Abstract
Epilepsy is a prevalent neurological condition, having a wide range of phenotypic traits, which complicate the diagnosis process. Next-generation sequencing (NGS) techniques have improved the diagnostics for unexplained epilepsies. Our goal was to evaluate the utility and impact of genetic testing in the clinical management of pediatric epilepsies. In addition, we aimed to identify clinical factors that could predict a genetic diagnosis. This was a retrospective study of 140 pediatric patients with epilepsy with or without other neurological conditions that underwent NGS testing (multigene panel, WES = whole exome sequencing and/or WGS = whole genome sequencing). A comparison between genetically diagnosed versus non-diagnosed children was performed based on different clinical features. Univariate and multivariate logistic regression analysis was performed to identify clinical predictors of a positive genetic diagnosis. Most children underwent gene panel testing, while 30 had exome sequencing and 3 had genome sequencing. The overall diagnostic yield of genetic testing was 28.6% (40/140) for more than 28 genes. The most frequently identified genes with causative variants were SCN1A (n = 4), SCN2A (n = 3), STXBP1 (n = 3), MECP2 (n = 2), KCNQ2 (n = 2), PRRT2 (n = 2), and NEXMIF (n = 2). Significant predictors from the logistic regression model were a younger age at seizure onset (p = 0.015), the presence of intellectual disability (p = 0.021), and facial dysmorphism (p = 0.049). A genetic diagnosis led to an impact on the choice or duration of medication in 85% (34/40) of the children, as well as the recommendation for screening of comorbidities or multidisciplinary referrals in 45% (18/40) of children. Epilepsy is a highly heterogeneous disorder, both genetically and phenotypically. Less than one third of patients had a genetic diagnosis identified using panels, exomes, and/or genomes. An early onset and syndromic features (including global developmental delay) were more likely to receive a diagnosis and benefit from optimized disease management.
1. Introduction
Epilepsy is a frequent neurological condition, with a global prevalence of 0.64% [1]. There is high genetic and phenotypic heterogeneity in epilepsy, which can make the diagnostic process more difficult [2,3]. According to current estimates, genetic factors contribute to 70–80% of epilepsy cases [4]. Genetic epilepsies can be broadly divided into rare monogenic epilepsies caused by single genetic variations and common epilepsies, characterized by a complex architecture of a multifactorial inheritance (multiple genetic variants together with environmental factors) [5,6].
Next-generation sequencing (NGS) improved the field of epilepsy genetics, with proven benefits in diagnosis, genetic counseling, and therapeutic approaches [7]. A genetic diagnosis in a patient with epilepsy can directly impact the choice of antiseizure medication, the eligibility for clinical trials, the cost of additional tests, the genetic testing for the relatives at risk, and the reproductive decision making [3].
The diagnostic yield of genetic tests for epilepsy can vary. A systematic review and meta-analysis that included 154 studies reported an overall diagnostic yield for genetic testing of 17% (48% for genome sequencing, 25% for exome sequencing, and 19% for gene panels) [8]. Stefanski A et al. in their systematic evidence review reported a 24% diagnostic rate for epilepsy, with the cohort of patients with intellectual disability and early-onset seizures having a higher diagnostic yield [9].
In Romania, NGS became available as part of the diagnostic workup relatively recently. In 2022, Riza et al. presented a case series of 36 Romanian patients with the early-onset developmental and epileptic encephalopathy (DEE) phenotype with an overall diagnostic rate of molecular genetic testing of 25% [10]. However, there is still a need for the evidence of utility and impact of genetic testing in the clinical management of epilepsy [8].
In our study, we assessed the utility of gene panels, exomes, and genome sequencing to diagnose the genetic etiology of Romanian children with epilepsy, with or without other neurological impairments. We aimed to evaluate the diagnostic yield of different genetic testing methods, as well as the impact on clinical management and the practice of precision medicine in the chosen cohort. In addition, we sought to assess the clinical predictors of a genetic diagnosis in NGS epilepsy testing.
2. Results
2.1. Patients’ Characteristics
The cohort consisted of 140 patients (male-to-female ratio 1:1) with a diagnosis of epilepsy/symptoms of seizures/modified EEG pattern with or without other neurological impairments, for which NGS genetic testing (multigene panels, WES, or WGS) was performed. The patients’ median age at the onset of seizures was 2.5 years (interquartile range IQR = 0.8–4.0). The median difference between the time at seizure onset and time of genetic testing was 2.1 years (IQR = 0–5.9). The age at seizure onset was statistically lower in the group of patients with a disease-causing variant compared to those without (p = 0.009), as well as the age at the time of genetic testing (p = 0.002). An epilepsy syndrome diagnosis, according to the 2022 ILAE classification [11], in all patients was not possible, as the seizure onset in categories of childhood onset, variable age onset, or idiopathic generalized epilepsy, could not be performed precisely; 35% of patients (n = 49) had diagnosed electroclinical epilepsy syndrome, out of which the most frequent included West syndrome (n = 7, 14.3%), genetic epilepsy with febrile seizures plus (n = 6, 12.2%), Dravet syndrome (n = 3, 6.1%), self-limited epilepsy with centrotemporal spikes (n = 3, 6.1%), and Lennox–Gastaut (n = 2, 4.1%). Epileptic encephalopathy or developmental and epileptic encephalopathy was present in 24 patients (17.0%). Fourteen patients (10%) had drug resistant epilepsy. Further details about patients’ phenotyping are available in Table 1.

Table 1.
Clinical characteristics of 140 pediatric patients with epilepsy and comparison between patients with and without a disease-causing genetic variant.
2.2. Genetic Findings
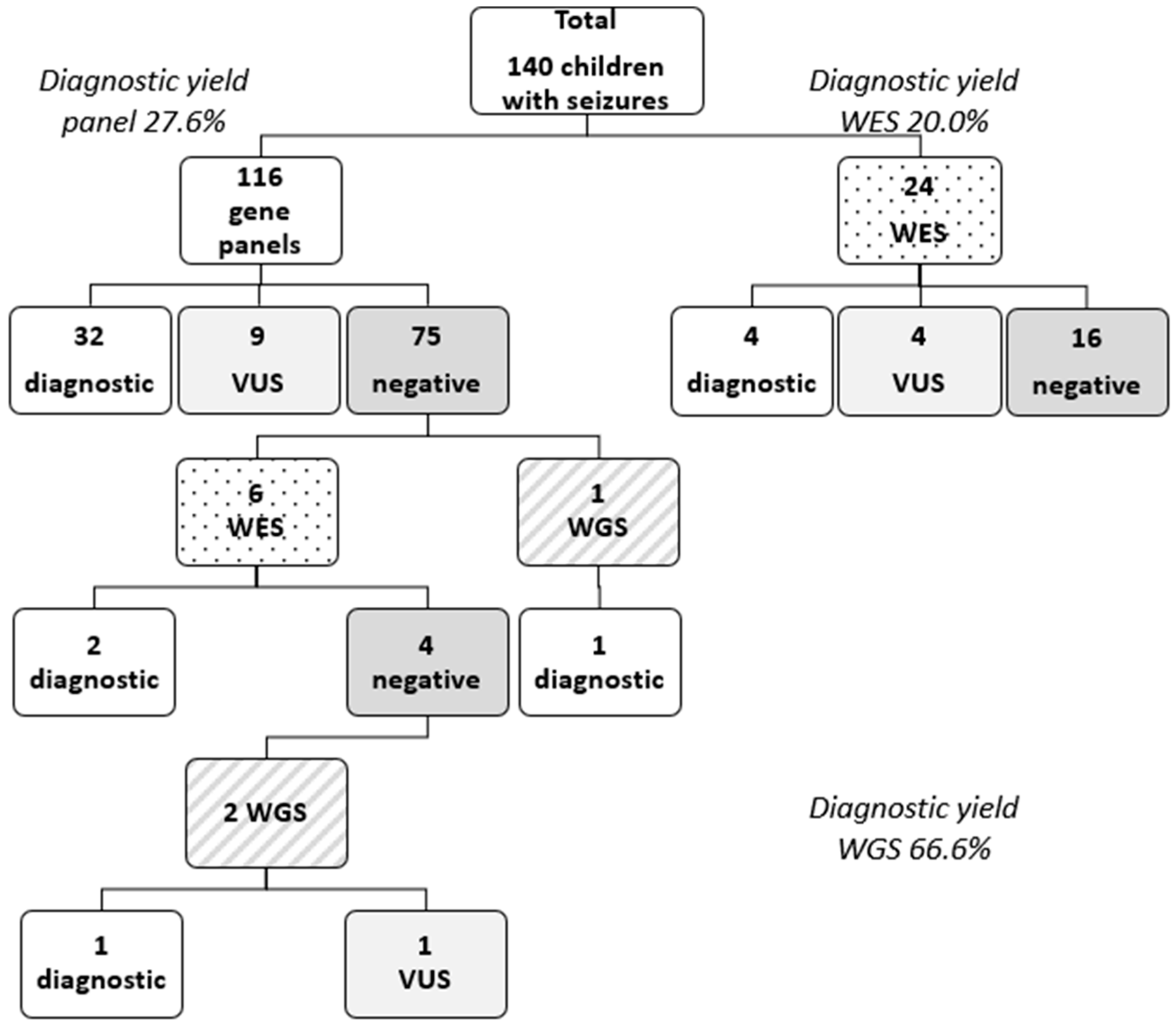
Out of the 140 patients, 116 patients underwent NGS gene panel testing, 30 patients underwent exome sequencing (19 solo WES, 10 trio WES, 1 quad WES), and 3 patients underwent WGS. A genetic etiology for epilepsy was identified in 40 patients, leading to an overall diagnostic yield of 28.6%. A positive result through multigene panel testing was obtained in 32 patients (27.6% diagnostic yield for multigene panel testing). From the subset of patients on which exome testing was performed, pathogenic or likely pathogenic variants were identified in 6 of them (20% diagnostic yield for exome sequencing). The WGS diagnostic yield was higher 66.6% (2/3) (see Figure 1 Patient diagnostic flowchart). However, most of the children from this cohort lacked an identifiable genetic diagnosis (n = 100, 71%). Fourteen unresolved cases (10%) are still under observation and need a further reevaluation of the VUS.
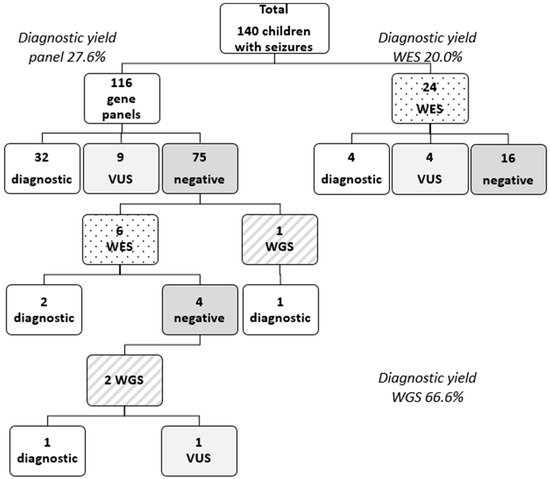
Figure 1.
Patient genetic testing and diagnostic flowchart. Legend: VUS = variant of unknown significance; WES = whole-exome sequencing; WGS = whole-genome sequencing.
Information about familial segregation was obtained for 52 children only (37.1%), mostly due to a lack of family compliance. However, family screening enabled variant reclassification from a variant of uncertain significance to likely pathogenic or a confirmation that variants were in trans in 10 children, offering an additional diagnostic yield for genetic testing (+7.1%).
The most frequently identified genes with pathogenic or likely pathogenic variants were SCN1A (n = 4), SCN2A (n = 3), STXBP1 (n = 3), MECP2 (n = 2), KCNQ2 (n = 2), PRRT2 (n = 2), and NEXMIF (n = 2). A description of the causative genes according to the NCBI Gene database [12] is presented in the Supplementary File. The causative genes identified in this study encompassed 28 causative genes, of which 15 were primary epilepsy genes (CACNA1A, KCNC1, KCNH5, KCNQ2, NRXN1, PRRT2, SCN1A, SCN2A, SCN8A, SLC2A1, STXBP1, SYNGAP1, TSC1, TSC2, WWOX) and 13 were syndromic epilepsy genes or syndromes (Phelan–McDermid Syndrome/22q13.33 deletion syndrome—ARSA, ALG12; chromosome 16p12.2p11.2 deletion syndrome—CLN3, EIF2B5, MECP2, MT-CYB, NEXMIF/KIAA2022, NFIA, NR2F1, P4HTM, PPP2R5D, PTCD3, PURA, RAI1, UBE3A). In relation to the gene disease mechanism, from the total of 28 causative genes, 10 were epilepsy genes (CACNA1A coding a calcium channel, SCN1A, SCN2A, and SCN8A coding sodium channels, KCNQ2 and KCNC1 coding potassium channels, SLC2A1 coding a glucose transporter, STXBP1 coding a protein involved in membrane trafficking, WWOX coding an enzyme, and the PRRT2 gene coding a transmembrane protein), 2 genes (TSC1 and TSC2 causing Tuberous Sclerosis) were neurodevelopment-associated genes, 7 genes were epilepsy-related genes (UBE3A, PPP2R5D, PURA, MECP2, ALG12, ARSA, CLN3), and 6 genes can be classified as potentially epilepsy-associated genes (KCNH5, NEXMIF, NFIA, P4HTM, PTCD3, EIF2B5).
Most of the causative variants were in a heterozygous state (35/40), four variants were in a compound heterozygous state and one mitochondrial variant had 5.3% heteroplasmy in the blood. A muscle biopsy was not accepted by the family for diagnosis. Among the positive results, the mode of inheritance of the causative genes was predominantly autosomal dominant (32/40), 4/40 had autosomal recessive inheritance, 3 variants were associated with dominant X-linked disorder (MECP2 gene and NEXMIF gene), and the MT-CYB variant had a mitochondrial mode of inheritance. Most of the disease-causing variants were point mutations/single nucleotide variants (SNVs; n = 32/40, 80%). Classifying SNVs by molecular consequence, most of them were missense variants (20/32), five were nonsense, four were frameshift, and two were stopgain. The NGS multigene panel testing identified eight children with pathogenic/likely pathogenic CNVs or indels: five children with a deletion of the entire coding sequence, one variant consisting of a deletion of exons 7–18 in the NRXN1 gene, one deletion of 12 bp of the STXBP1 gene, and one indel/copy number gain of exon 5 of the WWOX gene. For three of the previously mentioned eight children, a SNParray or array CGH was performed to confirm the deletion.
Comparing the proportion of children with a disease-causing variant with those without based on different phenotypic subsets, a statistically significant difference was observed for the presence of the following: EE/DEE (p = 0.000), age at genetic testing under 2 years (p = 0.000), developmental delay (p = 0.000), stationary evolution (p = 0.000), speech delay (p = 0.001), stationary evolution (p = 0.002), facial dysmorphism (p = 0.008), age at seizure onset below 2 years (p = 0.009), intellectual disability (p = 0.014), autism spectrum disorder (p = 0.035), female gender (p = 0.040), as well as antiepileptic monotherapy (p = 0.043). A high genetic diagnostic yield was obtained for the subset of patients with EE/DEE 15/24 (62.5%), followed by facial dysmorphism (59%), an age at genetic testing under 2 years 10/17 (58.8%), stationary evolution 20/37 (54.1%), autism spectrum disorder 13/28 (46.4%), an age at onset of seizures under 2 years 27/60 (45.0%), intellectual disability 33/74 (44.6%), and developmental delay 32/74 (43.2%).
2.3. Impact of Genetic Diagnosis on Clinical Management
Identifying a genetic diagnosis led to an impact in the choice or duration of medication in 85% of the diagnosed patients (n = 34/40). Multidisciplinary management was influenced by the genetic diagnosis in 45% of positive cases (n = 18/40). A genetic diagnosis led to the recommendation of a ketogenic diet in 17.5% of the diagnosed cases (n = 7/40). One case (2.5%) was referred for a clinical trial. Table 2 summarizes the impact of a genetic diagnosis divided into the four categories mentioned above, showing the causative genes/syndromes in each category. All patients with a causative genetic variant received genetic counseling. More than half (52.5%) of the diagnosed patients had an impact in three categories (n = 21/40), while 37.5% (n = 15/40) had an impact in two categories and 7.5% (n = 3/40) had an impact in four categories.
Table 2.
Impact of genetic testing in patients with a genetic diagnosis.
2.4. Clinical Predictors of a Genetic Diagnosis
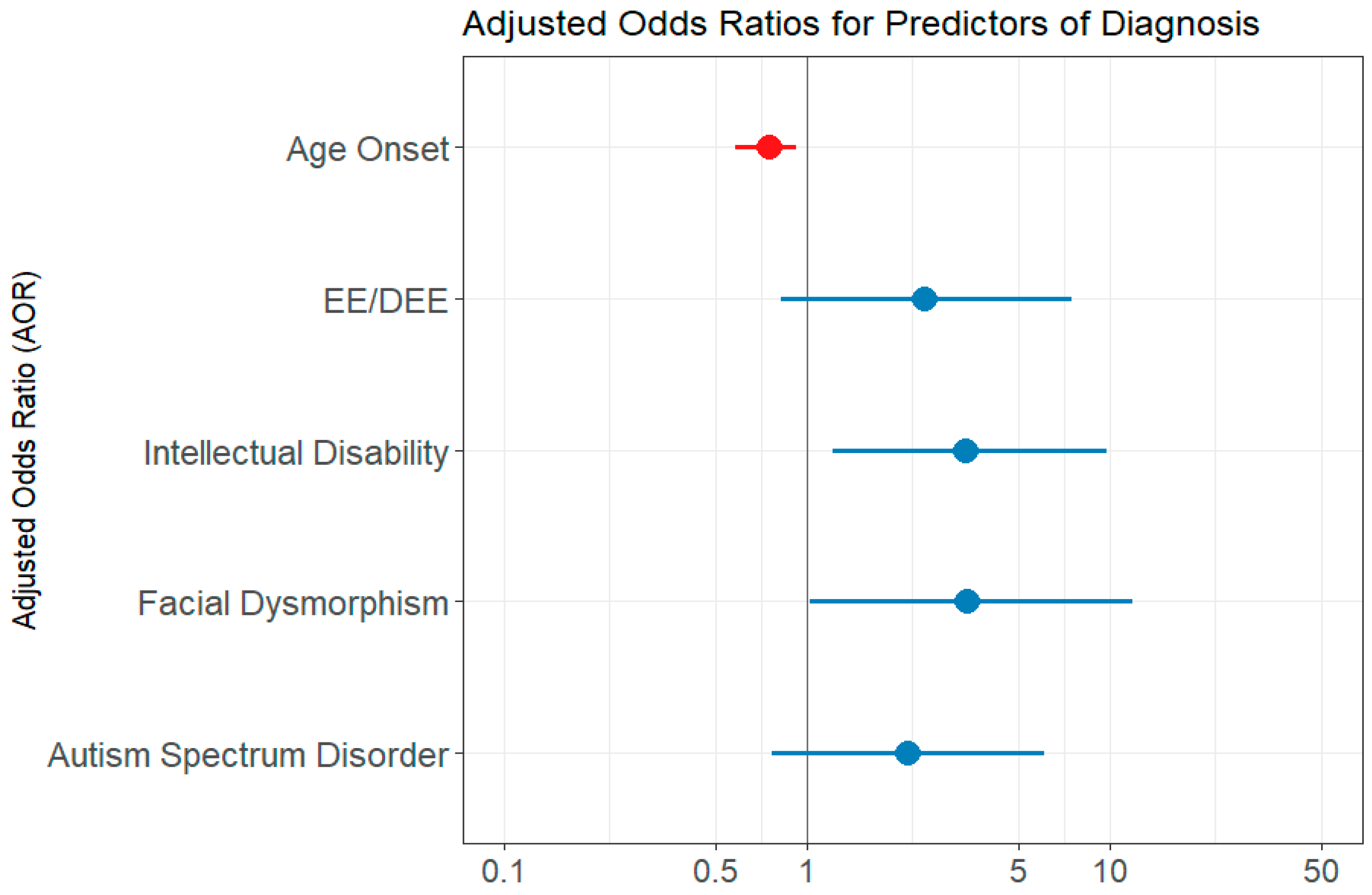
The univariate logistic regression analysis showed the following predictor variables as statistically significant: age at seizure onset, presence of EE/DEE, presence of intellectual disability, developmental delay, autism spectrum disorder, facial dysmorphism (Table 3). The variable number of genes included in the test performed did not reach statistical significance (p = 0.856). However, when computing the multivariate regression model, which first included all of the significant variables from the univariate analysis, each of the variables became a weaker predictor and did not reach statistical significance. We assumed that the variables under investigation contained resembling information regarding the outcome and that not all of them are needed to capture the information [13]. Indeed, when we assessed the correlation between age at seizure onset and the presence of EE/DEE, we found a significant strong negative correlation, using both the Kendall test (p= 0.003) and Spearman test (p = 0.018). Developmental delay and intellectual disability variables were also found to be intercorrelated (Kendall test p = 0.0183). Intellectual disability was also correlated with the presence of EE/DEE (Kendal test p = 0.007, Spearman test p = 0.007). The final multivariate regression model included the following variables: age at epilepsy onset, EE/DEE, intellectual disability, facial dysmorphism, and autism spectrum disorder. The AIC value was 136.85. Features predictive of a genetic diagnosis were the age at seizure onset, intellectual disability, and facial dysmorphism. Autism spectrum disorder and EE/DEE did not reach statistical significance (Table 3 and Figure 2). The chi-squared verisimilitude test revealed a p-value of 0.003, showing that the model is statistically valid. In addition, the Hoslem–Lemeshow concordance test showed that the model is good to describe the data (p = 0.964). The training accuracy through cross-validation was 0.74, and the predictive accuracy was 0.88.
Table 3.
Univariate and multivariate logistic regression models of phenotypic features as predictors of a positive genetic diagnosis.
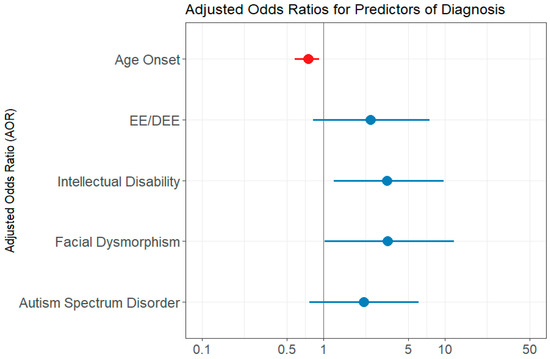
Figure 2.
Multivariate logistic regression model evaluation—forest plot of adjusted odds ratio (AOR). EE/DEE = epileptic encephalopathy/developmental and epileptic encephalopathy; ID = intellectual disability; DD = developmental delay; ASD = autism spectrum disorder. The forest plot displays multivariate logistic regression results examining associations between clinical features and the likelihood of a molecular diagnosis. Red points represent predictors with AOR < 1, indicating an inverse relationship, while blue points represent predictors with AOR > 1, indicating a direct association. The horizontal lines indicate 95% confidence intervals. Predictors are considered statistically significant if their 95% confidence interval does not include 1.
3. Discussion
This retrospective study evaluated 140 patients with epilepsy with or without other neurological conditions that underwent NGS testing. In 40 patients, pathogenic or likely pathogenic genetic variants were identified according to ACMG guidelines. The overall diagnostic yield was 28.6%, while gene panels had a diagnostic yield of 27.6% and exome sequencing had 20.0%. The highest diagnostic yield was found for genome sequencing (66.6%); however, only three children underwent WGS. Notably, six children for whom a gene panel was performed subsequently underwent WES. This is consistent with other previous studies, including a recent systematic review, which reported a 19% diagnostic yield for gene panel testing, 24% for exome sequencing, and 48% for genome sequencing in a cohort of 31,000 patients [8]. A. Stefanski et al., in their systematic review, reported an overall 24% diagnostic yield for sequencing testing (panels and exome) in epilepsy from 72 different studies [9]. Indeed the most comprehensive genetic test in epilepsy remains WGS, having the highest diagnostic yield of 48% in patients with epilepsy, as well as other neurodevelopmental disorders [8]. WGS has the advantage of detecting sequence variants, CNVs, and repeat expansion variations; however, it is not covered by the national insurance and interpretation is often hampered due to limited knowledge about the non-coding genome [14] On the other hand, multigene panel testing in epilepsies has the advantage of a lower cost and no incidental findings compared to wider testing; the drawbacks of panel testing in epilepsy might be that the newly discovered genes are not included in the panel. Furthermore, gene panels report a higher number of VUSs (depending on laboratory protocols), as panels are not usually performed in trio [14]. The cost-effectiveness of WES in the clinical practice of epilepsy has been proven before, leading to an overall reduction of costs due to shortening the time to diagnosis [15,16].
The diagnostic yield of genetic testing in epilepsy may vary, depending not only on the test performed, but also on the phenotype. For gene panels and WES, for example, the diagnostic yield may be highest for patients with early-onset DEE, ranging from 34% to 50% [14]. For multigene panels, the diagnostic yield is around 19%, but it might reach 54% when epilepsy is present along with structural brain abnormalities [8]. According to another study, patients with seizure onset before the age of 2 years had a 34% diagnostic yield using gene panels, whereas those with seizure onset after the age of 2 years had a 4% yield [17]. Other studies showed that the presence of developmental delay or intellectual disability may be predictive of a positive genetic diagnosis [18,19]. Rochtus et al. reported the highest diagnostic rate in patients with DEE (44%) [20]. In this cohort, we obtained a statistically significant diagnostic yield for the subset of patients that had developmental and/or epileptic encephalopathy, facial dysmorphism, developmental delay, seizure onset under 2 years, age at genetic testing under 2 years, intellectual disability, and autism spectrum disorder compared to those without. These findings suggest that patients with epilepsy and these characteristics should be swiftly referred to NGS testing to prevent diagnostic delays.
Although the panels had a variable number of genes included, this was not statistically correlated with the diagnostic yield in this study (logistic regression). The fact that the number of genes in the panel is not directly related to its yield has been previously highlighted [4,21,22]. Symonds et al. [23] concluded that SCN1A, KCNQ2, CDKL5, SCN2A, and STXBP1 are the genes that have been implicated in the most NGS research to date. Therefore, the number of frequently mutated genes included in the panel (not the total number) is thought to be a good indicator of the diagnostic yield. In addition, the appropriate selection of patients is an important aspect to be considered as highlighted by Blazekovic et al. [22]. The diagnostic yield of WES was 40% in a cohort of 125 patients of whom 70% had development and epileptic encephalopathy [20].
This study showed high phenotypic heterogeneity in epilepsy. The median age at the onset of seizures in the cohort (2.5 years) was statistically lower when comparing the proportion of patients with causative variants with those without. This is in line with other studies showing that the age at seizure onset is lower in positive cases [21,24]. Based on the seizure type, 38.8% of patients in this cohort had focal seizures, 29.5% had generalized seizures, and 31.8% had mixed (focal and generalized) seizures. There was no statistical significance regarding the type of seizure and the outcome of a positive result of genetic testing. However, statistical significance was reached if the patient had EE/DEE, with the highest diagnostic yield being achieved in the subset of patients with these characteristics (p = 0.0001, diagnostic yield = 62.5%).
In this cohort, forty causative variants were identified in 28 different genes, showing high genetic heterogeneity. As a result of the advancement in genetic testing availability and collaborative studies, an increasing number of genes linked to epilepsy has been discovered [25]. Indeed, a recent paper has curated a list of 926 genes that are associated with monogenic disorder involving epilepsy [26]. As Dunn et al. stated, genetic epilepsies can be categorized into two categories: primary genetic epilepsies and epilepsies as part of the symptoms of a genetic neurological disorder [4,27]. Jie Wang et al., in their review [25], identified 977 genes associated with epilepsy and classified them into four categories: 84 primary epilepsy genes, 74 genes associated with epilepsy and brain-developmental malformations, 536 epilepsy-related genes, and 284 genes that require further validation. Classifications of the causative genes identified in this study were made based on the reviews mentioned above and are presented in the results section. Another aspect to consider in terms of genetic heterogeneity refers to the type of genetic variation; 80% (32/40) of the total disease-causing variants were point mutations/single nucleotide variants. However, 20% (8/40) were copy number variants or indels. This highlights the importance of NGS bioinformatic pipelines to call for CNV events. It has been previously shown that copy number alterations have a significant role in clinical epilepsy [28,29]. A large genome-wide and phenome-wide association study of more than 700,000 individuals evaluated the importance of CNVs in epilepsy and seizure-associated disorders and identified 25 significant genome-wide loci, out of which 22 were novel; in addition, phenome-wide association analysis between individual CNVs and HPO terms showed, for six CNVs, 19 significant associations [30].
Although establishing a genetic diagnostic in monogenic epilepsy is often quite difficult, understanding the pathophysiological mechanisms may enable the use of precision medicine, as well as treatment optimization [21]. Ion channel diseases are a good example to understand the difference between gain-of-function versus loss-of-function variants when making treatment decisions [31]. In Dravet Syndrome caused by SCN1A-loss-of-function variants, sodium channel blockers should be avoided [31]. However, gain-of-function variants in the SCN2A gene are known to respond well to sodium channel blockers [31]. There are also other examples including retigabine in loss-of-function variants in KCNQ2 and KCNQ3 genes [32], carbamazepine in PRRT2-related disorders [33], and a ketogenic diet in SLC2A1 [33], among others. In addition, a genetic diagnosis for epilepsy may limit the need for additional tests, guide genetic counseling, inform the prognosis, or identify potential comorbidities [34]. Several studies evaluated the impact in the medical management of a genetic diagnosis in epilepsy [35,36,37,38] Haviland I et al. [7] reported an individualized direct medical impact in 72.4% of patients with a diagnostic result, with 45.4% of patients having an impact on treatment. In this cohort, 85% of the diagnosed patients had an impact in the choice or duration of the antiseizure medication. Two of the diagnosed individuals with a drug impact achieved seizure freedom (16p12.2p11.2 deletion syndrome, and the child with causative variant in KCNH5). However, six children with causative variants in the genes SCN2A, PRRT2, NEXMIF, SCN1A, and SYNGAP1 had refractory epilepsy. A few examples of the drug impact of the genetic diagnosis in this cohort include the avoidance of valproic acid in the case of MELAS syndrome due to possible toxicity of mitochondria [39], the administration of Everolimus in TSC1 Tuberous Sclerosis Complex [40], the recommendation of cannabidiol in developmental and epileptic encephalopathy SYNGAP1 [41], and the administration perampanel in NR2F1-related epilepsy [42]. Furthermore, negative genomic findings could also be considered helpful. It was difficult to assess the impact of negative results in this retrospective cohort; however, we cannot rule it out. While a negative result does not rule out all genetic contributions, it can provide some reassurance that a major known pathogenic variant was not found. It may prompt clinicians to explore alternative diagnoses, such as structural, metabolic, or autoimmune causes [43].
Prediction models are often used to help healthcare providers in the application of diagnostic tests [13]. A logistic regression model estimates the association between one or more independent variables (also called, predictors) with a binary dependent variable (also called, the outcome variable) [44]. In this study, univariate and multivariate logistic regression analysis was used to identify predictors for a positive genetic test result. Haviland et al. made a similar analysis, showing that an age at epilepsy onset under 2 years, focal motor seizures, developmental delay, and malformation of brain development are statistically significant predictors for the chosen outcome [7]. Benevides et al. [45] performed a multivariate analysis to determine predictors of a positive genetic diagnosis, showing that the first seizure in the context of fever and hypotonia were positively corelated with a genetic etiology, while atonic seizures were negatively associated. Wong et al. [46] used a classification and regression tree analysis to identify the phenotypic features associated with a genetic diagnosis in a cohort of 316 patients with neurodevelopmental disorders, showing that the female gender, history of motor delay, hypotonia, congenital heart disease, and early intervention were more likely to be associated with a genetic etiology. These studies show that prediction models based on phenotypic data could help clinicians to stratify patients for genetic testing.
While genetic testing in epilepsy led to a good diagnostic yield, there is still a high percentage of negative results in this cohort (71%). These unresolved genetic epilepsies may be due to several reasons, such as the following: complex polygenic inheritance, diseases that may be monogenic, but there are many genes that are not yet associated with a human disorder, the molecular defect could not be detected by the technique used (such as somatic mosaicism or complex structural rearrangements), the gene was not included in the panel, uninformative variant classification, such as VUS, due to limited data, the defect may be in the non-coding genome (regulatory elements, topologically associated domains, etc.). Potentially, the etiology of epilepsy might be uncovered by studying other omics (transcriptomics, epigenomics, or metabolomics) [43].
4. Material and Methods
4.1. Cohort Information
This retrospective study included pediatric patients diagnosed with epilepsy and/or other neurological disorders who underwent genetic testing between August 2019 and November 2023. The patients were referred to a tertiary care pediatric neurology clinic (Dr BACOS Cosma Iuliu Medical Center), with a minority being referred to the Regional Center of Medical Genetics Timis and Regional Center of Medical Genetics Dolj. All patients were examined by both a pediatric neurologist and a clinical geneticist.
The inclusion criteria were a clinical diagnosis of epilepsy, seizure-like symptoms or EEG (electroencephalogram) suggestive of epilepsy, and the availability of a genetic testing report (gene panels/WES/WGS). The exclusion criteria included patients who had epilepsy but did not undergo genetic testing, as well as those who underwent genetic testing but did not have a diagnosis of epilepsy, seizure symptoms, or EEG findings suggestive of epilepsy. A genetic testing decision was made on a case-by-case basis and initiated based on the neurologist’s or geneticist’s clinical judgment. In most cases, genetic testing was recommended after inconclusive imaging and EEG results. However, in some specific syndromic cases (e.g., suspected Tuberous Sclerosis Complex), genetic testing was used to confirm a suspected diagnosis based on clinical and imaging findings. Panel testing was most used as the first-tier test, with WES/WGS reserved for more complex or undiagnosed cases. However, patients who underwent panel testing were typically seen earlier in the study and often presented with more severe phenotypes. WES and WGS became more accessible later and were used for a broader spectrum of epilepsy types.
Data were retrospectively collected from the medical records of all patients, including demographic information, gender, age at genetic testing, age at epilepsy onset, associated comorbidities (intellectual disability, development delay, autism spectrum disorder, other comorbidities), seizure type, EEG findings, results of brain imaging (MRI-Magnetic Resonance Imaging or CT-Computed Tomography), and number of antiepileptic drugs used at last visit. The seizure type was subdivided into focal, generalized, focal, and generalized, with a special category of DEE [11]. The term DEE denotes “an epilepsy associated with developmental impairment that may be due to both the underlying etiology (development encephalopathy) and superimposed epileptic activity (epileptic encephalopathy)” [47]. In this cohort, individuals were considered as having DEE if they had a suspicion or confirmation of a genetic etiology, epilepsy onset <18 years, and an EEG pattern suggesting encephalopathy, as well as the presence of intellectual disability or development delay. We assessed whether the patient had drug-resistant epilepsy and a history of status epilepticus and whether the epilepsy was part of a genetic syndrome. To assess the severity of disease, the number of antiepileptic drugs and the frequency of seizures was reported (according to last medical visit). The frequency of seizures was classified into six categories: (a) one or fewer than one seizure per year, (b) one seizure per year, (c) 2–3 seizures per year, (d) monthly seizures, (e) one seizure per week, and (f) daily seizures. Drug-resistant epilepsy was defined as the “failure of adequate trials of two tolerated and appropriately chosen antiepileptic drugs schedules (whether as monotherapies or in combination) to achieve seizure freedom” [48]. Status epilepticus was defined considering revised guidelines from the Neurocritical Care Society as being a seizure lasting for five minutes or longer, presenting clinical and/or electrographic continuous seizure activity, or repeated seizure activity without recovery between episodes [49]. If possible, the epilepsy diagnosis was further specified according to the 2022 ILAE classification of epileptic syndromes [11]. Patients with idiopathic/benign epilepsies were included. The evolution of each patient’s disease was classified into three categories: favorable, stationary, and unfavorable/worsened evolution.
The study was approved by the Ethics Board of the Faculty of Medicine of University of Medicine and Pharmacy Victor Babes Timisoara (No. 64/8 November 2024). All patients included in the study gave their informed consent for anonymously using their clinical data and results of genetic testing for research purposes. This study was performed in accordance with the 1964 Declaration of Helsinki.
4.2. Genetic Testing
Prior to enrollment in the analysis, all the included patients underwent NGS genetic testing, either via Invitae Multigene Panel(s) testing (San Francisco, CA, USA), Blueprint Genetics WES (Espoo, Finland), or Centogene Whole Genome Sequencing (Rostock, Germany). All genetic tests included the detection of copy number variants/exonic deletions/exonic duplications. The number of genes included in the Invitae Epilepsy Panel varied from 133 to 964, depending on the time at genetic testing, as new genes were added to the panel throughout the study period. In some cases, depending on the phenotype and the genes available in the current epilepsy panel, one or more panels/genes were added. Besides the Invitae Epilepsy Panel, the additional panels/genes ordered by the clinician were as follows: Preliminary-evidence Genes for Epilepsy, Glycine Encephalopathy, FLNA gene, PTEN gene, RANBP2 gene, Tuberous Sclerosis Complex Panel, Rett and Angelman Syndromes and Related Disorders Panel, Supplemental Metabolic Newborn Screening Panel, Cerebral Palsy Spectrum Disorders Panel, Neurodevelopmental Disorders (NDD) Panel, Comprehensive Neurometabolic Disorders Panel.
Genetic variants were clinically interpreted considering the recommendation guidelines of the ACMG-AMP (American College of Medical Genetics and Genomics—Association for Molecular Pathology) published in 2015 [50]. Therefore, the genetic test result was categorized into one of the five ACMG-AMP categories: “pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign”, “benign”. Results were considered as disease-causing (positive) when identifying the presence of a pathogenic or likely pathogenic genetic variant: (1) in a heterozygous state associated with dominant conditions; (2) in a homozygous state or compound heterozygous state (in trans) associated with autosomal recessive conditions; and (3) in a hemizygous state associated with an X-linked recessive condition in males. All the other outcomes, including carrier results and variants of uncertain significance (VUS), were considered negative. In some cases of VUS that were highly suspected of pathogenicity based on the phenotype, we assessed the variant segregation in the patient’s family for a possible reclassification. Causative epilepsy genes identified were further classified based on existing classifications in the literature [4,25].
The clinical impact of genetic testing was evaluated in five categories: drug impact, dietary changes, clinical trials, multidisciplinary management, and genetic counseling. A direct impact on the choice of antiseizure medication was considered if the genetic diagnosis had an influence on the choice of the antiepileptic drug, had a specific drug contraindication, there was an influence regarding the duration of the medication, or there was a change in the medication. An impact in multidisciplinary management was considered if the genetic diagnosis led to recommendations for the screening of associated comorbidities or a referral for a multidisciplinary dispensary other than neurology.
4.3. Data Analysis
Clinical patient data were inserted into a Microsoft Excel sheet [51]. Further statistics and data analysis were performed using R-4.4.2 and R-Studio 2024.12.0+467 [52,53]. The diagnostic rate was computed as the number of genetically diagnosed patients in a specific subset (e.g., subset of patients with seizure onset under two years) and then reported as the percentage of the total number of patients in that category. Pearson’s chi-squared test statistic or a Fisher test were used to compare the proportion of the positive results versus negative results (R package stats, functions prop.test/fisher.test [52]). The Anderson normality test was used to asses whether the data had a normal distribution (function ad.test, R package nortest [54]. The Wilcoxon rank sum test was used to compare two independent continuous variables that did not follow a normal distribution (function wilcox.test). A logistic regression model was used to assess clinical factors predictive of a positive genetic diagnosis. Before creating the multivariate logistic regression model, univariate logistic regression was used to examine each predictor separately. A background selection approach was used to build the multivariate logistic regression model. The predictors in the final model were chosen based on the model’s Akaike information criterion (AIC) value and taking into consideration if there was a correlation between variables. The correlation of two variables that did not follow a normal distribution was assessed using the Kendall rank and Spearman rank correlation test (function cor.test). The evaluation of the logistic regression model included a quality evaluation of each predictor using the p-value and odds ratio, goodness of fit, and accuracy. A chi-squared verisimilitude test was used to check if the model was statistically validated (function lrtest, R package lmtest [55]. The Hosmer–Lemeshow test was used to check if the model was good to describe the data (function hoslem.test, R package ResourceSelection [56]. A forest plot was built to visualize the predictors ‘adjusted odds ratio of the model (function plot_model R package sjPlot [57]. The R package boot [58] was used to compute the model’s training and predicted accuracy. Statistical significance was defined as a value of p < 0.05.
5. Limitations
There are limitations to this observational retrospective study. First, the sample size was relatively small, which may have limited the statistical power. Perhaps, with higher a power rate, other predictors would have reached significance when evaluating the clinical predictors of a genetic diagnosis through genetic testing. Second, patient selection bias is possible given the evolving genetic testing practices over the study period. This study was conducted when the practice of genetic testing for epilepsy was evolving. In early years, genetic testing was most probably affected by selection bias; the most severely affected patients were proposed for genetic testing, which mainly consisted of multigene panel testing. On the other hand, in more recent years, a broader group of epilepsy patients underwent genetic testing, which consisted of exome sequencing. However, the Invitae laboratory regularly reevaluates variant classification and informs the physician and patient about reclassification. Even more, Invitae offers WES reanalysis every year and phenotypic data are requested to the physician. Test selection bias (for gene panel or WES) might be the reason for a lower diagnostic yield of exome sequencing compared to multigene panels. Another limitation would be that the study was performed on a Romanian population only, which may restrict its generalization. Nonetheless, this population has very few published studies investigating the genotype and phenotype in children with seizures. Regarding data collection, although all efforts were made to reduce missing data, some variable values remained incomplete. Epilepsy syndromes were classified using the ILAE 2022 [11] framework when possible. However, due to retrospective data collection and evolving diagnostic documentation, we could not systematically subcategorize all patients prior to testing. In addition, due to sample size constraints, we did not analyze benign or idiopathic epilepsies or developmental and epileptic encephalopathies separately, but it will be worth exploring in future studies. Furthermore, due to a lack of family compliance and other factors regarding regular visits and recommendations, segregation analysis was not performed on all children where necessary. A significant limitation of the study is that the impact of genetic testing was only evaluated in the patients with a genetic diagnosis. In addition, the numbers and percentages reported might be underestimated due to a lack of follow-up documentation, as some of the patients were followed elsewhere.
6. Conclusions
The genetic diagnosis of epilepsy in Romanian children was highly heterogeneous, both phenotypically and genetically, with 40 variants identified in 28 genes. Fewer than one third (28.6%) of patients had a genetic diagnosis identified using panels, exomes, and/or genomes, leading to changes in medical management, including treatment recommendations and surveillance for comorbidities. Early-onset and syndromic features (including global developmental delay) were more likely to receive a diagnosis and benefit from optimized disease management.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms26104843/s1.
Author Contributions
Conceptualization, I.M.S., I.S.B.-C. and A.C.-E.; Methodology, I.M.S., M.P. and A.C.-E.; Software, I.M.S. and B.D.; Validation, I.S.B.-C. and A.C.-E.; Formal analysis, I.M.S. and B.D.; Investigation, I.M.S., I.S.B.-C., M.P. and A.C.-E.; Resources, I.S.B.-C., I.S., M.P. and A.C.-E.; Data curation, I.M.S. and I.S.; Writing—original draft, I.M.S.; Writing—review & editing, A.C.-E.; Visualization, I.M.S. and A.C.-E.; Supervision, A.C.-E.; Project administration, A.C.-E. All authors have read and agreed to the published version of the manuscript.
Funding
We would like to acknowledge the Victor Babes University of Medicine and Pharmacy Timișoara for their support in covering the costs of publication for this research paper.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of UNIVERSITY OF MEDICINE AND PHARMACY “VICTOR BABES” TIMISOARA (No. 64/8 November 2024).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
The data supporting the findings of this study are available in the Supplementary Materials. Additional information may be provided by the corresponding author upon reasonable request.
Acknowledgments
We thank the patients and families for participating.
Conflicts of Interest
The authors declare no conflicts of interest. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Smith, L.; Malinowski, J.; Ceulemans, S.; Peck, K.; Walton, N.; Sheidley, B.R.; Lippa, N. Genetic testing and counseling for the unexplained epilepsies: An evidence-based practice guideline of the National Society of Genetic Counselors. J. Genet. Couns. 2023, 32, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.; Albury, C.L.; Maksemous, N.; Benton, M.C.; Sutherland, H.G.; Smith, R.A.; Haupt, L.M.; Griffiths, L.R. Next Generation Sequencing Methods for Diagnosis of Epilepsy Syndromes. Front. Genet. 2018, 9, 20. [Google Scholar] [CrossRef]
- Lesca, G.; Baumgartner, T.; Monin, P.; De Dominicis, A.; Kunz, W.S.; Specchio, N. Genetic causes of rare and common epilepsies: What should the epileptologist know? Eur. J. Med. Genet. 2022, 65, 104570. [Google Scholar] [CrossRef] [PubMed]
- Thakran, S.; Guin, D.; Singh, P.; Singh, P.; Kukal, S.; Rawat, C.; Yadav, S.; Kushwaha, S.S.; Srivastava, A.K.; Hasija, Y.; et al. Genetic Landscape of Common Epilepsies: Advancing towards Precision in Treatment. Int. J. Mol. Sci. 2020, 21, 7784. [Google Scholar] [CrossRef] [PubMed]
- Haviland, I.; Daniels, C.I.; Greene, C.A.; Drew, J.; Love-Nichols, J.A.; Swanson, L.C.; Smith, L.; Nie, D.A.; Benke, T.; Sheidley, B.R.; et al. Genetic Diagnosis Impacts Medical Management for Pediatric Epilepsies. Pediatr. Neurol. 2023, 138, 71–80. [Google Scholar] [CrossRef]
- Sheidley, B.R.; Malinowski, J.; Bergner, A.L.; Bier, L.; Gloss, D.S.; Mu, W.; Mulhern, M.M.; Partack, E.J.; Poduri, A. Genetic testing for the epilepsies: A systematic review. Epilepsia 2022, 63, 375–387. [Google Scholar] [CrossRef]
- Stefanski, A.; Calle-López, Y.; Leu, C.; Pérez-Palma, E.; Pestana-Knight, E.; Lal, D. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta-analysis. Epilepsia 2021, 62, 143–151. [Google Scholar] [CrossRef]
- Riza, A.L.; Streață, I.; Roza, E.; Budișteanu, M.; Iliescu, C.; Burloiu, C.; Dobrescu, M.A.; Dorobanțu, S.; Dragoș, A.; Grigorescu, A.; et al. Phenotypic and Genotypic Spectrum of Early-Onset Developmental and Epileptic Encephalopathies-Data from a Romanian Cohort. Genes 2022, 13, 1253. [Google Scholar] [CrossRef]
- Wirrell, E.C.; Nabbout, R.; Scheffer, I.E.; Alsaadi, T.; Bogacz, A.; French, J.A.; Hirsch, E.; Jain, S.; Kaneko, S.; Riney, K.; et al. Methodology for classification and definition of epilepsy syndromes with list of syndromes: Report of the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. Gene [Internet]. 2004. Available online: https://www.ncbi.nlm.nih.gov/gene/ (accessed on 15 January 2025).
- Shipe, M.E.; Deppen, S.A.; Farjah, F.; Grogan, E.L. Developing prediction models for clinical use using logistic regression: An overview. J. Thorac. Dis. 2019, 11, S574–S584. [Google Scholar] [CrossRef] [PubMed]
- Habela, C.W.; Schatz, K.; Kelley, S.A. Genetic Testing in Epilepsy: Improving Outcomes and Informing Gaps in Research. Epilepsy Curr. 2024. [Google Scholar] [CrossRef]
- Howell, K.B.; Eggers, S.; Dalziel, K.; Riseley, J.; Mandelstam, S.; Myers, C.T.; McMahon, J.M.; Schneider, A.; Carvill, G.L.; Mefford, H.C.; et al. A population-based cost-effectiveness study of early genetic testing in severe epilepsies of infancy. Epilepsia 2018, 59, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.E.; Schofield, D.; Shrestha, R.; Kandula, T.; Macintosh, R.; Lawson, J.A.; Andrews, I.; Sampaio, H.; Johnson, A.M.; Farrar, M.A.; et al. Integrating exome sequencing into a diagnostic pathway for epileptic encephalopathy: Evidence of clinical utility and cost effectiveness. Mol. Genet. Genom. Med. 2018, 6, 186–199. [Google Scholar] [CrossRef]
- Oates, S.; Tang, S.; Rosch, R.; Lear, R.; Hughes, E.F.; Williams, R.E.; Larsen, L.H.G.; Hao, Q.; Dahl, H.A.; Møller, R.S.; et al. Incorporating epilepsy genetics into clinical practice: A 360°evaluation. NPJ Genom. Med. 2018, 3, 13. [Google Scholar] [CrossRef]
- McKnight, D.; Morales, A.; Hatchell, K.E.; Bristow, S.L.; Bonkowsky, J.L.; Perry, M.S.; Berg, A.T.; Borlot, F.; Esplin, E.D.; Moretz, C.; et al. Genetic Testing to Inform Epilepsy Treatment Management from an International Study of Clinical Practice. JAMA Neurol. 2022, 79, 1267–1276. [Google Scholar] [CrossRef]
- Benson, K.A.; White, M.; Allen, N.M.; Byrne, S.; Carton, R.; Comerford, E.; Costello, D.; Doherty, C.; Dunleavey, B.; El-Naggar, H.; et al. A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. Eur. J. Hum. Genet. 2020, 28, 1066–1077. [Google Scholar] [CrossRef]
- Rochtus, A.; Olson, H.E.; Smith, L.; Keith, L.G.; El Achkar, C.; Taylor, A.; Mahida, S.; Park, M.; Kelly, M.; Shain, C.; et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia 2020, 61, 249–258. [Google Scholar] [CrossRef]
- Horák, O.; Burešová, M.; Kolář, S.; Španělová, K.; Jeřábková, B.; Gaillyová, R.; Česká, K.; Réblová, K.; Šoukalová, J.; Zídková, J.; et al. Next-generation sequencing in children with epilepsy: The importance of precise genotype-phenotype correlation. Epilepsy Behav. 2022, 128, 108564. [Google Scholar] [CrossRef]
- Blazekovic, A.; Gotovac Jercic, K.; Meglaj, S.; Duranovic, V.; Prpic, I.; Lozic, B.; Malenica, M.; Markovic, S.; Lujic, L.; Petelin Gadze, Z.; et al. Genetics of Pediatric Epilepsy: Next-Generation Sequencing in Clinical Practice. Genes 2022, 13, 1466. [Google Scholar] [CrossRef]
- Symonds, J.D.; McTague, A. Epilepsy and developmental disorders: Next generation sequencing in the clinic. Eur. J. Paediatr. Neurol. 2020, 24, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Costain, G.; Cordeiro, D.; Matviychuk, D.; Mercimek-Andrews, S. Clinical Application of Targeted Next-Generation Sequencing Panels and Whole Exome Sequencing in Childhood Epilepsy. Neuroscience 2019, 418, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.W.; Liang, X.Y.; Wang, J.; Gao, L.D.; Liao, H.J.; He, Y.H.; Yi, Y.H.; He, N.; Liao, W.P. Epilepsy-associated genes: An update. Seizure 2024, 116, 4–13. [Google Scholar] [CrossRef]
- Oliver, K.L.; Scheffer, I.E.; Bennett, M.F.; Grinton, B.E.; Bahlo, M.; Berkovic, S.F. Genes4Epilepsy: An epilepsy gene resource. Epilepsia 2023, 64, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Poduri, A.; Lowenstein, D. Epilepsy genetics--past, present, and future. Curr Opin. Genet. Dev. 2011, 21, 325–332. [Google Scholar] [CrossRef]
- Olson, H.; Shen, Y.; Avallone, J.; Sheidley, B.R.; Pinsky, R.; Bergin, A.M.; Berry, G.T.; Duffy, F.H.; Eksioglu, Y.; Harris, D.J.; et al. Copy number variation plays an important role in clinical epilepsy. Ann. Neurol. 2014, 75, 943–958. [Google Scholar] [CrossRef]
- Pérez-Palma, E.; Helbig, I.; Klein, K.M.; Anttila, V.; Horn, H.; Reinthaler, E.M.; Gormley, P.; Ganna, A.; Byrnes, A.; Pernhorst, K.; et al. Heterogeneous contribution of microdeletions in the development of common generalised and focal epilepsies. J. Med. Genet. 2017, 54, 598–606. [Google Scholar] [CrossRef]
- Montanucci, L.; Lewis-Smith, D.; Collins, R.L.; Niestroj, L.M.; Parthasarathy, S.; Xian, J.; Ganesan, S.; Macnee, M.; Brünger, T.; Thomas, R.H.; et al. Genome-wide identification and phenotypic characterization of seizure-associated copy number variations in 741,075 individuals. Nat. Commun. 2023, 14, 4392. [Google Scholar] [CrossRef]
- Bayat, A.; Bayat, M.; Rubboli, G.; Møller, R.S. Epilepsy Syndromes in the First Year of Life and Usefulness of Genetic Testing for Precision Therapy. Genes 2021, 12, 1051. [Google Scholar] [CrossRef]
- Millichap, J.J.; Park, K.L.; Tsuchida, T.; Ben-Zeev, B.; Carmant, L.; Flamini, R.; Joshi, N.; Levisohn, P.M.; Marsh, E.; Nangia, S.; et al. KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurol. Genet. 2016, 2, e96. [Google Scholar] [CrossRef]
- Helbig, I.; Ellis, C. A Personalized medicine in genetic epilepsies—Possibilities, challenges, and new frontiers. Neuropharmacology 2020, 172, 107970. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Toffa, D.H.; Lefèbvre, M.; Tétreault, M.; Cossette, P.; Samarut, É.; Nguyen, D.K. Usage of Genetic Panels in an Adult Epilepsy Clinic. Can. J. Neurol. Sci. 2023, 50, 411–417. [Google Scholar] [CrossRef] [PubMed]
- McKnight, D.; Bristow, S.L.; Truty, R.M.; Morales, A.; Stetler, M.; Westbrook, M.J.; Robinson, K.; Riethmaier, D.; Borlot, F.; Kellogg, M.; et al. Multigene Panel Testing in a Large Cohort of Adults with Epilepsy: Diagnostic Yield and Clinically Actionable Genetic Findings. Neurol. Genet. 2021, 8, e650. [Google Scholar] [CrossRef]
- Iglesias, A.; Anyane-Yeboa, K.; Wynn, J.; Wilson, A.; Truitt Cho, M.; Guzman, E.; Sisson, R.; Egan, C.; Chung, W.K. The usefulness of whole-exome sequencing in routine clinical practice. Genet. Med. 2014, 16, 922–931. [Google Scholar] [CrossRef]
- Valencia, C.A.; Husami, A.; Holle, J.; Johnson, J.A.; Qian, Y.; Mathur, A.; Wei, C.; Indugula, S.R.; Zou, F.; Meng, H.; et al. Clinical Impact and Cost-Effectiveness of Whole Exome Sequencing as a Diagnostic Tool: A Pediatric Center’s Experience. Front. Pediatr. 2015, 3, 67. [Google Scholar] [CrossRef] [PubMed]
- Matias, M.; Wusik, K.; Neilson, D.; Zhang, X.; Valencia, C.A.; Collins, K. Comparison of medical management and genetic counseling options pre- and post-whole exome sequencing for patients with positive and negative results. J. Genet. Couns. 2019, 28, 182–193. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Northrup, H.; Aronow, M.E.; Bebin, E.M.; Darling, T.N.; de Vries, P.J.; Frost, M.D.; Gosnell, E.S.; Gupta, N.; Jansen, A.C.; Jóźwiak, S.; et al. International Tuberous Sclerosis Complex Consensus Group Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr. Neurol. 2021, 123, 50–66. [Google Scholar] [CrossRef]
- Kuchenbuch, M.; D’Onofrio, G.; Chemaly, N.; Barcia, G.; Teng, T.; Nabbout, R. Add-on cannabidiol significantly decreases seizures in 3 patients with SYNGAP1 developmental and epileptic encephalopathy. Epilepsia Open 2020, 5, 496–500. [Google Scholar] [CrossRef]
- Li, X.; Gao, K.; Li, Y.; Zhang, Y.; Zhang, H.; Jiang, Y. Effective treatment of NR2F1-related epilepsy with perampanel. Acta Epileptol. 2024, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Johannesen, K.M.; Tümer, Z.; Weckhuysen, S.; Barakat, T.S.; Bayat, A. Solving the unsolved genetic epilepsies—Current and future perspectives. Epilepsia 2023, 64, 3143–3154. [Google Scholar] [CrossRef] [PubMed]
- Schober, P.; Vetter, T.R. Logistic Regression in Medical Research. Anesth. Analg. 2021, 132, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Luiza Benevides, M.; de Moraes, H.T.; Granados, D.M.M.; Bonadia, L.C.; Sauma, L.; Augusta Montenegro, M.; Guerreiro, M.M.; Lopes-Cendes, Í.; Carolina Coan, A. Predictors of genetic diagnosis in individuals with developmental and epileptic encephalopathies. Epilepsy Behav. 2024, 155, 109762. [Google Scholar] [CrossRef]
- Wong, N.R.; Klomhaus, A.; Adams, D.J.; Schneider, B.N.; Mehta, S.; DiStefano, C.; Wilson, R.B.; Martinez-Agosto, J.A.; Jeste, S.S.; Besterman, A.D. Clinical factors associated with genetic diagnosis in suspected neurogenetic disorders in a tertiary care clinic. Genet. Med. 2024, 27, 101252. [Google Scholar] [CrossRef]
- Wirrell, E.; Tinuper, P.; Perucca, E.; Moshé, S.L. Introduction to the epilepsy syndrome papers. Epilepsia 2022, 63, 1330–1332. [Google Scholar] [CrossRef]
- Jobst, B.C. Consensus Over Individualism: Validation of the ILAE Definition for Drug Resistant Epilepsy. Epilepsy Curr. 2015, 15, 172–173. [Google Scholar] [CrossRef]
- Wylie, T.; Sandhu, D.S.; Murr, N.I. Status Epilepticus [Internet]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Microsoft Corporation. Microsoft Excel 2018. Available online: https://office.microsoft.com/excel (accessed on 21 January 2025).
- R Core Team. _R: A Language and Environment for Statistical Computing_ [Internet]. Vienna, Austria: R Foundation for Statistical Computing. 2024. Available online: https://www.R-project.org/ (accessed on 21 January 2025).
- RStudio Team. RStudio: Integrated Development for R [Internet]. Boston, MA, USA. 2020. Available online: http://www.rstudio.com (accessed on 21 January 2025).
- Gross, J.; Ligges, U. _nortest: Tests for Normality_ [Internet]. 2015. Available online: https://CRAN.R-project.org/package=nortest (accessed on 21 January 2025).
- Zeileis, A.; Hothorn, T. Diagnostic Checking in Regression Relationships. R News 2002, 3, 7–10. [Google Scholar]
- Lele, S.R.; Keim, J.L.; Solymos, P. _ResourceSelection: Resource Selection (Probability) Functions for Use-Availability Data_ [Internet]. 2023. Available online: https://CRAN.R-project.org/package=ResourceSelection (accessed on 21 January 2025).
- Lüdecke, D. sjPlot:Data Visualization for Statistics in Social Science[R package] Version 2.8.15. 2024. Available online: https://CRAN.R-project.org/package=sjPlot (accessed on 21 January 2025).
- Canty, A.; Ripley, B. boot: Bootstrap R (S-Plus) Functions [R package]. Version 1.3-28. 2024. Available online: https://CRAN.R-project.org/package=boot (accessed on 21 January 2025).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).