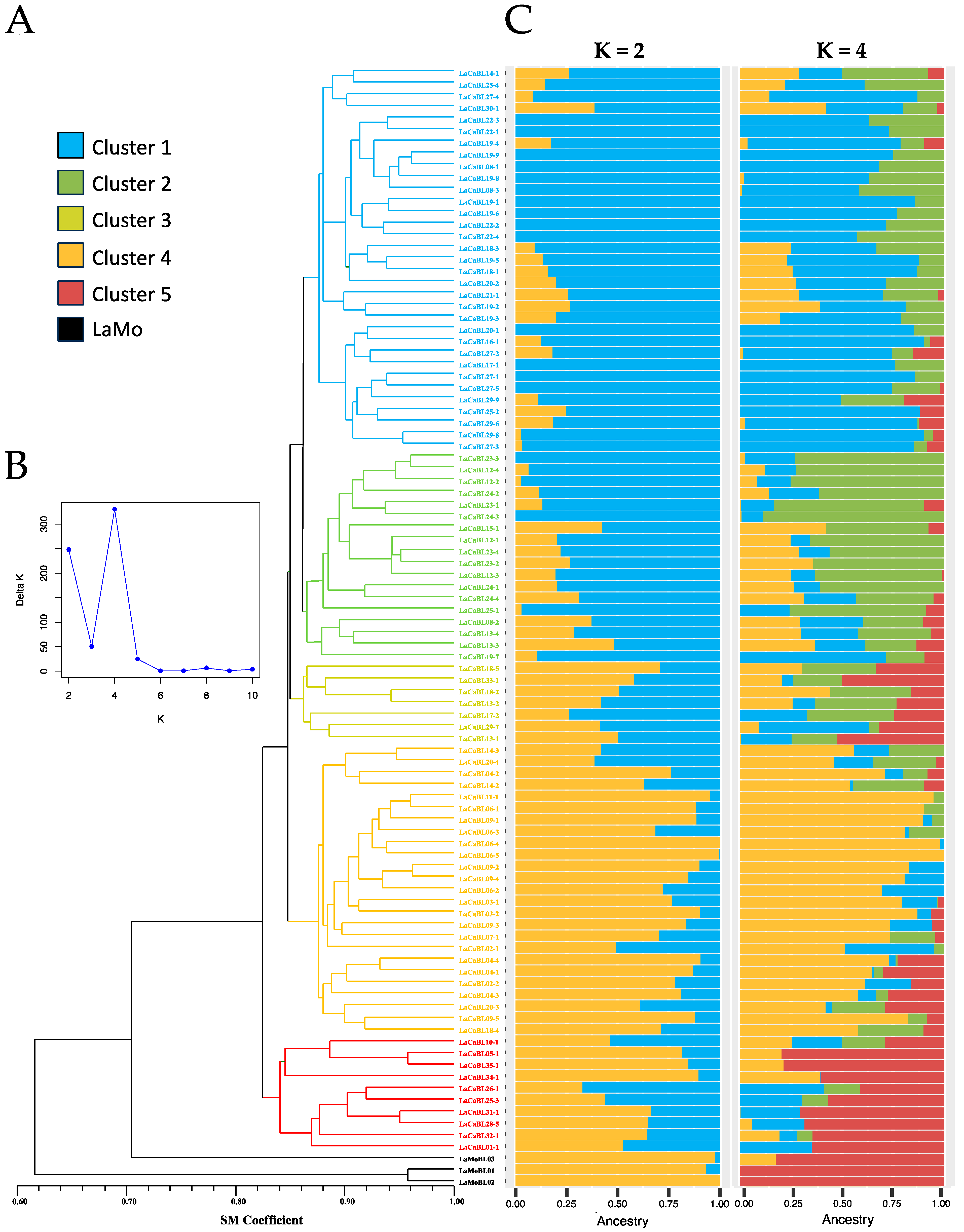
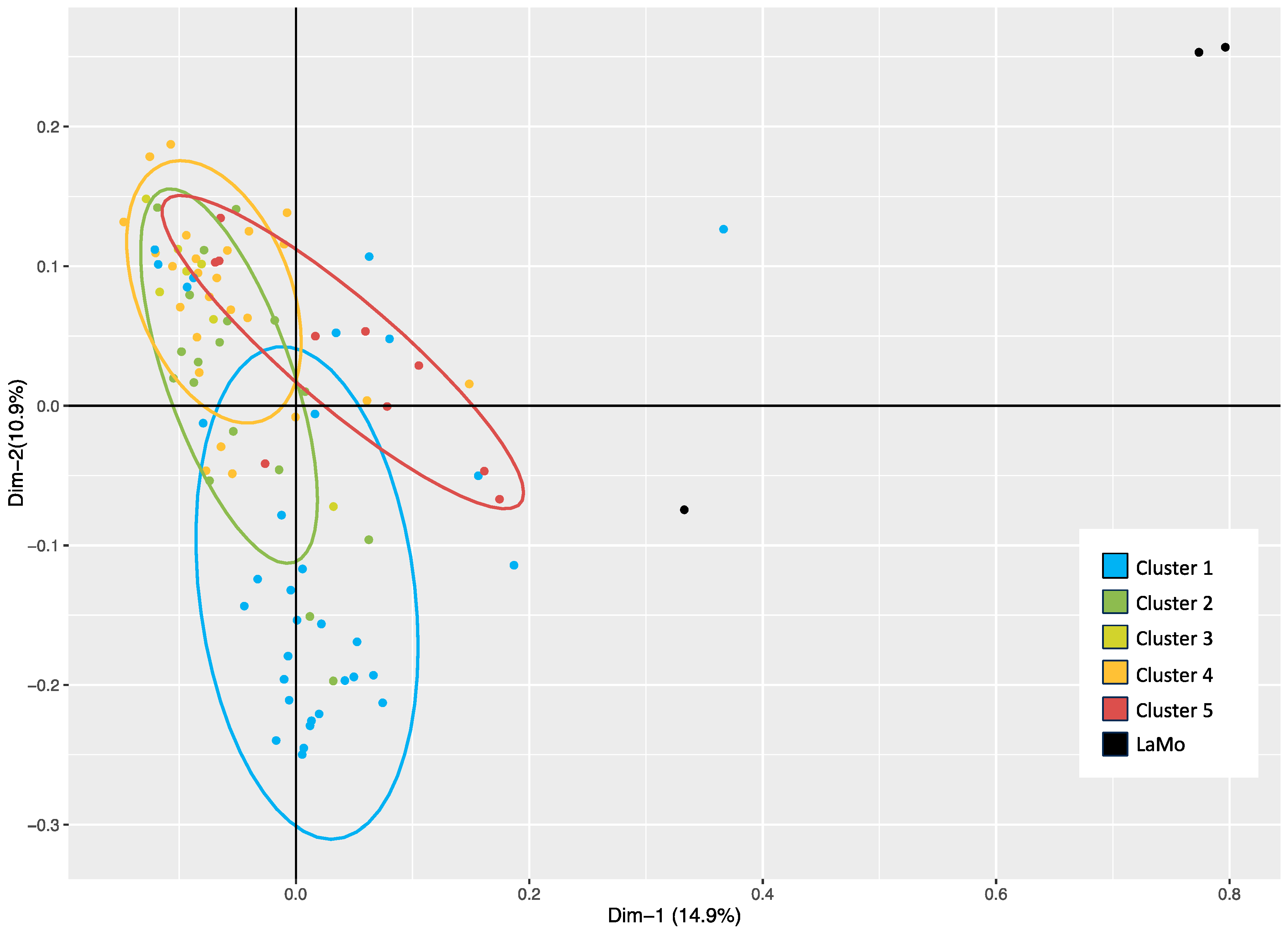
2.1. Genetic Similarity Estimates and Genetic Structure Analysis of the Breeding Population
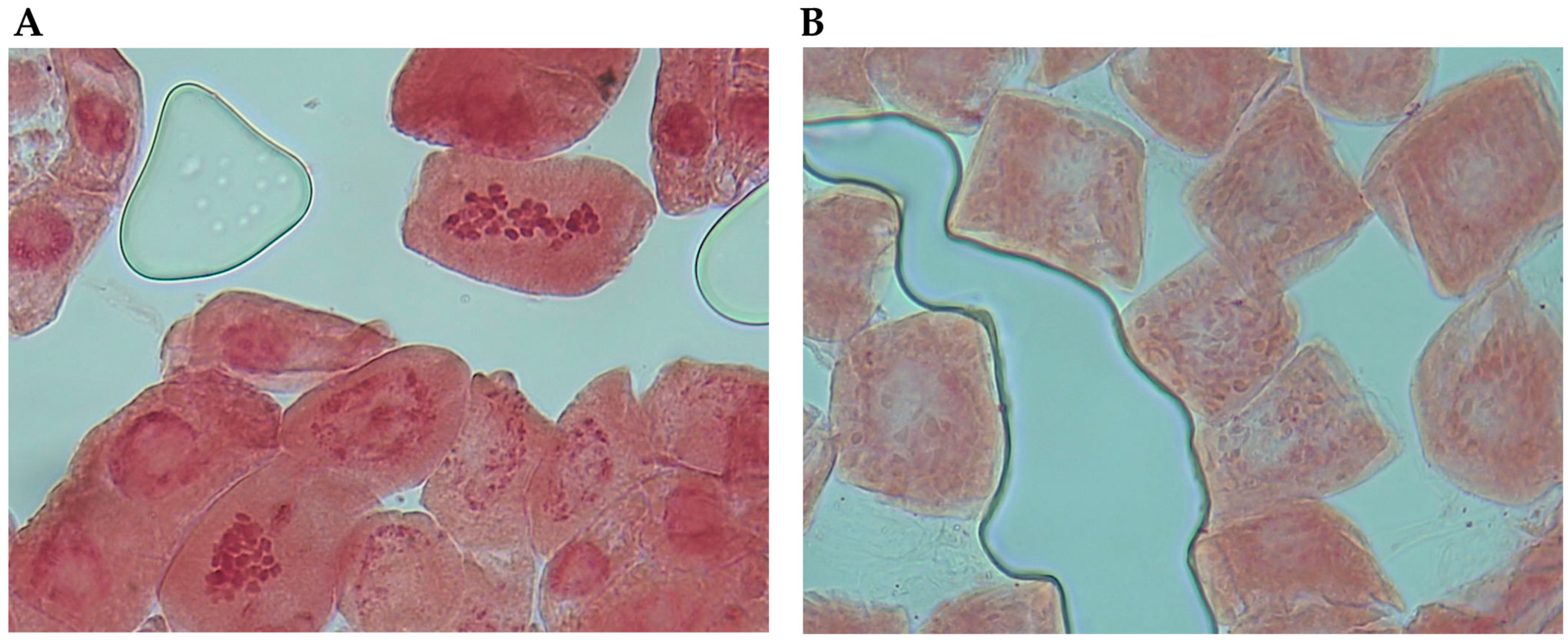
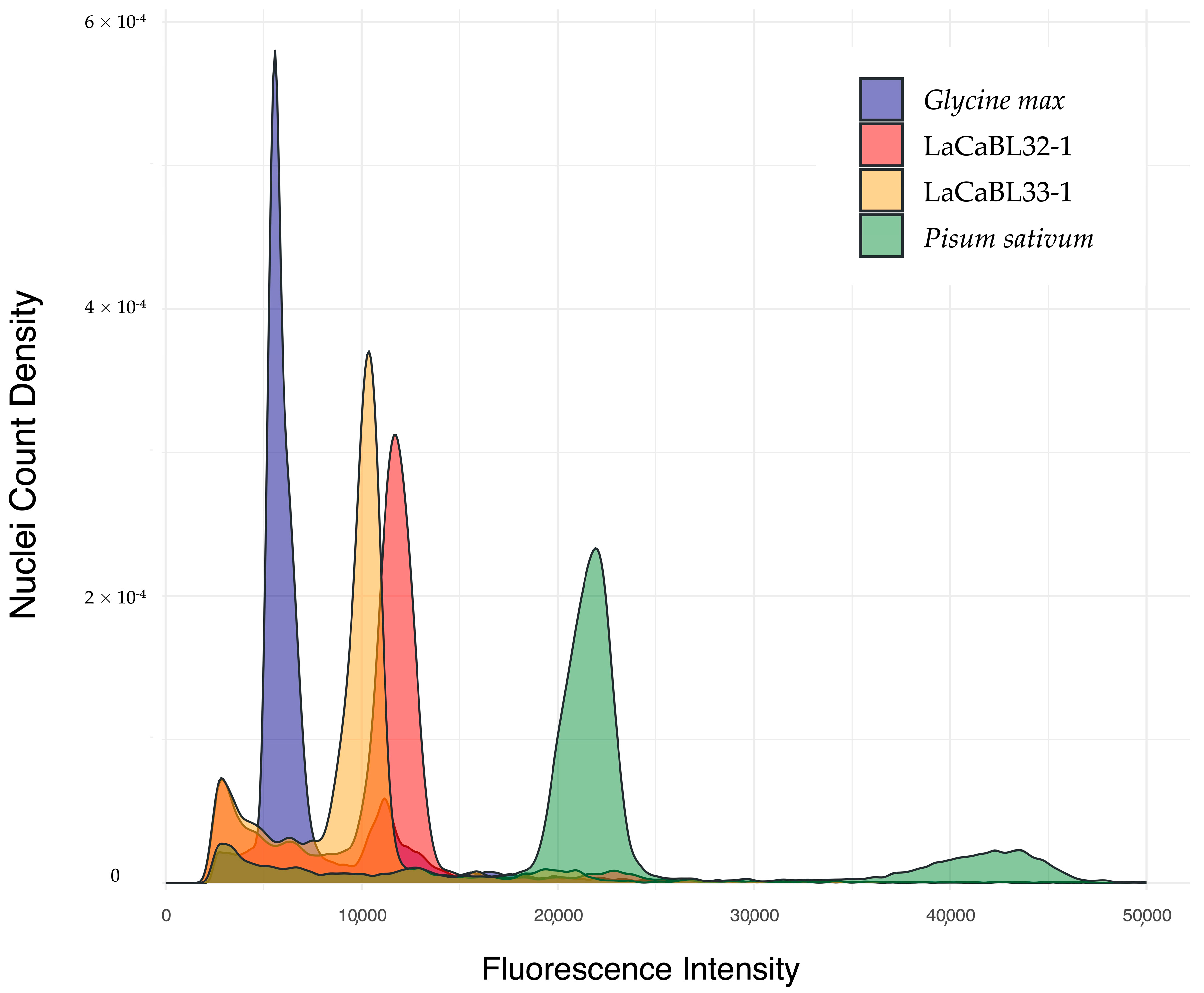
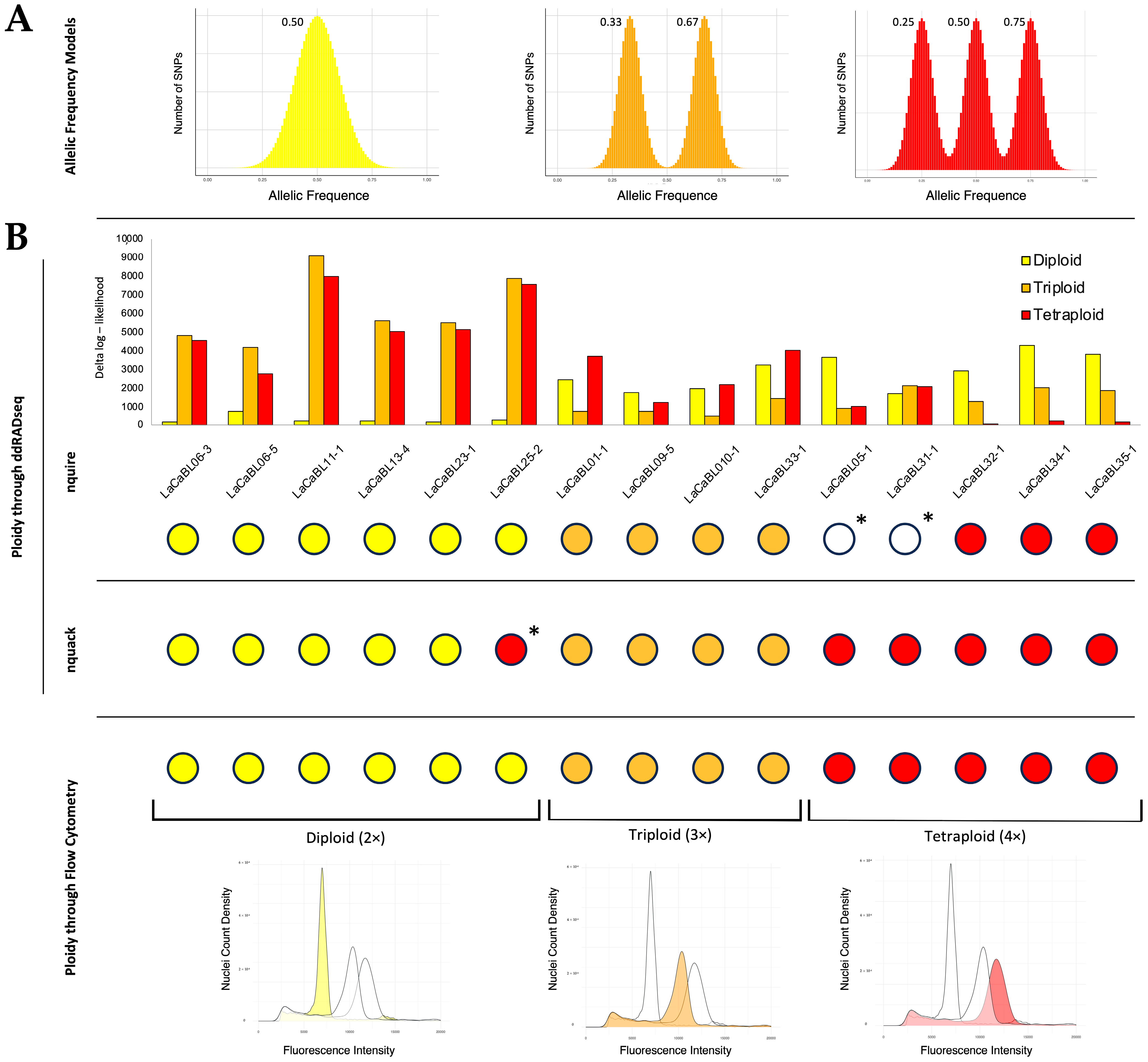
2.2. Ploidy Assessment by Flow Cytometry
2.3. Ploidy Prediction Using ddRADseq Data
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. ddRADseq Library Preparation
4.3. ddRADseq Data Analysis for Assessing Genetic Diversity in Lantana Clonal Lines
4.4. Flow Cytometry, Chromosome Counting and ddRADseq Data Analysis for Ploidy Assessment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Day, M.; Wileym, C.; Playford, J.; Zalucki, M. Lantana: Current Management, Status and Future Prospects. Available online: https://www.researchgate.net/publication/227364997_Lantana_Current_Management_Status_and_Future_Prospects (accessed on 26 October 2024).
- Priyanka, N.; Joshi, P. A Review of Lantana Camara Studies in India. Int. J. Sci. Res. Publ. 2013, 3. [Google Scholar]
- Taylor, S.; Kumar, L.; Reid, N. Impacts of Climate Change and Land-Use on the Potential Distribution of an Invasive Weed: A Case Study of Lantana camara in Australia. Weed Res. 2012, 52, 391–401. [Google Scholar] [CrossRef]
- Ghisalberti, E.L. Lantana camara L. (Verbenaceae). Fitoterapia 2000, 71, 467–486. [Google Scholar] [CrossRef]
- Mansoori, A.; Singh, N.; Dubey, S.K.; Thakur, T.K.; Alkan, N.; Das, S.N.; Kumar, A. Phytochemical Characterization and Assessment of Crude Extracts From Lantana camara L. for Antioxidant and Antimicrobial Activity. Front. Agron. 2020, 2, 582268. [Google Scholar] [CrossRef]
- Sousa, E.O.; Miranda, C.M.B.A.; Nobre, C.B.; Boligon, A.A.; Athayde, M.L.; Costa, J.G.M. Phytochemical Analysis and Antioxidant Activities of Lantana camara and Lantana Montevidensis Extracts. Ind. Crops. Prod. 2015, 70, 7–15. [Google Scholar] [CrossRef]
- Kumar, S.; Sandhir, R.; Ojha, S. Evaluation of Antioxidant Activity and Total Phenol in Different Varieties of Lantana camara Leaves. BMC Res. Notes 2014, 7, 560. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.; Shackleton, C.M.; Uma Shaanker, R. Reconstructing the History of Introduction and Spread of the Invasive Species, Lantana, at Three Spatial Scales in India. Biol. Invasions 2013, 15, 1287–1302. [Google Scholar] [CrossRef]
- Kato-Noguchi, H.; Kurniadie, D. Allelopathy of Lantana camara as an Invasive Plant. Plants 2021, 10, 1028. [Google Scholar] [CrossRef]
- Taylor, S.; Kumar, L.; Reid, N.; Kriticos, D.J. Climate Change and the Potential Distribution of an Invasive Shrub, Lantana camara L. PLoS ONE 2012, 7, e35565. [Google Scholar] [CrossRef]
- Simelane, D.O.; Katembo, N.; Mawela, K.V. Current Status of Biological Control of Lantana camara L. (sensu lato) in South Africa. Afr. Entomol. 2021, 29, 775–783. [Google Scholar] [CrossRef]
- Lowe, S.; Browne, M.; Boudjelas, S.; De Poorter, M. 100 of the World’s Worst Invasive Alien Species: A Selection from the Global Invasive Species Database. Auckl. Invasive Species Spec. Group 2000, 12, 12. [Google Scholar]
- Deng, Z.; Wilson, S.B.; Ying, X.; Chen, C.; Freyre, R.; Zayas, V.; Czarnecki, D.M. ‘UF-1013-1’: An Infertile Cultivar of Lantana Camara. HortScience 2020, 55, 953–958. [Google Scholar] [CrossRef]
- Sanders, R.W. Identity of Lantana Depressa and L. Ovatifolia (Verbenaceae) of Florida and the Bahamas. Syst. Bot. 1987, 12, 44. [Google Scholar] [CrossRef]
- Maschinski, J.; Sirkin, E.; Fant, J. Using Genetic and Morphological Analysis to Distinguish Endangered Taxa from Their Hybrids with the Cultivated Exotic Pest Plant Lantana Strigocamara (Syn: Lantana camara). Conserv. Genet. 2010, 11, 1607–1621. [Google Scholar] [CrossRef]
- Rambuda, T.D.; Johnson, S.D. Breeding Systems of Invasive Alien Plants in South Africa: Does Baker’s Rule Apply? Divers. Distrib. 2004, 10, 409–416. [Google Scholar] [CrossRef]
- Mathur, G.; Mohan Ram, H.Y. Significance of Petal Colour in Thrips-Pollinated Lantana camara L. Ann. Bot. 1978, 42, 1473–1476. [Google Scholar] [CrossRef]
- Negi, G.C.S.; Sharma, S.; Vishvakarma, S.C.R.; Samant, S.S.; Maikhuri, R.K.; Prasad, R.C.; Palni, L.M.S. Ecology and Use of Lantana camara in India. Bot. Rev. 2019, 85, 109–130. [Google Scholar] [CrossRef]
- Praveen, P.; Gopal, R.; Ramakrishnan, U. The Population Structure of Invasive Lantana Camara Is Shaped by Its Mating System. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ojha, B.M.; Dayal, N. Cytological Investigations in the Genus Lantana in India. Cytologia 1992, 57, 9–13. [Google Scholar] [CrossRef]
- Czarnecki, D.M.; Hershberger, A.J.; Robacker, C.D.; Clark, D.G.; Deng, Z. Ploidy Levels and Pollen Stainability of Lantana Camara Cultivars and Breeding Lines. HortScience 2014, 49, 1271–1276. [Google Scholar] [CrossRef]
- Czarnecki, D.M.; Deng, Z. Assessment of the Female Fertility of 26 Commercial Lantana Camara Cultivars and Six Experimental Lines. HortScience 2020, 55, 709–715. [Google Scholar] [CrossRef]
- Czarnecki, D.M.; Deng, Z. Occurrence of Unreduced Female Gametes Leads to Sexual Polyploidization in Lantana. J. Am. Soc. Hortic. Sci. 2009, 134, 560–566. [Google Scholar] [CrossRef]
- Parrish, S.B.; Deng, Z. The First High-Quality Genome Assembly and Annotation of Lantana Camara, an Important Ornamental Plant and a Major Invasive Species. Hortic. Adv. 2024, 2, 14. [Google Scholar] [CrossRef]
- Parrish, S.B.; Paudel, D.; Deng, Z. Transcriptome Analysis of Lantana Camara Flower Petals Reveals Candidate Anthocyanin Biosynthesis Genes Mediating Red Flower Color Development. G3 Genes Genomes Genet. 2023, 14, jkad259. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Alharby, H.F.; Hakeem, K.R.; Ali, N.; Rahman, I.U.; Munawar, M.; Anwar, Y. De novo Transcriptome Analysis of Lantana camara L. Revealed Candidate Genes Involved in Phenylpropanoid Biosynthesis Pathway. Sci. Rep. 2020, 10, 13726. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Bhattarai, K.; Parajuli, S.; Cao, Z.; Deng, Z. Transcriptome Analysis of Young Ovaries Reveals Candidate Genes Involved in Gamete Formation in Lantana camara. Plants 2019, 8, 263. [Google Scholar] [CrossRef]
- Explanatory Notes on Essentially Derived Varieties under the 1991 Act of the UPOV Convention. Available online: https://www.upov.int/edocs/expndocs/en/upov_exn_edv.pdf (accessed on 4 May 2025).
- Yu, J.K.; Chung, Y.S. Plant Variety Protection: Current Practices and Insights. Genes 2021, 12, 1127. [Google Scholar] [CrossRef]
- CPVO Legislation in Force. Available online: https://cpvo.europa.eu/en/about-us/law-and-practice/legislation-in-force (accessed on 27 October 2024).
- UPOV Lex. Available online: https://upovlex.upov.int/en/convention (accessed on 27 October 2024).
- Button, P. New Developments in the International Union for the Protection of New Varieties of Plants (UPOV). Acta Hortic 2006, 714, 195–210. [Google Scholar] [CrossRef]
- Ekvad, M. The CPVO and the EU-Wide System of Plant Variety Protection. Stockh. Intellect. Prop. Law Rev. 2021, 1, 12–13. [Google Scholar]
- CPVO National Guidelines. Available online: https://cpvo.europa.eu/en/applications-and-examinations/technical-examinations/technical-protocols/national-guidelines?t=lantana&field_crop_sector_tid=All (accessed on 26 October 2024).
- CPVO Table of Characteristics for the Conduct of Tests for Distinctness, Uniformity and Stability—Lantana L. Available online: https://cpvo.europa.eu/sites/default/files/documents/TP/National_Guidelines/Lantana_DE.pdf (accessed on 4 May 2025).
- Gilliland, T.J.; Gensollen, V. Review of the Protocols Used for Assessment of DUS and VCU in Europe—Perspectives. In Proceedings of the Sustainable use of Genetic Diversity in Forage and Turf Breeding; Huyghe, C., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 261–275. [Google Scholar] [CrossRef]
- Farinati, S.; Betto, A.; Palumbo, F.; Scariolo, F.; Vannozzi, A.; Barcaccia, G. The New Green Challenge in Urban Planning: The Right Genetics in the Right Place. Horticulturae 2022, 8, 761. [Google Scholar] [CrossRef]
- Yu, Q.; Ling, Y.; Xiong, Y.; Zhao, W.; Xiong, Y.; Dong, Z.; Yang, J.; Zhao, J.; Zhang, X.; Ma, X. RAD-Seq as an Effective Strategy for Heterogenous Variety Identification in Plants—A Case Study in Italian Ryegrass (Lolium multiflorum). BMC Plant Biol. 2022, 22, 231. [Google Scholar] [CrossRef] [PubMed]
- Karikari, B.; Li, S.; Bhat, J.A.; Cao, Y.; Kong, J.; Yang, J.; Gai, J.; Zhao, T. Genome-Wide Detection of Major and Epistatic Effect QTLs for Seed Protein and Oil Content in Soybean Under Multiple Environments Using High-Density Bin Map. Int. J. Mol. Sci. 2019, 20, 979. [Google Scholar] [CrossRef] [PubMed]
- Seki, K.; Komatsu, K.; Hiraga, M.; Tanaka, K.; Uno, Y.; Matsumura, H. Development of PCR-Based Marker for Resistance to Fusarium Wilt Race 2 in Lettuce (Lactuca sativa L.). Euphytica 2021, 217, 126. [Google Scholar] [CrossRef]
- Krishnan, G.S.; Singh, A.K.; Waters, D.L.E.; Henry, R.J. Molecular Markers for Harnessing Heterosis. Mol. Markers Plants 2012, 119–136. [Google Scholar] [CrossRef]
- Bohra, A.; Jha, U.C.; Kishor, K.B.K.; Pandey, S.; Singh, N.P. Genomics and Molecular Breeding in Lesser Explored Pulse Crops: Current Trends and Future Opportunities. Biotechnol. Adv. 2014, 32, 1410–1428. [Google Scholar] [CrossRef]
- Simko, I.; Jia, M.; Venkatesh, J.; Kang, B.C.; Weng, Y.; Barcaccia, G.; Lanteri, S.; Bhattarai, G.; Foolad, M.R. Genomics and Marker-Assisted Improvement of Vegetable Crops. CRC Crit. Rev. Plant Sci. 2021, 40, 303–365. [Google Scholar] [CrossRef]
- Park, J.S.; Kang, M.Y.; Shim, E.J.; Oh, J.H.; Seo, K.I.; Kim, K.S.; Sim, S.C.; Chung, S.M.; Park, Y.; Lee, G.P.; et al. Genome-Wide Core Sets of SNP Markers and Fluidigm Assays for Rapid and Effective Genotypic Identification of Korean Cultivars of Lettuce (Lactuca sativa L.). Hortic. Res. 2022, 9, uhac119. [Google Scholar] [CrossRef]
- Smulders, M.J.M.; Arens, P. New Developments in Molecular Techniques for Breeding in Ornamentals. In Ornamental Crops. Handbook of Plant Breeding; Van Huylenbroeck, J., Ed.; Springer: Cham, Switzerland, 2018; Volume 11, pp. 213–230. ISBN 978-3-319-90698-0. [Google Scholar]
- Yagi, M.; Shirasawa, K.; Waki, T.; Kume, T.; Isobe, S.; Tanase, K.; Yamaguchi, H. Construction of an SSR and RAD Marker-Based Genetic Linkage Map for Carnation (Dianthus caryophyllus L.). Plant Mol. Biol. Rep. 2017, 35, 110–117. [Google Scholar] [CrossRef]
- CPVO Guidance on the Use of Biochemical and Molecular Markers in the Examination of Distinctiveness, Uniformity and Stability (DUS). Available online: https://www.upov.int/edocs/tgpdocs/en/tgp_15.pdf (accessed on 4 May 2025).
- Akbarzadeh, M.; Van Laere, K.; Leus, L.; De Riek, J.; Van Huylenbroeck, J.; Werbrouck, S.P.O.; Dhooghe, E. Can Knowledge of Genetic Distances, Genome Sizes and Chromosome Numbers Support Breeding Programs in Hardy Geraniums? Genes 2021, 12, 730. [Google Scholar] [CrossRef]
- Trojak-Goluch, A.; Kawka-Lipińska, M.; Wielgusz, K.; Praczyk, M. Polyploidy in Industrial Crops: Applications and Perspectives in Plant Breeding. Agronomy 2021, 11, 2574. [Google Scholar] [CrossRef]
- Zenil-Ferguson, R.; Burleigh, J.G.; Freyman, W.A.; Igić, B.; Mayrose, I.; Goldberg, E.E. Interaction among Ploidy, Breeding System and Lineage Diversification. New Phytol. 2019, 224, 1252–1265. [Google Scholar] [CrossRef]
- Eeckhaut, T.; Van der Veken, J.; Dhooghe, E.; Leus, L.; Van Laere, K.; Van Huylenbroeck, J. Ploidy Breeding in Ornamentals. Ornam. Crops 2018, 11, 145–173. [Google Scholar] [CrossRef]
- de Resende, K.F.M. Karyotype Evolution: Concepts and Applications. Chromosome Struct. Aberrations 2017, 11, 181–200. [Google Scholar] [CrossRef]
- Sliwinska, E.; Loureiro, J.; Leitch, I.J.; Šmarda, P.; Bainard, J.; Bureš, P.; Chumová, Z.; Horová, L.; Koutecký, P.; Lučanová, M.; et al. Application-Based Guidelines for Best Practices in Plant Flow Cytometry. Cytom. Part A 2021, 101, 749–781. [Google Scholar] [CrossRef] [PubMed]
- Viruel, J.; Hidalgo, O.; Pokorny, L.; Forest, F.; Gravendeel, B.; Wilkin, P.; Leitch, I.J. A Bioinformatic Pipeline to Estimate Ploidy Level from Target Capture Sequence Data Obtained from Herbarium Specimens. Methods Mol Biol. 2023, 2672, 115–126. [Google Scholar] [CrossRef]
- Viruel, J.; Conejero, M.; Hidalgo, O.; Pokorny, L.; Powell, R.F.; Forest, F.; Kantar, M.B.; Soto Gomez, M.; Graham, S.W.; Gravendeel, B.; et al. A Target Capture-Based Method to Estimate Ploidy From Herbarium Specimens. Front. Plant Sci. 2019, 10, 467104. [Google Scholar] [CrossRef]
- Gaynor, M.L.; Landis, J.B.; O’Connor, T.K.; Laport, R.G.; Doyle, J.J.; Soltis, D.E.; Ponciano, J.M.; Soltis, P.S. NQuack: An R Package for Predicting Ploidal Level from Sequence Data Using Site-Based Heterozygosity. Appl. Plant Sci. 2024, 12, e11606. [Google Scholar] [CrossRef]
- Weib, C.L.; Pais, M.; Cano, L.M.; Kamoun, S.; Burbano, H.A. NQuire: A Statistical Framework for Ploidy Estimation Using next Generation Sequencing. BMC Bioinform. 2018, 19, 122. [Google Scholar] [CrossRef]
- Doležel, J.; Greilhuber, J.; Suda, J. Estimation of Nuclear DNA Content in Plants Using Flow Cytometry. Nat. Protoc. 2007, 2, 2233–2244. [Google Scholar] [CrossRef]
- De Riek, J. Are Molecular Markers Strengthening Plant Variety Registration and Protection? Acta Hortic 2001, 552, 215–223. [Google Scholar] [CrossRef]
- Gilliland, T.J.; Annicchiarico, P.; Julier, B.; Ghesquière, M. A Proposal for Enhanced EU Herbage VCU and DUS Testing Procedures. Grass Forage Sci. 2020, 75, 227–241. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Zhigunov, A.V.; Ulianich, P.S.; Lebedeva, M.V.; Chang, P.L.; Nuzhdin, S.V.; Potokina, E.K. Development of F1 Hybrid Population and the High-Density Linkage Map for European Aspen (Populus tremula L.) Using RADseq Technology. BMC Plant Biol. 2017, 17, 180. [Google Scholar] [CrossRef]
- Scariolo, F.; Palumbo, F.; Farinati, S.; Barcaccia, G. Pipeline to Design Inbred Lines and F1 Hybrids of Leaf Chicory (Radicchio) Using Male Sterility and Genotyping-by-Sequencing. Plants 2023, 12, 1242. [Google Scholar] [CrossRef] [PubMed]
- Bohling, J. Evaluating the Effect of Reference Genome Divergence on the Analysis of Empirical RADseq Datasets. Ecol. Evol. 2020, 10, 7585–7601. [Google Scholar] [CrossRef]
- Dittberner, H.; Becker, C.; Jiao, W.B.; Schneeberger, K.; Hölzel, N.; Tellier, A.; de Meaux, J. Strengths and Potential Pitfalls of Hay Transfer for Ecological Restoration Revealed by RAD-Seq Analysis in Floodplain Arabis Species. Mol. Ecol. 2019, 28, 3887–3901. [Google Scholar] [CrossRef]
- Huang, P.H.; Wang, T.R.; Li, M.; Fang, O.Y.; Su, R.P.; Meng, H.H.; Song, Y.G.; Li, J. Different Reference Genomes Determine Different Results: Comparing SNP Calling in RAD-Seq of Engelhardia Roxburghiana Using Different Reference Genomes. Plant Sci. 2024, 344, 112109. [Google Scholar] [CrossRef]
- Shafer, A.B.A.; Peart, C.R.; Tusso, S.; Maayan, I.; Brelsford, A.; Wheat, C.W.; Wolf, J.B.W. Bioinformatic Processing of RAD-Seq Data Dramatically Impacts Downstream Population Genetic Inference. Methods Ecol. Evol. 2016, 8, 907–917. [Google Scholar] [CrossRef]
- Chiurugwi, T.; Kemp, S.; Powell, W.; Hickey, L.T. Speed Breeding Orphan Crops. Theor. Appl. Genet. 2019, 132, 607–616. [Google Scholar] [CrossRef]
- Russell, J.; Hackett, C.; Hedley, P.; Liu, H.; Milne, L.; Bayer, M.; Marshall, D.; Jorgensen, L.; Gordon, S.; Brennan, R. The Use of Genotyping by Sequencing in Blackcurrant (Ribes Nigrum): Developing High-Resolution Linkage Maps in Species without Reference Genome Sequences. Mol. Breed. 2014, 33, 835–849. [Google Scholar] [CrossRef]
- Johnson, M.; Deshpande, S.; Vetriventhan, M.; Upadhyaya, H.D.; Wallace, J.G. Genome-Wide Population Structure Analyses of Three Minor Millets: Kodo Millet, Little Millet, and Proso Millet. Plant Genome 2019, 12, 190021. [Google Scholar] [CrossRef] [PubMed]
- Lu-Irving, P.; Encinas-Viso, F.; Callander, J.; Day, M.D.; Le Roux, J. New Insights from Population Genomics into the Invasive Lantana Camara L. Species Complex. In Proceedings of the 21st Australasian Weeds Conference, Adelaide, Australia, 25–29 September 2022; p. 45. [Google Scholar]
- Ray, A.; Quader, S. Genetic Diversity and Population Structure of Lantana Camara in India Indicates Multiple Introductions and Gene Flow. Plant Biol. 2014, 16, 651–658. [Google Scholar] [CrossRef]
- Ray, A.; Sumangala, R.C.; Ravikanth, G.; Uma Shaanker, R.; Quader, S. Isolation and Characterization of Polymorphic Microsatellite Loci from the Invasive Plant Lantana camara L. Conserv. Genet. Resour. 2012, 4, 171–173. [Google Scholar] [CrossRef]
- de Sena Filho, J.G.; Rabbani, A.R.C.; dos Santos Silva, T.R.; da Silva, A.V.C.; Souza, I.A.; Santos, M.J.B.A.; de Jesus, J.R.; de Nogueira, P.C.L.; Duringer, J.M. Chemical and Molecular Characterization of Fifteen Species from the Lantana (Verbenaceae) Genus. Biochem. Syst. Ecol. 2012, 45, 130–137. [Google Scholar] [CrossRef]
- Spies, J.J.; du Plessis, H. Sterile Lantana Camara: Fact or Theory. S. Afr. J. Plant Soil 1987, 4, 171–174. [Google Scholar]
- Goulson, D.; Derwent, L.C. Synergistic Interactions between an Exotic Honeybee and an Exotic Weed: Pollination of Lantana camara in Australia. Weed Res. 2004, 44, 195–202. [Google Scholar] [CrossRef]
- Petanidou, T.; Godfree, R.C.; Song, D.S.; Kantsa, A.; Dupont, Y.L.; Waser, N.M. Self-Compatibility and Plant Invasiveness: Comparing Species in Native and Invasive Ranges. Perspect Plant Ecol. Evol. Syst. 2012, 14, 3–12. [Google Scholar] [CrossRef]
- Palmer, W.A.; Pullen, K.R. The Phytophagous Arthropods Associated with Lantana camara L., Hirsuta, L., Urticifolia, and L. Urticoides (Verbenaceae) in North America. Biol. Control. 1995, 5, 54–72. [Google Scholar] [CrossRef]
- Goyal, N.; Sharma, G.P. Lantana camara L. (Sensu Lato): An Enigmatic Complex. NeoBiota 2015, 25, 15–26. [Google Scholar] [CrossRef]
- de Sousa, E.O.; de Almeida, S.C.X.; Damasceno, C.B.; Nobre, S.S.; da Costa, J.G.M. Lantana camara L. and Lantana Montevidensis (Spreng.) Briq. In Medicinal and Aromatic Plants of the World; Springer: Berlin/Heidelberg, Germany, 2018; Volume 5, pp. 275–288. ISBN 978-94-024-1552-0. [Google Scholar]
- Johnson, S.B.; Lisle, S.D. The Problem with Lantana Montevidensis (Creeping Lantana). In Proceedings of the Fifteenth Australian Weeds Conference, Orange, Australia, 24–28 September 2006; 2006; pp. 727–730. [Google Scholar]
- Parrish, S.B.; Qian, R.; Deng, Z. Genome Size and Karyotype Studies in Five Species of Lantana (Verbenaceae). HortScience 2021, 56, 352–356. [Google Scholar] [CrossRef]
- Ohri, D.; Bhargava, A.; Chatterjee, A. Nuclear DNA Amounts in 112 Species of Tropical Hardwoods—New Estimates. Plant Biol. 2004, 6, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.S.; Roy, R.P. Meiotic Studies and Taxonomic Considerations in Some Taxa of Verbenaceae. Cytologia 1982, 47, 771–777. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the Power of RADseq for Ecological and Evolutionary Genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Puritz, J.B.; Hollenbeck, C.M.; Gold, J.R. DDocent: A RADseq, Variant-Calling Pipeline Designed for Population Genomics of Non-Model Organisms. PeerJ 2014, 2, e431. [Google Scholar] [CrossRef]
- Jantzen, J.R.; Guimarães, P.J.F.; Pederneiras, L.C.; Oliveira, A.L.F.; Soltis, D.E.; Soltis, P.S. Phylogenomic Analysis of Tibouchina s.s. (Melastomataceae) Highlights the Evolutionary Complexity of Neotropical Savannas. Bot. J. Linn. Soc. 2022, 199, 372–411. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An Analysis Tool Set for Population Genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-Pc: Numerical Taxonomy and Multivariate Analysis System; Applied Biostatistics, I. & Exeter Software (Firm): Setauket, NY, USA, 2009. [Google Scholar]
- Dray, S.; Dufour, A.B. The Ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef]
- Goudet, J. Hierfstat, a Package for r to Compute and Test Hierarchical F-Statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A Website and Program for Visualizing STRUCTURE Output and Implementing the Evanno Method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Battaglia, E. Accorgimenti Nella Tecnica Citologica Relativa Alla Reazione Di Feulgen. Caryologia 1951, 3, 381–383. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | GS | |||||
|---|---|---|---|---|---|---|
| Cluster 1 | ||||||
| Cluster 2 | 89.85% | |||||
| Cluster 3 | 88.61% | 91.35% | ||||
| Cluster 4 | 85.34% | 85.69% | 86.31% | |||
| Cluster 5 | 88.14% | 93.61% | 91.20% | 85.60% | ||
| LaMo | 55.15% | 58.42% | 56.45% | 60.55% | 62.39% | |
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | LaMo | |
| Cluster | N | Na | Ne | Ho (%) | He (%) | Fis | PA (%) | PL (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 33 | 1.14 | 1.12 | 14.34% | 15.31% | 0.06 | 4.91% | 73.05% |
| 2 | 18 | 1.15 | 1.06 | 14.79% | 15.30% | 0.03 | 1.68% | 69.87% |
| 3 | 7 | 1.13 | 1.08 | 13.35% | 14.26% | 0.06 | 2.66% | 72.05% |
| 4 | 25 | 1.19 | 1.15 | 18.99% | 18.00% | -0.05 | 3.36% | 70.06% |
| 5 | 10 | 1.14 | 1.05 | 13.85% | 14.20% | 0.02 | 0.45% | 62.47% |
| LaMo | 3 | 1.07 | 1.51 | 28.29% | 39.23% | 0.28 | 11.27% | 2.76% |
| Avg | 16 | 1.14 | 1.16 | 17.27% | 19.38% | 0.07 | 4.05% | 58.38% |
| Sample ID | Reference | Sample | Reference | Ref. Genome Size (pg) | Sample Genome Size (pg) | Avg Sample Genome Size (pg) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Events | % of Vis | Mean | GeoMean | Median | CV | Events | % of Vis | Mean | GeoMean | Median | CV | |||||
| LaCaBL33-1 | G. max | 3273 | 27.3 | 10,480.4 | 10,470.1 | 10,500 | 4.4 | 11,233 | 53.1 | 5692.4 | 5683.4 | 5647 | 5.7 | 2.5 | 4.6 | 4.5 |
| P. sativum | 3477 | 22.9 | 10,458.9 | 10,451.6 | 10,493 | 3.6 | 7080 | 44.7 | 21,771.6 | 21,762.6 | 21,791 | 2.9 | 9.1 | 4.4 | ||
| LaCaBL32-1 | G. max | 2173 | 14.1 | 12,588.6 | 12,585.2 | 12,602 | 2.3 | 7591 | 49.2 | 5022.2 | 5018.7 | 5021 | 3.7 | 2.5 | 6.3 | 6.1 |
| P. sativum | 1821 | 23.3 | 13,023.8 | 13,015.1 | 13,080 | 3.6 | 2756 | 35.3 | 20,045.1 | 20,034.1 | 20,101 | 3.3 | 9.1 | 5.9 | ||
| Sample ID | Sample | Control (LaCaBL32-1) | Median Sample/ Control | Ploidy | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Events | % of Vis | Mean | GeoMean | Median | CV | Events | % of Vis | Mean | GeoMean | Median | CV | |||
| LaCaBL06-3 | 2240.0 | 21.5 | 4221.8 | 4214.7 | 4217 | 5.8 | 1987.0 | 19.1 | 8365.9 | 8359.6 | 8368 | 3.9 | 2.0 | Diploid |
| LaCaBL06-5 | 9917.0 | 21.3 | 4395.4 | 4381.2 | 4391 | 8.0 | 10,634.0 | 22.8 | 8621.7 | 8608.4 | 8611 | 5.6 | 2.0 | Diploid |
| LaCaBL13-4 | 10,056.0 | 28.5 | 5693.3 | 5682.3 | 5695 | 6.2 | 3401.0 | 9.7 | 11,292.4 | 11,283.7 | 11,301 | 3.9 | 2.0 | Diploid |
| LaCaBL23-1 | 6753.0 | 25.2 | 4601.4 | 4589.7 | 4601 | 7.1 | 3254.0 | 12.1 | 9147.1 | 9134.2 | 9155 | 5.3 | 2.0 | Diploid |
| LaCaBL25-2 | 3612.0 | 20.8 | 4608.8 | 4597.4 | 4609 | 7.0 | 2277.0 | 13.1 | 8690.1 | 8682.6 | 8677 | 4.2 | 1.9 | Diploid |
| LaCaBL11-1 | 4573.0 | 14.8 | 5230.2 | 5227.0 | 5236 | 3.5 | 7765.0 | 25.2 | 10,500.1 | 10,496.7 | 10,515 | 2.5 | 2.0 | Diploid |
| LaCaBL01-1 | 3249.0 | 31.6 | 6762.2 | 6759.0 | 6760 | 3.1 | 1558.0 | 15.2 | 8877.2 | 8874.3 | 8881 | 2.6 | 1.3 | Triploid |
| LaCaBL09-5 | 1357.0 | 14.5 | 5810.5 | 5807.3 | 5805 | 3.3 | 1439.0 | 15.4 | 7437.3 | 7434.4 | 7430 | 2.8 | 1.3 | Triploid |
| LaCaBL010-1 | 748.0 | 21.5 | 8367.1 | 8363.8 | 8362 | 4.3 | 2125.0 | 21.6 | 10,863.9 | 10,861.1 | 10,871 | 2.3 | 1.3 | Triploid |
| LaCaBL33-1 | 642.0 | 31.3 | 7311.4 | 7307.5 | 7309 | 3.2 | 277.0 | 13.5 | 9882.7 | 9880.9 | 9884 | 1.9 | 1.4 | Triploid |
| LaCaBL05-1 | 2793.0 | 15.0 | 11,179.0 | 11,172.4 | 11,188 | 3.4 | - | - | - | - | - | - | - | Tetraploid |
| LaCaBL31-1 | 1355.0 | 48.1 | 12,151.0 | 12,145.8 | 12,157 | 2.9 | - | - | - | - | - | - | - | Tetraploid |
| LaCaBL32-1 | 1548.0 | 40.7 | 10,373.4 | 10,368.8 | 10,372 | 3.0 | - | - | - | - | - | - | - | Tetraploid |
| LaCaBL34-1 | 4089.0 | 41.0 | 11,885.2 | 11,880.9 | 11,876 | 2.7 | - | - | - | - | - | - | - | Tetraploid |
| LaCaBL35-1 | 866.0 | 49.6 | 11,795.9 | 11,790.4 | 11,774 | 3.1 | - | - | - | - | - | - | - | Tetraploid |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betto, A.; Palumbo, F.; Riommi, D.; Vannozzi, A.; Barcaccia, G. Harnessing Genomics for Breeding Lantana camara L.: Genotyping and Ploidy Testing of Clonal Lines Through ddRADseq Applications. Int. J. Mol. Sci. 2025, 26, 4898. https://doi.org/10.3390/ijms26104898
Betto A, Palumbo F, Riommi D, Vannozzi A, Barcaccia G. Harnessing Genomics for Breeding Lantana camara L.: Genotyping and Ploidy Testing of Clonal Lines Through ddRADseq Applications. International Journal of Molecular Sciences. 2025; 26(10):4898. https://doi.org/10.3390/ijms26104898
Chicago/Turabian StyleBetto, Angelo, Fabio Palumbo, Damiano Riommi, Alessandro Vannozzi, and Gianni Barcaccia. 2025. "Harnessing Genomics for Breeding Lantana camara L.: Genotyping and Ploidy Testing of Clonal Lines Through ddRADseq Applications" International Journal of Molecular Sciences 26, no. 10: 4898. https://doi.org/10.3390/ijms26104898
APA StyleBetto, A., Palumbo, F., Riommi, D., Vannozzi, A., & Barcaccia, G. (2025). Harnessing Genomics for Breeding Lantana camara L.: Genotyping and Ploidy Testing of Clonal Lines Through ddRADseq Applications. International Journal of Molecular Sciences, 26(10), 4898. https://doi.org/10.3390/ijms26104898