
Sotatercept: A Crosstalk Between Pathways and Activities in the Pulmonary Circulation and Blood


{kind=link}
{kind=link}
Abstract
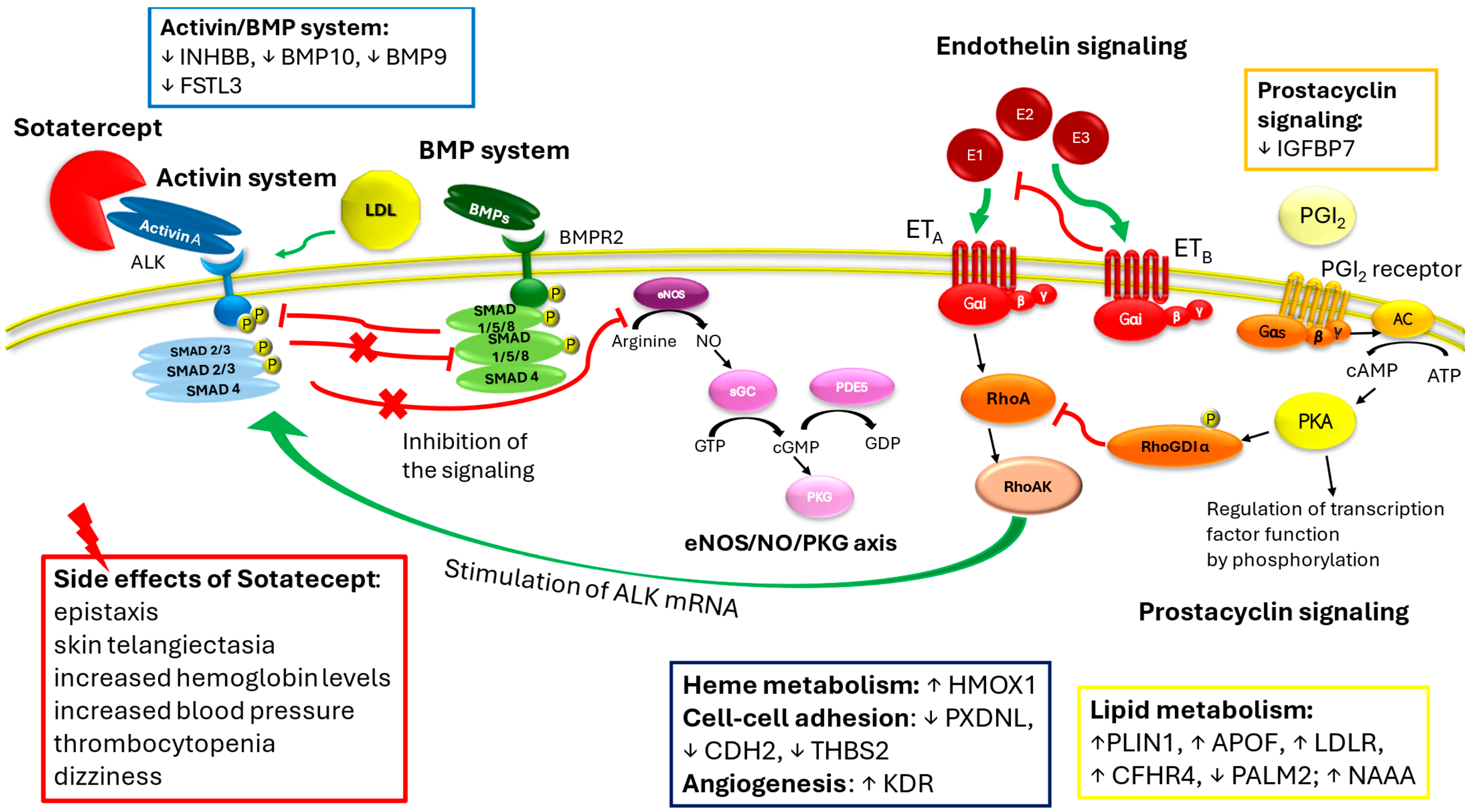
1. Sotatercept in Pulmonary Arterial Hypertension: Between Efficacy and Side Effects
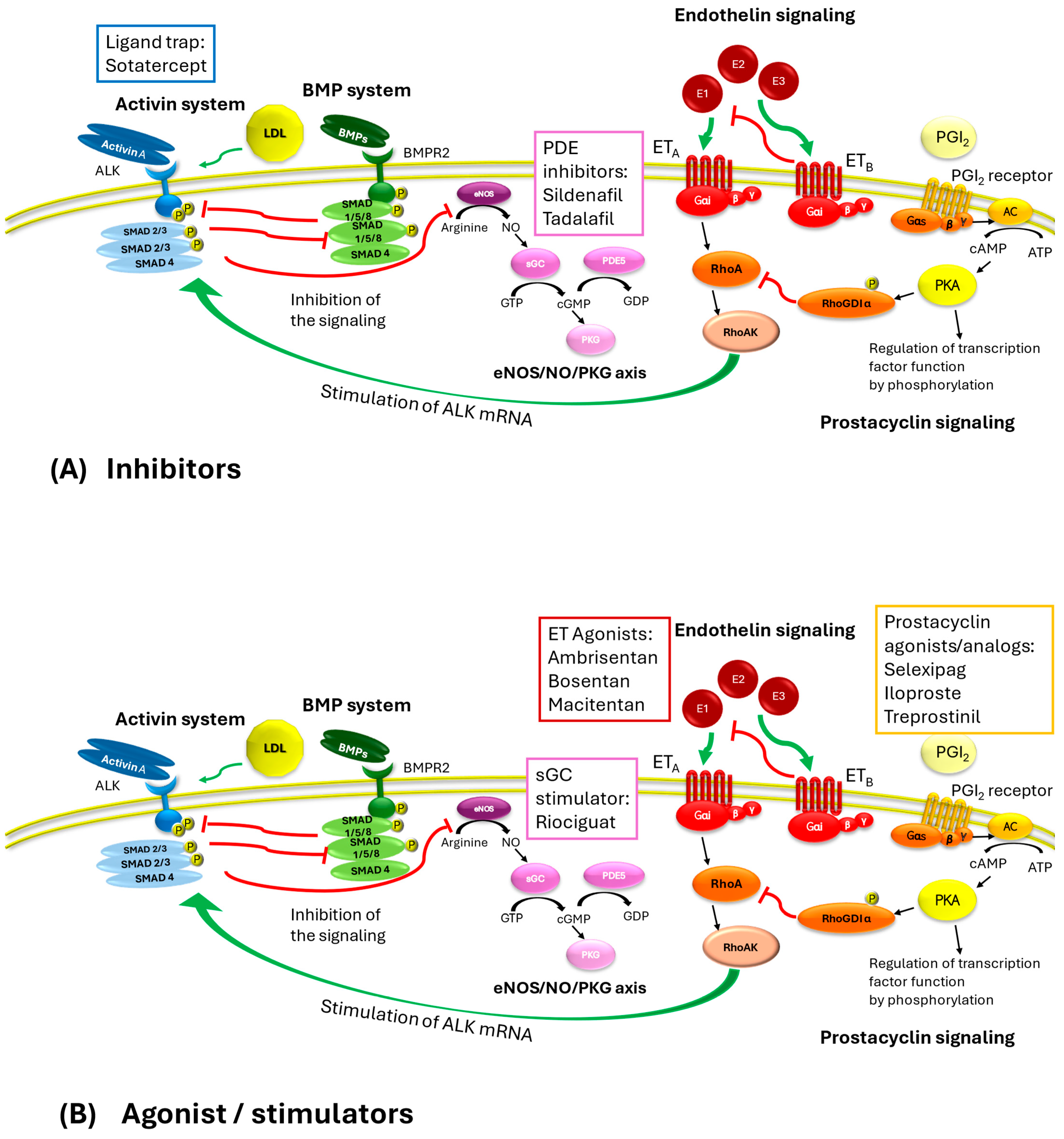
2. Crosstalk Between the Activin System and Other Signaling
2.1. The Activin System
2.2. The Bone Morphogenetic Protein System
2.3. Endothelin Receptor Signaling
2.4. CGMP/Phosphodiesterase Signaling
2.5. Prostacyclin System
3. Sotatercept Beyond the Anti-Remodeling Effect: Impact on Cell Metabolism and Regulation
4. Hematological and Vascular Effects of Sotatercept
5. Effects of RAP-011, the Murine Orthologue of Sotatercept, in Experimental Models of Hematologic Disorders
6. Sotatercept in Hematological Clinical Studies: A Summary of Findings
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. Eur. Respir. Soc. 2023, 61, 2200879. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Waxman, A.B.; Grünig, E.; Kopeć, G.; et al. Effects of sotatercept on haemodynamics and right heart function: Analysis of the STELLAR trial. Eur. Respir. J. 2023, 62, 2301107. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Ghelardoni, S. Sotatercept in pulmonary hypertension and beyond. Eur. J. Clin. Invest. 2025, 55, e14386. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Biondi, F. Sotatercept: New drug on the horizon of pulmonary hypertension. Vasc. Pharmacol. 2024, 157, 107442. [Google Scholar] [CrossRef]
- Yung, L.-M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.-S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12, eaaz5660. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Blanco, F.J.; Garrido-Martin, E.M.; Sanz-Rodriguez, F.; del Pozo, M.A.; Bernabeu, C. Caveolin-1 interacts and cooperates with the transforming growth factor-beta type I receptor ALK1 in endothelial caveolae. Cardiovasc. Res. 2008, 77, 791–799. [Google Scholar] [CrossRef]
- Yong, H.E.; Murthi, P.; Wong, M.H.; Kalionis, B.; Cartwright, J.E.; Brennecke, S.P.; Keogh, R.J. Effects of normal and high circulating concentrations of activin A on vascular endothelial cell functions and vasoactive factor production. Pregnancy Hypertens. 2015, 5, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Yokokawa, T.; Misaka, T.; Kaneshiro, T.; Yamada, S.; Yoshihisa, A.; Nakazato, K.; Takeishi, Y. Endothelin-1 Upregulates Activin Receptor-Like Kinase-1 Expression via Gi/RhoA/Sp-1/Rho Kinase Pathways in Human Pulmonary Arterial Endothelial Cells. Front. Cardiovasc. Med. 2021, 8, 648981. [Google Scholar] [CrossRef]
- Santini, V. Of blood and bone: The sotatercept adventure. Lancet Haematol. 2018, 5, e54–e55. [Google Scholar] [CrossRef] [PubMed]
- Meunier, H.; Cajander, S.B.; Roberts, V.J.; Rivier, C.; Sawchenko, P.E.; Hsueh, A.J.W.; Vale, W. Rapid changes in the expression of inhibin alpha-, beta A-, and beta B-subunits in ovarian cell types during the rat estrous cycle. Mol. Endocrinol. 1988, 2, 1352–1363. [Google Scholar] [CrossRef]
- Olsen, O.E.; Hella, H.; Elsaadi, S.; Jacobi, C.; Martinez-Hackert, E.; Holien, T. Activins as Dual Specificity TGF-β Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors. Biomolecules 2020, 10, 519. [Google Scholar] [CrossRef] [PubMed]
- Stump, B.; Waxman, A.B. Pulmonary Arterial Hypertension and TGF-β Superfamily Signaling: Focus on Sotatercept. BioDrugs 2024, 38, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Namwanje, M.; Brown, C.W. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a021881. [Google Scholar] [CrossRef]
- Ye, Q.; Taleb, S.J.; Zhao, J.; Zhao, Y. Emerging role of BMPs/BMPR2 signaling pathway in treatment for pulmonary fibrosis. Biomed. Pharmacother. 2024, 178, 117178. [Google Scholar] [CrossRef]
- Gkarmiris, K.I.; Lindbäck, J.; Alexander, J.H.; Granger, C.B.; Kastner, P.; Lopes, R.D.; Ziegler, A.; Oldgren, J.; Siegbahn, A.; Wallentin, L.; et al. Repeated Measurement of the Novel Atrial Biomarker BMP10 (Bone Morphogenetic Protein 10) Refines Risk Stratification in Anticoagulated Patients With Atrial Fibrillation: Insights From the ARISTOTLE Trial. J. Am. Heart Assoc. 2024, 13, e033720. [Google Scholar] [CrossRef]
- Liu, L.; Liang, Y.; Lan, Q.-G.; Chen, J.-Z.; Wang, R.; Zhao, J.-H.; Liang, B. Bone morphogenetic protein 10 and atrial fibrillation. IJC Heart Vasc. 2024, 51, 101376. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, X.; Sun, H.; Yu, H.; Du, B.; Fan, Q.; Jia, B.; Zhang, Z. Bone morphogenetic protein 10, a rising star in the field of diabetes and cardiovascular disease. J. Cell. Mol. Med. 2024, 28, e18324. [Google Scholar] [CrossRef]
- Capasso, T.L.; Li, B.; Volek, H.J.; Khalid, W.; Rochon, E.R.; Anbalagan, A.; Herdman, C.; Yost, H.J.; Villanueva, F.S.; Kim, K.; et al. BMP10-mediated ALK1 signaling is continuously required for vascular development and maintenance. Angiogenesis 2020, 23, 203–220. [Google Scholar] [CrossRef]
- Yu, J.; Dolter, K.E. Production of activin A and its roles in inflammation and hematopoiesis. Cytokines Cell. Mol. Ther. 1997, 3, 169–177. [Google Scholar]
- Dussiot, M.; Maciel, T.T.; Fricot, A.; Chartier, C.; Negre, O.; Veiga, J.; Grapton, D.; Paubelle, E.; Payen, E.; Beuzard, Y.; et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat. Med. 2014, 20, 398–407. [Google Scholar] [CrossRef]
- De Rosa, G.; Andolfo, I.; Marra, R.; Manna, F.; Rosato, B.E.; Iolascon, A.; Russo, R. RAP-011 Rescues the Disease Phenotype in a Cellular Model of Congenital Dyserythropoietic Anemia Type II by Inhibiting the SMAD2-3 Pathway. Int. J. Mol. Sci. 2020, 21, 5577. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Quigley, K. Bone morphogenetic protein signalling in pulmonary arterial hypertension: Revisiting the BMPRII connection. Biochem. Soc. Trans. 2024, 52, 1515–1528. [Google Scholar] [CrossRef] [PubMed]
- Jerkic, M.; Kabir, M.G.; Davies, A.; Yu, L.X.; McIntyre, B.A.; Husain, N.W.; Enomoto, M.; Sotov, V.; Husain, M.; Henkelman, M.; et al. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc. Res. 2011, 92, 375–384. [Google Scholar] [CrossRef]
- Girerd, B.; Montani, D.; Coulet, F.; Sztrymf, B.; Yaici, A.; Jaïs, X.; Tregouet, D.; Reis, A.; Drouin-Garraud, V.; Fraisse, A.; et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am. J. Respir. Crit. Care Med. 2010, 181, 851–861. [Google Scholar] [CrossRef]
- Eswaran, H.; Kasthuri, R.S. Potential and emerging therapeutics for HHT. Hematology 2024, 2024, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Aslam, S.; Sadiq, H.Z.; Ali, S.; Syed, R.; Panjiyar, B.K. New and Emerging Therapeutic Drugs for the Treatment of Pulmonary Arterial Hypertension: A Systematic Review. Cureus 2024, 16, e68117. [Google Scholar] [CrossRef]
- Frantz, R.P.; Desai, S.S.; Ewald, G.; Franco, V.; Hage, A.; Horn, E.M.; LaRue, S.J.; Mathier, M.A.; Mandras, S.; Park, M.H.; et al. SOPRANO: Macitentan in patients with pulmonary hypertension following left ventricular assist device implantation. Pulm. Circ. 2024, 14, e12446. [Google Scholar] [CrossRef]
- Shihoya, W.; Sano, F.K.; Nureki, O. Structural insights into endothelin receptor signalling. J. Biochem. 2023, 174, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.H.; Kar, S.; Kirkpatrick, P. Ambrisentan. Nat. Rev. Drug Discov. 2007, 6, 697–698. [Google Scholar] [CrossRef]
- Madonna, R.; Cocco, N.; De Caterina, R. Pathways and Drugs in Pulmonary Arterial Hypertension - Focus on the Role of Endothelin Receptor Antagonists. Cardiovasc Drugs Ther. 2015, 29, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Dhaun, N.; Webb, D.J. Endothelins in cardiovascular biology and therapeutics. Nat. Rev. Cardiol. 2019, 16, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Gawrys, O.; Kala, P.; Sadowski, J.; Melenovský, V.; Sandner, P.; Červenka, L. Soluble guanylyl cyclase stimulators and activators: Promising drugs for the treatment of hypertension? Eur. J. Pharmacol. 2025, 987, 177175. [Google Scholar] [CrossRef] [PubMed]
- Weatherald, J.; Varughese, R.A.; Liu, J.; Humbert, M. Management of Pulmonary Arterial Hypertension. Semin. Respir. Crit. Care Med. 2023, 44, 746–761. [Google Scholar] [CrossRef]
- Savale, L.; Tu, L.; Normand, C.; Boucly, A.; Sitbon, O.; Montani, D.; Olsson, K.M.; Park, D.-H.; Fuge, J.; Kamp, J.C.; et al. Effect of sotatercept on circulating proteomics in pulmonary arterial hypertension. Eur. Respir. J. 2024, 64, 2401483. [Google Scholar] [CrossRef]
- Torres, G.; Lancaster, A.C.; Yang, J.; Griffiths, M.; Brandal, S.; Damico, R.; Vaidya, D.; Simpson, C.E.; Martin, L.J.; Pauciulo, M.W.; et al. Low-affinity insulin-like growth factor binding protein 7 and its association with pulmonary arterial hypertension severity and survival. Pulm. Circ. 2023, 13, e12284. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, M.; Cai, W.; Sun, W.; Shi, X.; Liu, D.; Song, W.; Yan, Y.; Chen, T.; Bao, Q.; et al. Prostaglandin I2 signaling prevents angiotensin II-induced atrial remodeling and vulnerability to atrial fibrillation in mice. Cell. Mol. Life Sci. 2024, 81, 264. [Google Scholar] [CrossRef]
- Zeng, C.; Liu, J.; Zheng, X.; Hu, X.; He, Y. Prostaglandin and prostaglandin receptors: Present and future promising therapeutic targets for pulmonary arterial hypertension. Respir. Res. 2023, 24, 263. [Google Scholar] [CrossRef] [PubMed]
- Mismetti, V.; Delavenne, X.; Montani, D.; Bezzeghoud, S.; Delezay, O.; Hodin, S.; Launay, D.; Marchand-Adam, S.; Nunes, H.; Ollier, E.; et al. Proteomic biomarkers for survival in systemic sclerosis-associated pulmonary hypertension. Respir. Res. 2023, 24, 273. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, P.; Deng, J.; Zhang, Q.; Feng, H.; Zhang, Y.; Lu, Y.; Han, L.; Yang, P.; Deng, Z. Additional Use of Prostacyclin Analogs in Patients With Pulmonary Arterial Hypertension: A Meta-Analysis. Front. Pharmacol. 2022, 13, 817119. [Google Scholar] [CrossRef]
- Joshi, S.R.; Liu, J.; Bloom, T.; Atabay, E.K.; Kuo, T.-H.; Lee, M.; Belcheva, E.; Spaits, M.; Grenha, R.; Maguire, M.C.; et al. Sotatercept analog suppresses inflammation to reverse experimental pulmonary arterial hypertension. Sci. Rep. 2022, 12, 7803. [Google Scholar] [CrossRef]
- Kraehling, J.R.; Chidlow, J.H.; Rajagopal, C.; Sugiyama, M.G.; Fowler, J.W.; Lee, M.Y.; Zhang, X.; Ramírez, C.M.; Park, E.J.; Tao, B.; et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat. Commun. 2016, 7, 13516. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.Y.; Ho, T.W.W.; Xu, Z.; Neculai, D.; Beauchemin, C.A.; Lee, W.L.; Fairn, G.D. Apolipoprotein A1 and high-density lipoprotein limit low-density lipoprotein transcytosis by binding SR-B1. J. Lipid Res. 2024, 65, 100530. [Google Scholar] [CrossRef]
- Lan, Z.; Lv, Z.; Zuo, W.; Xiao, Y. From bench to bedside: The promise of sotatercept in hematologic disorders. Biomed. Pharmacother. 2023, 165, 115239. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E.; et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Subias, P.E.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef]
- Liu, Q.; Han, Z.; Li, T.; Meng, J.; Zhu, C.; Wang, J.; Wang, J.; Zhang, Z.; Wu, H. Microglial HO-1 aggravates neuronal ferroptosis via regulating iron metabolism and inflammation in the early stage after intracerebral hemorrhage. Int. Immunopharmacol. 2025, 147, 113942. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Lambein, F.; Kusama-Eguchi, K. Vascular insult accompanied by overexpressed heme oxygenase-1 as a pathophysiological mechanism in experimental neurolathyrism with hind-leg paraparesis. Biochem. Biophys. Res. Commun. 2012, 428, 160–166. [Google Scholar] [CrossRef]
- Chen-Roetling, J.; Kamalapathy, P.; Cao, Y.; Song, W.; Schipper, H.M.; Regan, R.F. Astrocyte heme oxygenase-1 reduces mortality and improves outcome after collagenase-induced intracerebral hemorrhage. Neurobiol. Dis. 2017, 102, 140–146. [Google Scholar] [CrossRef]
- Gao, Q.; Su, Z.; Pang, X.; Chen, J.; Luo, R.; Li, X.; Zhang, C.; Zhao, Y. Overexpression of Heme Oxygenase 1 Enhances the Neuroprotective Effects of Exosomes in Subarachnoid Hemorrhage by Suppressing Oxidative Stress and Endoplasmic Reticulum Stress. Mol. Neurobiol. 2024, 62, 6088–6101. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Chen, W.-C.; Tseng, C.-K.; Chen, Y.-H.; Lin, C.-K.; Lee, J.-C. Heme oxygenase-1 inhibits DENV-induced endothelial hyperpermeability and serves as a potential target against dengue hemorrhagic fever. FASEB J. 2022, 36, e22110. [Google Scholar] [CrossRef]
- Tian, S.; Xu, X.; Yang, X.; Fan, L.; Jiao, Y.; Zheng, M.; Zhang, S. Roles of follistatin-like protein 3 in human non-tumor pathophysiologies and cancers. Front. Cell Dev. Biol. 2022, 10, 953551. [Google Scholar] [CrossRef] [PubMed]
- Kelaini, S.; Vilà-González, M.; Caines, R.; Campbell, D.; Eleftheriadou, M.; Tsifaki, M.; Magee, C.; Cochrane, A.; O’Neill, K.; Yang, C.; et al. Follistatin-Like 3 Enhances the Function of Endothelial Cells Derived from Pluripotent Stem Cells by Facilitating β-Catenin Nuclear Translocation Through Inhibition of Glycogen Synthase Kinase-3β Activity. Stem Cells 2018, 36, 1033–1044. [Google Scholar] [CrossRef]
- Péterfi, Z.; Tóth, Z.E.; Kovács, H.A.; Lázár, E.; Sum, A.; Donkó, A.; Sirokmány, G.; Shah, A.M.; Geiszt, M. Peroxidasin-like protein: A novel peroxidase homologue in the human heart. Cardiovasc. Res. 2014, 101, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Pearsall, R.S.; Canalis, E.; Cornwall-Brady, M.; Underwood, K.W.; Haigis, B.; Ucran, J.; Kumar, R.; Pobre, E.; Grinberg, A.; Werner, E.D.; et al. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc. Natl. Acad. Sci. USA 2008, 105, 7082–7087. [Google Scholar] [CrossRef] [PubMed]
- Carrancio, S.; Markovics, J.; Wong, P.; Leisten, J.; Castiglioni, P.; Groza, M.C.; Raymon, H.K.; Heise, C.; Daniel, T.; Chopra, R.; et al. An activin receptor IIA ligand trap promotes erythropoiesis resulting in a rapid induction of red blood cells and haemoglobin. Br. J. Haematol. 2014, 165, 870–882. [Google Scholar] [CrossRef]
- Ear, J.; Hsueh, J.; Nguyen, M.; Zhang, Q.; Sung, V.; Chopra, R.; Sakamoto, K.M.; Lin, S. A Zebrafish Model of 5q-Syndrome Using CRISPR/Cas9 Targeting RPS14 Reveals a p53-Independent and p53-Dependent Mechanism of Erythroid Failure. J. Genet. Genom. 2016, 43, 307–318. [Google Scholar] [CrossRef]
- Ear, J.; Huang, H.; Wilson, T.; Tehrani, Z.; Lindgren, A.; Sung, V.; Laadem, A.; Daniel, T.O.; Chopra, R.; Lin, S. RAP-011 improves erythropoiesis in zebrafish model of Diamond-Blackfan anemia through antagonizing lefty1. Blood 2015, 126, 880–890. [Google Scholar] [CrossRef]
- Iancu-Rubin, C.; Mosoyan, G.; Wang, J.; Kraus, T.; Sung, V.; Hoffman, R. Stromal cell-mediated inhibition of erythropoiesis can be attenuated by Sotatercept (ACE-011), an activin receptor type II ligand trap. Exp. Hematol. 2013, 41, 155–166. [Google Scholar] [CrossRef]
- Williams, M.J.; Sugatani, T.; Agapova, O.A.; Fang, Y.; Gaut, J.P.; Faugere, M.-C.; Malluche, H.H.; Hruska, K.A. The activin receptor is stimulated in the skeleton, vasculature, heart, and kidney during chronic kidney disease. Kidney Int. 2018, 93, 147–158. [Google Scholar] [CrossRef]
- Sugatani, T.; Agapova, O.A.; Fang, Y.; Berman, A.G.; Wallace, J.M.; Malluche, H.H.; Faugere, M.-C.; Smith, W.; Sung, V.; Hruska, K.A. Ligand trap of the activin receptor type IIA inhibits osteoclast stimulation of bone remodeling in diabetic mice with chronic kidney disease. Kidney Int. 2017, 91, 86–95. [Google Scholar] [CrossRef]
- Fajardo, R.J.; Manoharan, R.K.; Pearsall, R.S.; Davies, M.V.; Marvell, T.; Monnell, T.E.; Ucran, J.A.; Pearsall, A.E.; Khanzode, D.; Kumar, R.; et al. Treatment with a soluble receptor for activin improves bone mass and structure in the axial and appendicular skeleton of female cynomolgus macaques (Macaca fascicularis). Bone 2010, 46, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Morse, A.; Cheng, T.L.; Peacock, L.; Mikulec, K.; Little, D.G.; Schindeler, A. RAP-011 augments callus formation in closed fractures in rats. J. Orthop. Res. 2016, 34, 320–330. [Google Scholar] [CrossRef]
- Sherman, M.L.; Borgstein, N.G.; Mook, L.; Wilson, D.; Yang, Y.; Chen, N.; Kumar, R.; Kim, K.; Laadem, A. Multiple-dose, safety, pharmacokinetic, and pharmacodynamic study of sotatercept (ActRIIA-IgG1), a novel erythropoietic agent, in healthy postmenopausal women. J. Clin. Pharmacol. 2013, 53, 1121–1130. [Google Scholar] [CrossRef]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulos, H.; Laadem, A.; Hesketh, P.J.; Goldschmidt, J.; Gabrail, N.; Osborne, C.; Ali, M.; Sherman, M.L.; Wang, D.; Glaspy, J.A.; et al. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: Results from two phase 2 studies. Support. Care Cancer 2016, 24, 1517–1525. [Google Scholar] [CrossRef]
- Bose, P.; Masarova, L.; Pemmaraju, N.; Bledsoe, S.D.; Daver, N.G.; Jabbour, E.J.; Kadia, T.M.; Estrov, Z.; Kornblau, S.M.; Andreeff, M.; et al. Sotatercept for anemia of myelofibrosis: A phase II investigator-initiated study. Haematologica 2024, 109, 2660–2664. [Google Scholar] [CrossRef]
- Komrokji, R.; Garcia-Manero, G.; Ades, L.; Prebet, T.; Steensma, D.P.; Jurcic, J.G.; Sekeres, M.A.; Berdeja, J.; Savona, M.R.; Beyne-Rauzy, O.; et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: A phase 2, dose-ranging trial. Lancet Haematol. 2018, 5, e63–e72. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madonna, R.; Ghelardoni, S. Sotatercept: A Crosstalk Between Pathways and Activities in the Pulmonary Circulation and Blood. Int. J. Mol. Sci. 2025, 26, 4851. https://doi.org/10.3390/ijms26104851
Madonna R, Ghelardoni S. Sotatercept: A Crosstalk Between Pathways and Activities in the Pulmonary Circulation and Blood. International Journal of Molecular Sciences. 2025; 26(10):4851. https://doi.org/10.3390/ijms26104851
Chicago/Turabian StyleMadonna, Rosalinda, and Sandra Ghelardoni. 2025. "Sotatercept: A Crosstalk Between Pathways and Activities in the Pulmonary Circulation and Blood" International Journal of Molecular Sciences 26, no. 10: 4851. https://doi.org/10.3390/ijms26104851
APA StyleMadonna, R., & Ghelardoni, S. (2025). Sotatercept: A Crosstalk Between Pathways and Activities in the Pulmonary Circulation and Blood. International Journal of Molecular Sciences, 26(10), 4851. https://doi.org/10.3390/ijms26104851