
Structural Characterisation of TetR/AcrR Regulators in Streptomyces fildesensis So13.3: An In Silico CRISPR-Based Strategy to Influence the Suppression of Actinomycin D Production

 , , ,
, , , 
Abstract
1. Introduction
2. Results
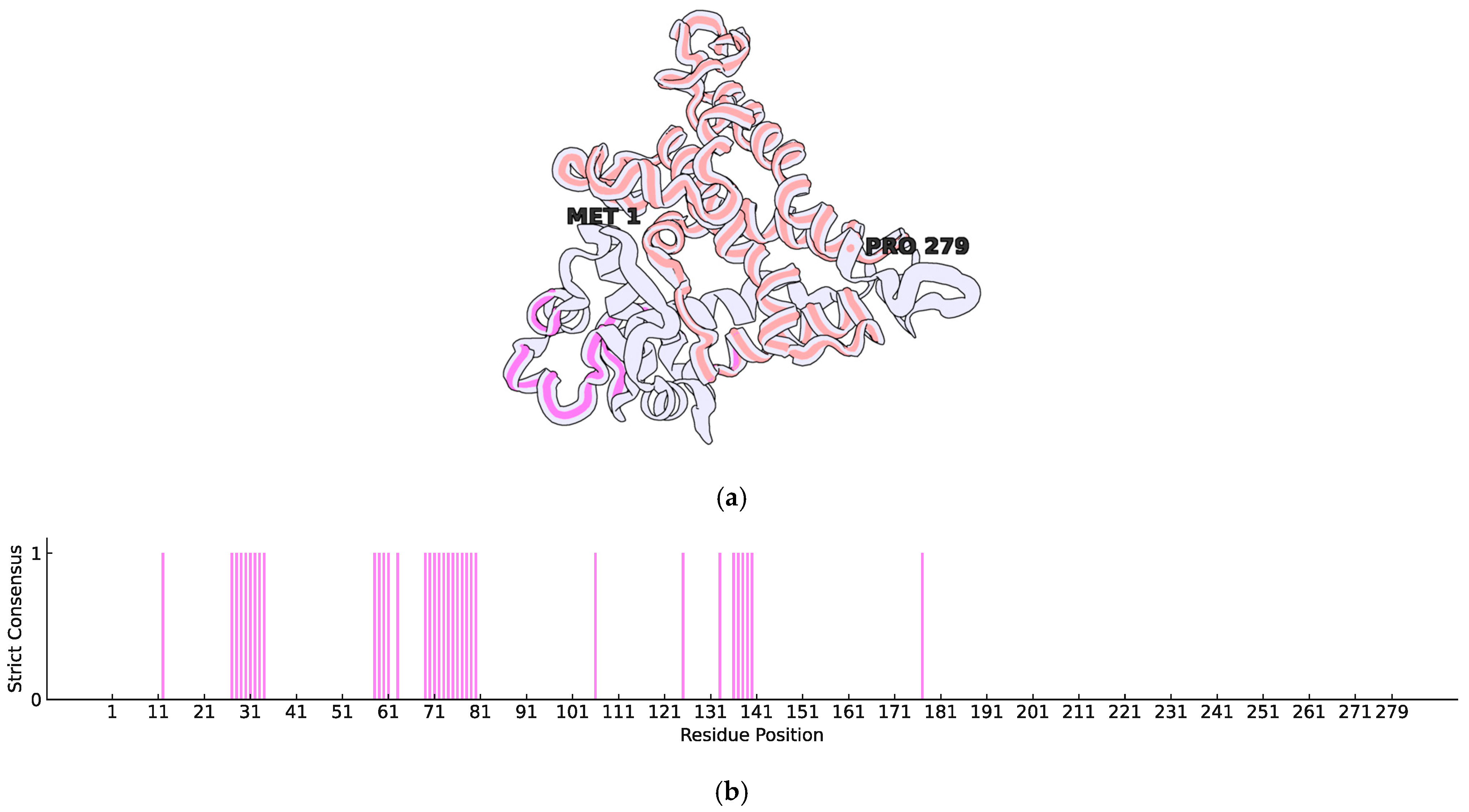
2.1. Structural Characterisation and Refinement of TetR Transcription Factors in the Actinomycin D Biosynthetic Cluster of Streptomyces fildesensis So13.3
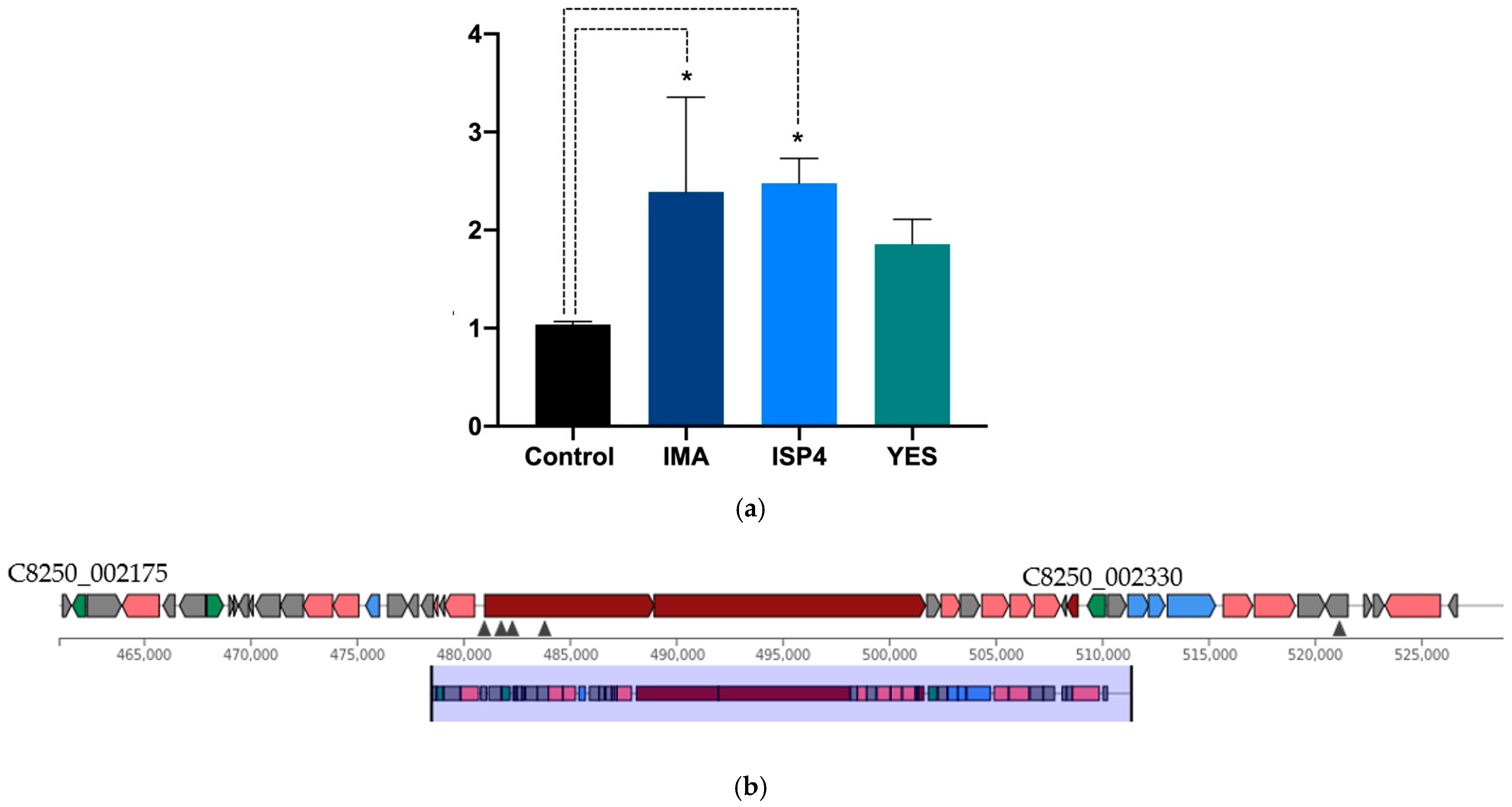
2.2. Nutritional Regulation of TetR-279 Suggests Its Role as a Key Activator of the Actinomycin D Biosynthetic Cluster
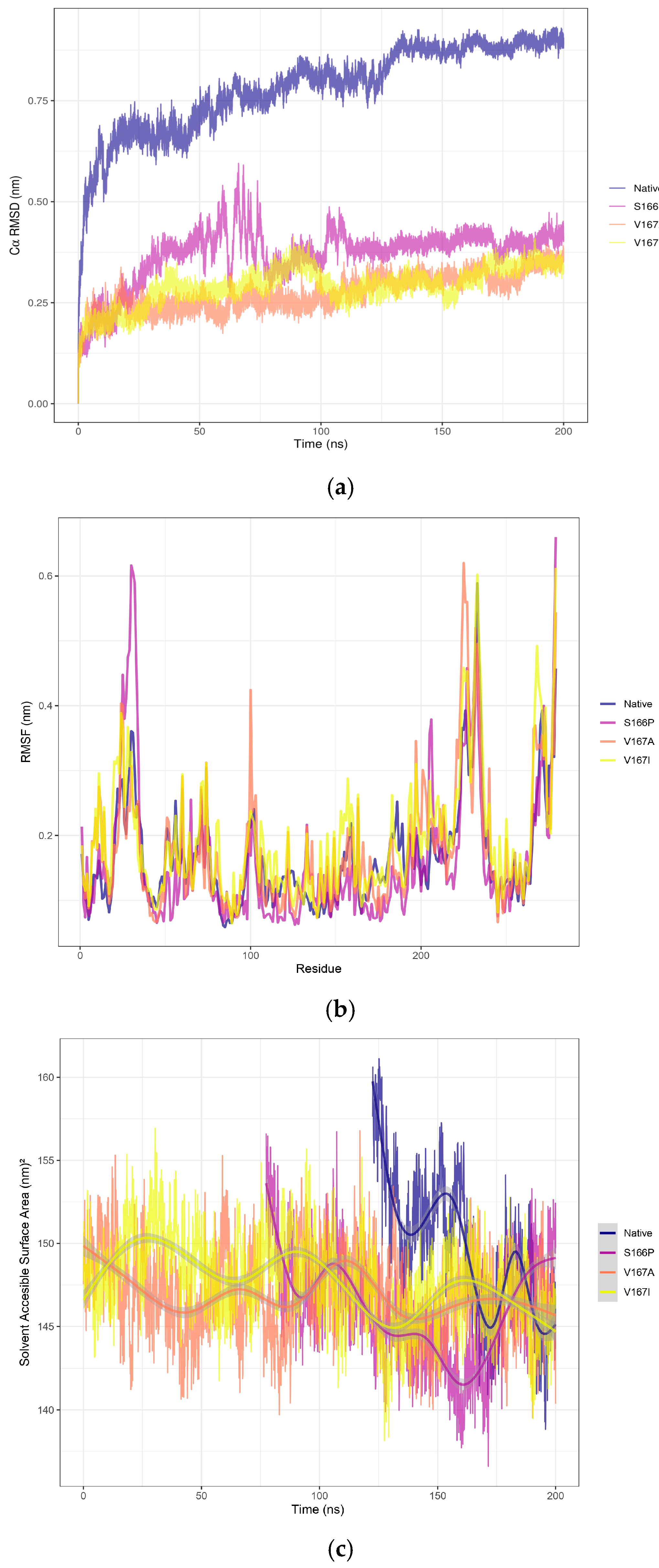
2.3. Structural Effects of Point Mutations in TetR-279 via Molecular Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Identification and Structural Analysis of the Protein
4.2. Cultivation Conditions of Streptomyces fildesensis So13.3
4.3. Gene Expression Analysis of TetR-279 Under Different Nutritional Conditions
4.4. sgRNA Design Targeting Transcriptional Regulators of the Actinomycin D Cluster
4.5. Homology Modelling, Refinement, and Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BGC | Biosynthetic Gene Cluster |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| MD | Molecular Dynamics |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| SASA | Solvent Accessible Surface Area |
| TF | Transcription Factor |
| ISP4 | International Streptomyces Project Medium 4 |
| YES | Yeast Extract Sucrose Medium |
| HTH | Helix-Turn-Helix |
| SNP | Sodium Nitroprusside |
| LPS | Lipopolysaccharide |
| qPCR | Quantitative Polymerase Chain Reaction |
| PDB | Protein Data Bank |
| pLDDT | Predicted Local Distance Difference Test |
| VMD | Visual Molecular Dynamics |
| PBC | Periodic Boundary Condition |
| PME | Particle Mesh Ewald |
| NVT | Constant Number, Volume, Temperature Ensemble |
| NPT | Constant Number, Pressure, Temperature Ensemble |
| LINCS | Linear Constraint Solver |
| EMSA | Electrophoretic Mobility Shift Assay |
| DSB | Double-Strand Break |
| NRPS | Non-Ribosomal Peptide Synthetase |
| CRISPR-BEST | CRISPR Base Editing SysTem |
| EXPosition | EXpression Prediction from Sequence Mutations |
References
- Vaithinathan, A.G.; Vanitha, A. WHO global priority pathogens list on antibiotic resistance: An urgent need for action to integrate One Health data. Perspect. Public Health 2018, 138, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Nisa, T.T.; Nakatani, D.; Kaneko, F.; Takeda, T.; Nakata, K. Antimicrobial resistance patterns of WHO priority pathogens isolated in hospitalized patients in Japan: A tertiary center observational study. PLoS ONE 2024, 19, e0294229. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.; Medina-Lara, A.; Smith, R. Antimicrobial resistance and the COVID-19 pandemic. Bull. World Health Organ. 2022, 100, 295. [Google Scholar] [CrossRef]
- Núñez-Montero, K.; Barrientos, L. Advances in Antarctic Research for Antimicrobial Discovery: A Comprehensive Narrative Review of Bacteria from Antarctic Environments as Potential Sources of Novel Antibiotic Compounds Against Human Pathogens and Microorganisms of Industrial Importance. Antibiotics 2018, 7, 90. [Google Scholar] [CrossRef]
- Thompson, T.P.; Gilmore, B.F. Exploring halophilic environments as a source of new antibiotics. Crit. Rev. Microbiol. 2024, 50, 341–370. [Google Scholar] [CrossRef]
- Coppola, D.; Lauritano, C.; Zazo, G.; Nuzzo, G.; Fontana, A.; Ianora, A.; Costantini, M.; Verde, C.; Giordano, D. Biodiversity of UV-Resistant Bacteria in Antarctic Aquatic Environments. J. Mar. Sci. Eng. 2023, 11, 968. [Google Scholar] [CrossRef]
- Lugg, D.J.; Roy, C.R. Ultraviolet radiation and health effects in the Antarctic. Polar Res. 1999, 18, 353–359. [Google Scholar] [CrossRef]
- Silva, L.J.; Crevelin, E.J.; Souza, D.T.; Lacerda-Júnior, G.V.; de Oliveira, V.M.; Ruiz, A.L.T.G.; Rosa, L.H.; Moraes, L.A.B. Actinobacteria from Antarctica as a source for anticancer discovery. Sci. Rep. 2020, 10, 13870. [Google Scholar] [CrossRef]
- Benaud, N.; Edwards, R.J.; Amos, T.G.; D’Agostino, P.M.; Gutiérrez-Chávez, C.; Montgomery, K.; Nicetic, I.; Ferrari, B.C. Antarctic desert soil bacteria exhibit high novel natural product potential, evaluated through long-read genome sequencing and comparative genomics. Environ. Microbiol. 2021, 23, 3646–3664. [Google Scholar] [CrossRef]
- Lebedeva, J.; Jukneviciute, G.; Čepaitė, R.; Vickackaite, V.; Pranckutė, R.; Kuisiene, N. Genome Mining and Characterization of Biosynthetic Gene Clusters in Two Cave Strains of Paenibacillus sp. Front. Microbiol. 2021, 11, 612483. [Google Scholar] [CrossRef]
- Russell, A.; Lacret, R.; Truman, A. Genome-led discovery of novel microbial natural products. Access Microbiol. 2019, 1. [Google Scholar] [CrossRef]
- Núñez-Montero, K.; Lamilla, C.; Abanto, M.; Maruyama, F.; Jorquera, M.A.; Santos, A.; Martinez-Urtaza, J.; Barrientos, L. Antarctic Streptomyces fildesensis So13.3 strain as a promising source for antimicrobials discovery. Sci. Rep. 2019, 9, 7488. [Google Scholar] [CrossRef]
- Núñez-Montero, K.; Quezada-Solís, D.; Khalil, Z.G.; Capon, R.J.; Andreote, F.D.; Barrientos, L. Genomic and Metabolomic Analysis of Antarctic Bacteria Revealed Culture and Elicitation Conditions for the Production of Antimicrobial Compounds. Biomolecules 2020, 10, 673. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Diamond, S.; Butterfield, C.N.; Thomas, B.C.; Banfield, J.F. Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 2018, 558, 440–444. [Google Scholar] [CrossRef]
- Scherlach, K.; Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 2021, 12, 3864. [Google Scholar] [CrossRef]
- Leal, K.; Rojas, E.; Madariaga, D.; Contreras, M.J.; Nuñez-Montero, K.; Barrientos, L.; Goméz-Espinoza, O.; Iturrieta-González, I. Unlocking Fungal Potential: The CRISPR-Cas System as a Strategy for Secondary Metabolite Discovery. J. Fungi 2024, 10, 748. [Google Scholar] [CrossRef]
- Tong, Y.; Whitford, C.M.; Blin, K.; Jørgensen, T.S.; Weber, T.; Lee, S.Y. CRISPR–Cas9, CRISPRi and CRISPR-BEST-mediated genetic manipulation in streptomycetes. Nat. Protoc. 2020, 15, 2470–2502. [Google Scholar] [CrossRef]
- Park, W.; Woo, J.-K.; Shin, J.; Oh, K.-B. nonG, a constituent of the nonactin biosynthetic gene cluster, regulates nocardamine synthesis in Streptomyces albus J1074. Biochem. Biophys. Res. Commun. 2017, 490, 664–669. [Google Scholar] [CrossRef]
- Liang, Y.; Lu, H.; Tang, J.; Ye, X.; Wei, Y.; Liao, B.; Liu, L.; Xu, H. ActO, a positive cluster-situated regulator for actinomycins biosynthesis in Streptomyces antibioticus ZS. Gene 2025, 933, 148962. [Google Scholar] [CrossRef]
- Lei, Y.; Asamizu, S.; Ishizuka, T.; Onaka, H. Regulation of Multidrug Efflux Pumps by TetR Family Transcriptional Repressor Negatively Affects Secondary Metabolism in Streptomyces coelicolor A3(2). Appl. Environ. Microbiol. 2023, 89, e0182222. [Google Scholar] [CrossRef]
- Yu, D.; Lin, H.; Bechthold, A.; Yu, X.; Ma, Z. RS24090, a TetR family transcriptional repressor, negatively affects the rimocidin biosynthesis in Streptomyces rimosus M527. Int. J. Biol. Macromol. 2025, 285, 138043. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Alkhnbashi, O.S. Current Bioinformatics Tools to Optimize CRISPR/Cas9 Experiments to Reduce Off-Target Effects. Int. J. Mol. Sci. 2023, 24, 6261. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Whitford, C.M.; Robertsen, H.L.; Blin, K.; Jørgensen, T.S.; Klitgaard, A.K.; Gren, T.; Jiang, X.; Weber, T.; Lee, S.Y. Highly efficient DSB-free base editing for streptomycetes with CRISPR-BEST. Proc. Natl. Acad. Sci. USA 2019, 116, 20366–20375. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, M.; Zhao, R.; Pan, Y.; Wu, P.; Zhang, C.; Chi, X.; Zhang, B.; Wu, H. TetR family regulator AbrT controls lincomycin production and morphological development in Streptomyces lincolnensis. Microb. Cell Fact. 2024, 23, 223. [Google Scholar] [CrossRef]
- Inahashi, Y.; Shiraishi, T.; Také, A.; Matsumoto, A.; Takahashi, Y.; Ōmura, S.; Kuzuyama, T.; Nakashima, T. Identification and heterologous expression of the actinoallolide biosynthetic gene cluster. J. Antibiot. 2018, 71, 749–752. [Google Scholar] [CrossRef]
- Liu, M.-S.; Gong, S.; Yu, H.-H.; Jung, K.; Johnson, K.A.; Taylor, D.W. Taylor, Basis for discrimination by engineered CRISPR/Cas9 enzymes. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cohen, S.; Bergman, S.; Lynn, N.; Tuller, T. A tool for CRISPR-Cas9 gRNA evaluation based on computational models of gene expression. bioRxiv 2024. [Google Scholar] [CrossRef]
- Yuan, S. Mitigating the Off-target Effects in CRISPR/Cas9-mediated Genetic Editing with Bioinformatic Technologies. Trans. Mater. Biotechnol. Life Sci. 2024, 3, 318–326. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, N.; Jeong, Y.; Lee, Y.; Kim, W.; Cho, S.; Palsson, B.O.; Cho, B.-K. Primary transcriptome and translatome analysis determines transcriptional and translational regulatory elements encoded in the Streptomyces clavuligerus genome. Nucleic Acids Res. 2019, 47, 6114–6129. [Google Scholar] [CrossRef]
- Hussein, Z.A.; Al-Kazaz, A.A. Bioinformatics Evaluation of Crisp2 Gene Snps and Their Impacts on Protein. Iraqi J. Agric. sciences 2023, 54, 369–377. [Google Scholar] [CrossRef]
- Chellapandi, P. Structural-functional integrity of hypothetical proteins identical to ADPribosylation superfamily upon point mutations. Protein Pept. Lett. 2014, 21, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Chen, J.; Blundell, T.L.; Ascher, D.B. Ascher, In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity. Sci. Rep. 2016, 6, 19848. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhao, Y.; Guo, C.; Gan, Y.; Huang, H. The role of proline substitutions within flexible regions on thermostability of luciferase. Biochim. Biophys. Acta 2015, 1854, 65–72. [Google Scholar] [CrossRef]
- Atsavapranee, B.; Sunden, F.; Herschlag, D.; Fordyce, P.M. Quantifying protein unfolding kinetics with a high-throughput microfluidic platform. bioRxiv 2025. [Google Scholar] [CrossRef]
- Kellis, J.T.; Nyberg, K.; Fersht, A.R. Energetics of complementary side-chain packing in a protein hydrophobic core. Biochemistry 1989, 28, 4914–4922. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Montero, K.; Rojas-Villalta, D.; Hernández-Moncada, R.; Esquivel, A.; Barrientos, L. Genome Sequence of Pseudomonas sp. Strain So3.2b, Isolated from a Soil Sample from Robert Island (Antarctic Specially Protected Area 112), Antarctic. Microbiol. Resour. Announc. 2023, 12, e01167-22. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lin, S.; Zou, Z.; Zhou, C.; Zhang, H.; Cai, Z. Transcriptome Analysis Reveals the Molecular Mechanisms Underlying Adenosine Biosynthesis in Anamorph Strain of Caterpillar Fungus. Biomed Res. Int. 2019, 2019, 1864168. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Pedersen, L.E.; Weber, T.; Lee, S.Y. CRISPy-web: An online resource to design sgRNAs for CRISPR applications. Synth. Syst. Biotechnol. 2016, 1, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Berendsen, H.J.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Tomasello, G.; Armenia, I.; Molla, G. The Protein Imager: A full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 2020, 36, 2909–2911. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2; Springer: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- MODELLER A Program for Protein Structure Modeling Release 10.6, r12888. Available online: https://salilab.org/modeller/10.6/manual/?utm_source=chatgpt.com (accessed on 14 May 2025).

{kind=link}
{kind=link}
{kind=link}
| Locus Tag | Product | Length | Function | |
|---|---|---|---|---|
| NT | AA | |||
| C8250_002175 | TetR/AcrR family transcriptional regulator | 621 | 206 | Regulatory |
| C8250_002330 | TetR/AcrR family transcriptional regulator | 840 | 279 | Regulatory |
| Medium | Components (Per Litre) |
|---|---|
| IMA | Yeast extract (4 g), Malt extract (10 g), Glucose (4 g), Mannitol (40 g) |
| YES | Sucrose (150 g), Yeast extract (20 g), MgSO4·7H2O (0.5 g), ZnSO4·7H2O (0.01 g), CuSO4·5H2O (0.005 g) |
| ISP-4 | Starch (10 g), CaCO3 (2 g), (NH4)2SO4 (2 g), K2HPO4 (1 g), MgSO4·7H2O (1 g), NaCl (1 g), FeSO4·7H2O (1 mg), MnCl2·7H2O (1 mg), ZnSO4·7H2O (1 mg) |
| M2 | Mannitol (40 g), Maltose (40 g), Yeast extract (10 g), K2HPO4 (2 g), MgSO4·7H2O (0.5 g), FeSO4·7H2O (0.01 g) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leal, K.; Machuca, J.; Gajardo, H.; Palma, M.; Contreras, M.J.; Nuñez-Montero, K.; Gutiérrez, Á.; Barrientos, L. Structural Characterisation of TetR/AcrR Regulators in Streptomyces fildesensis So13.3: An In Silico CRISPR-Based Strategy to Influence the Suppression of Actinomycin D Production. Int. J. Mol. Sci. 2025, 26, 4839. https://doi.org/10.3390/ijms26104839
Leal K, Machuca J, Gajardo H, Palma M, Contreras MJ, Nuñez-Montero K, Gutiérrez Á, Barrientos L. Structural Characterisation of TetR/AcrR Regulators in Streptomyces fildesensis So13.3: An In Silico CRISPR-Based Strategy to Influence the Suppression of Actinomycin D Production. International Journal of Molecular Sciences. 2025; 26(10):4839. https://doi.org/10.3390/ijms26104839
Chicago/Turabian StyleLeal, Karla, Juan Machuca, Humberto Gajardo, Matías Palma, María José Contreras, Kattia Nuñez-Montero, Álvaro Gutiérrez, and Leticia Barrientos. 2025. "Structural Characterisation of TetR/AcrR Regulators in Streptomyces fildesensis So13.3: An In Silico CRISPR-Based Strategy to Influence the Suppression of Actinomycin D Production" International Journal of Molecular Sciences 26, no. 10: 4839. https://doi.org/10.3390/ijms26104839
APA StyleLeal, K., Machuca, J., Gajardo, H., Palma, M., Contreras, M. J., Nuñez-Montero, K., Gutiérrez, Á., & Barrientos, L. (2025). Structural Characterisation of TetR/AcrR Regulators in Streptomyces fildesensis So13.3: An In Silico CRISPR-Based Strategy to Influence the Suppression of Actinomycin D Production. International Journal of Molecular Sciences, 26(10), 4839. https://doi.org/10.3390/ijms26104839