
Targeting Myostatin as an Adjunct Treatment for the Preservation of Cardiometabolic and Skeletal Muscle Function in Type 1 Diabetes
 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Results
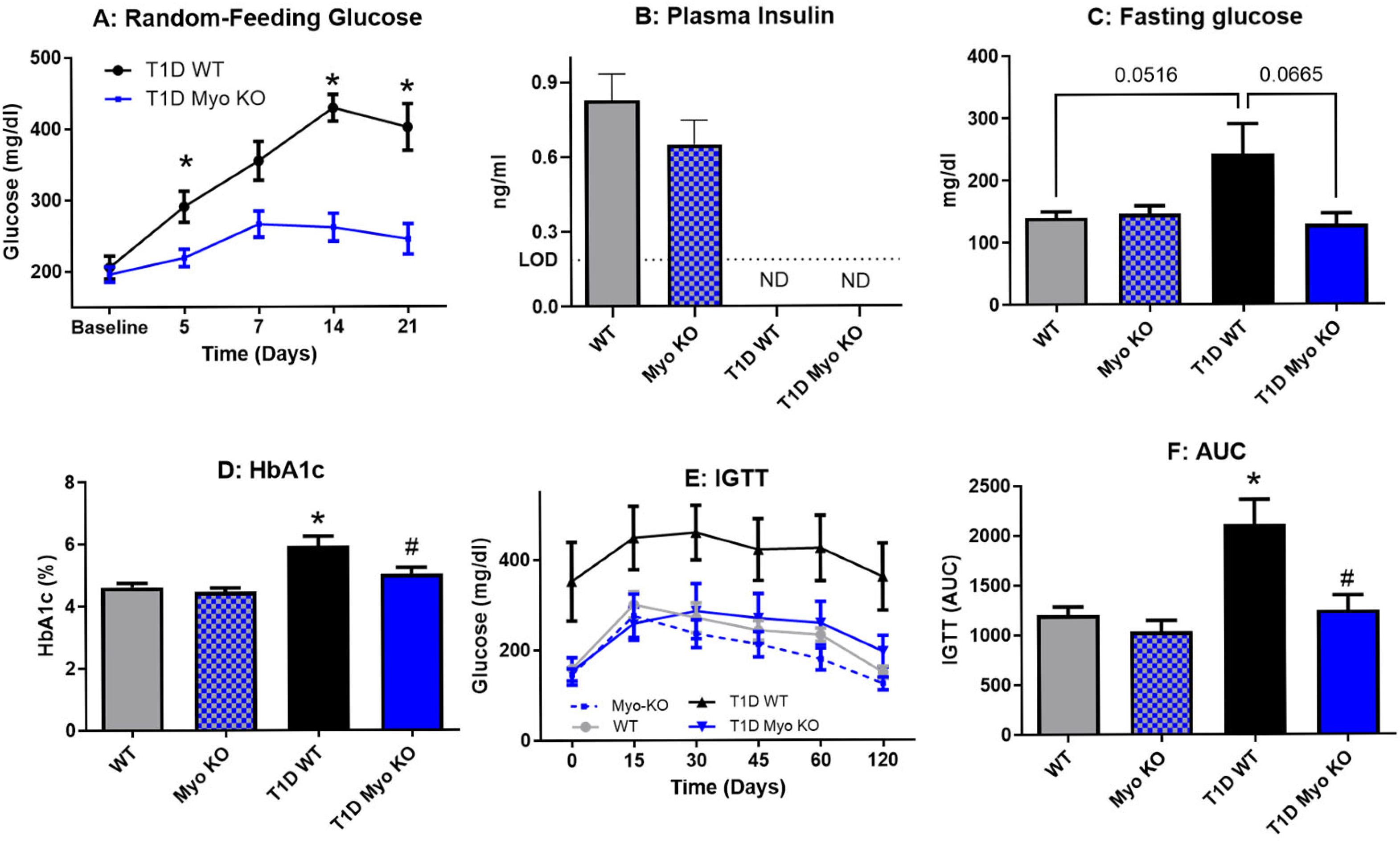
2.1. Deletion of Myostatin Preserves Glucose Homeostasis in Type 1 Diabetes
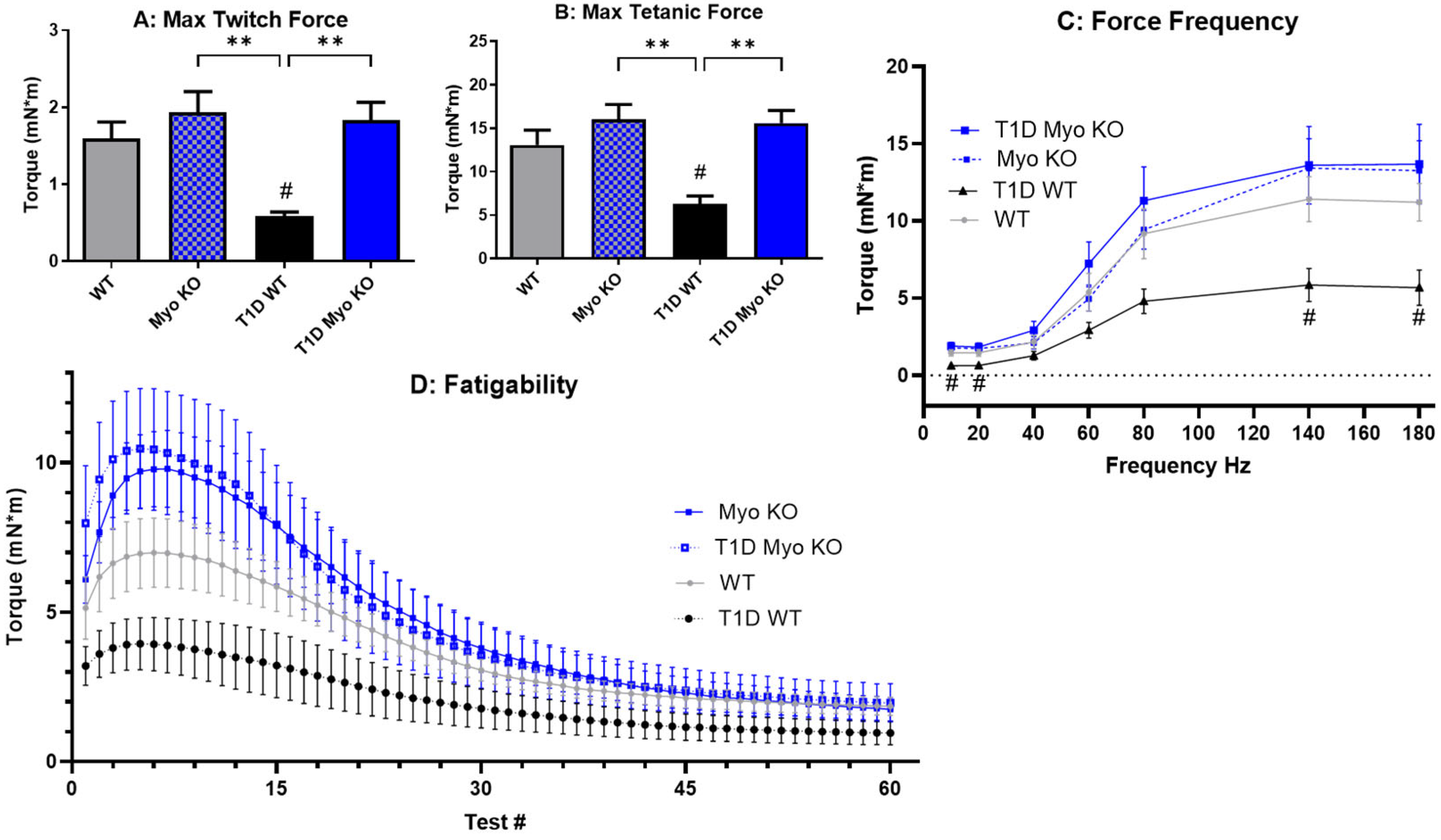
2.2. Deletion of Myostatin Preserves Muscle Function in Type 1 Diabetes
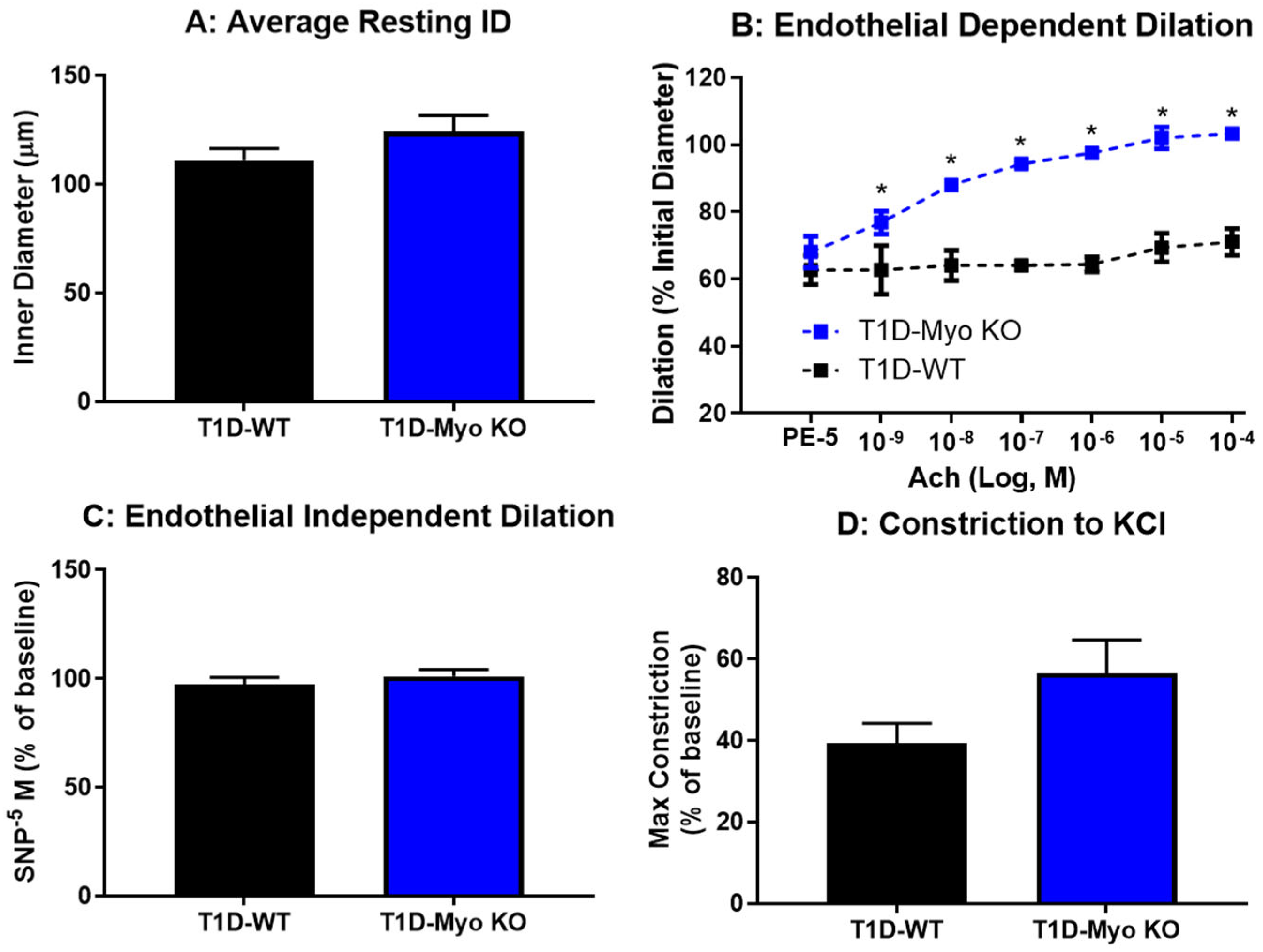
2.3. Deletion of Myostatin Protects Against Endothelial Dysfunction in Type 1 Diabetes
2.4. Deletion of Myostatin Protects Against Metabolic Dysfunction in Type 1 Diabetes
3. Discussion
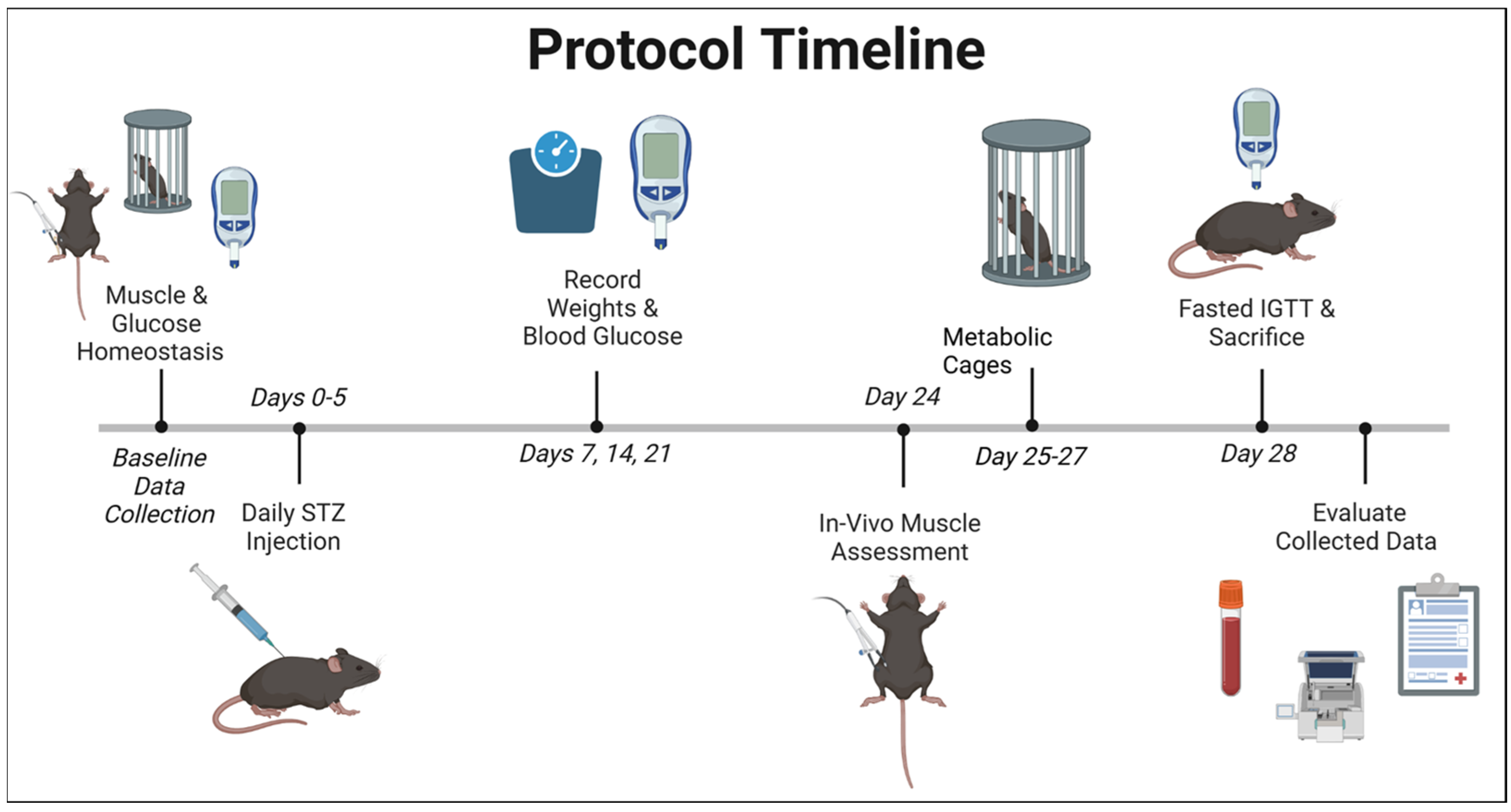
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| T1D | Type 1 Diabetes Mellitus |
| STZ | Streptozotocin |
| Myo | Myostatin |
References
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes-2023. Diabetes Care 2023, 46, S19–S40. [Google Scholar] [CrossRef] [PubMed]
- Leslie, R.D.; Evans-Molina, C.; Freund-Brown, J.; Buzzetti, R.; Dabelea, D.; Gillespie, K.M.; Goland, R.; Jones, A.G.; Kacher, M.; Phillips, L.S.; et al. Adult-Onset Type 1 Diabetes: Current Understanding and Challenges. Diabetes Care 2021, 44, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Gong, B.; Yang, W.; Xing, Y.; Lai, Y.; Shan, Z. Global, Regional, and National Burden of Type 1 Diabetes in Adolescents and Young Adults. Pediatr. Res. 2024, 97, 568–576. [Google Scholar] [CrossRef]
- Burahmah, J.; Zheng, D.; Leslie, R.D. Adult-Onset Type 1 Diabetes: A Changing Perspective. Eur. J. Intern. Med. 2022, 104, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report Website. 2022. Available online: https://www.cdc.gov/diabetes/php/data-research/index.html (accessed on 1 January 2025).
- Arffman, M.; Hakkarainen, P.; Keskimäki, I.; Oksanen, T.; Sund, R. Long-Term and Recent Trends in Survival and Life Expectancy for People with Type 1 Diabetes in Finland. Diabetes Res. Clin. Pract. 2023, 198, 110580. [Google Scholar] [CrossRef]
- Rawshani, A.; Sattar, N.; Franzén, S.; Rawshani, A.; Hattersley, A.T.; Svensson, A.-M.; Eliasson, B.; Gudbjörnsdottir, S. Excess Mortality and Cardiovascular Disease in Young Adults with Type 1 Diabetes in Relation to Age at Onset: A Nationwide, Register-Based Cohort Study. Lancet 2018, 392, 477–486. [Google Scholar] [CrossRef]
- Colom, C.; Rull, A.; Sanchez-Quesada, J.L.; Pérez, A. Cardiovascular Disease in Type 1 Diabetes Mellitus: Epidemiology and Management of Cardiovascular Risk. J. Clin. Med. 2021, 10, 1798. [Google Scholar] [CrossRef]
- Bhutta, Z.A.; Salam, R.A.; Gomber, A.; Lewis-Watts, L.; Narang, T.; Mbanya, J.C.; Alleyne, G. A Century Past the Discovery of Insulin: Global Progress and Challenges for Type 1 Diabetes among Children and Adolescents in Low-Income and Middle-Income Countries. Lancet 2021, 398, 1837–1850. [Google Scholar] [CrossRef]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.-Y.C.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group Intensive Diabetes Treatment and Cardiovascular Disease in Patients with Type 1 Diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [CrossRef]
- Stettler, C.; Allemann, S.; Jüni, P.; Cull, C.A.; Holman, R.R.; Egger, M.; Krähenbühl, S.; Diem, P. Glycemic Control and Macrovascular Disease in Types 1 and 2 Diabetes Mellitus: Meta-Analysis of Randomized Trials. Am. Heart J. 2006, 152, 27–38. [Google Scholar] [CrossRef]
- Anselmino, M.; Ohrvik, J.; Malmberg, K.; Standl, E.; Rydén, L. Euro Heart Survey Investigators Glucose Lowering Treatment in Patients with Coronary Artery Disease is Prognostically Important not only in Established but Also in Newly Detected Diabetes Mellitus: A Report from the Euro Heart Survey on Diabetes and the Heart. Eur. Heart J. 2008, 29, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Arboleda-Velasquez, J.F.; Valdez, C.N.; Marko, C.K.; D’Amore, P.A. From Pathobiology to the Targeting of Pericytes for the Treatment of Diabetic Retinopathy. Curr. Diabetes Rep. 2015, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.N.; Arboleda-Velasquez, J.F.; Amarnani, D.S.; Kim, L.A.; D’Amore, P.A. Retinal Microangiopathy in a Mouse Model of Inducible Mural Cell Loss. Am. J. Pathol. 2014, 184, 2618–2626. [Google Scholar] [CrossRef] [PubMed]
- Vergès, B. Cardiovascular Disease in Type 1 Diabetes, an Underestimated Danger: Epidemiological and Pathophysiological Data. Atherosclerosis 2024, 394, 117158. [Google Scholar] [CrossRef]
- Perticone, F.; Ceravolo, R.; Pujia, A.; Ventura, G.; Iacopino, S.; Scozzafava, A.; Ferraro, A.; Chello, M.; Mastroroberto, P.; Verdecchia, P.; et al. Prognostic Significance of Endothelial Dysfunction in Hypertensive Patients. Circulation 2001, 104, 191–196. [Google Scholar] [CrossRef]
- Knapp, M.; Tu, X.; Wu, R. Vascular Endothelial Dysfunction, a Major Mediator in Diabetic Cardiomyopathy. Acta Pharmacol. Sin. 2019, 40, 1–8. [Google Scholar] [CrossRef]
- Rebalka, I.A.; Noguchi, K.S.; Bulyovsky, K.R.; Badour, M.I.; Juracic, E.S.; Barrett, K.; Brahmbhatt, A.; Al-Khazraji, B.; Punthakee, Z.; Perry, C.G.R.; et al. Targeting Skeletal Muscle Health with Exercise in People with Type 1 Diabetes: A Protocol for HOMET1D, a Prospective Observational Trial with Matched Controls. PLoS ONE 2024, 19, e0303448. [Google Scholar] [CrossRef]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral Advanced Glycation Endproducts (AGEs) Promote Insulin Resistance and Diabetes by Depleting the Antioxidant Defenses AGE Receptor-1 and Sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef]
- Qiu, Y.; Fernández-García, B.; Lehmann, H.I.; Li, G.; Kroemer, G.; López-Otín, C.; Xiao, J. Exercise Sustains the Hallmarks of Health. J. Sport Health Sci. 2023, 12, 8–35. [Google Scholar] [CrossRef]
- Bird, S.R.; Hawley, J.A. Update on the Effects of Physical Activity on Insulin Sensitivity in Humans. BMJ Open Sport Exerc. Med. 2017, 2, e000143. [Google Scholar] [CrossRef]
- Coderre, L.; Kandror, K.V.; Vallega, G.; Pilch, P.F. Identification and Characterization of an Exercise-Sensitive Pool of Glucose Transporters in Skeletal Muscle. J. Biol. Chem. 1995, 270, 27584–27588. [Google Scholar] [CrossRef]
- Roy, D.; Marette, A. Exercise Induces the Translocation of GLUT4 to Transverse Tubules from an Intracellular Pool in Rat Skeletal Muscle. Biochem. Biophys. Res. Commun. 1996, 223, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Holten, M.K.; Zacho, M.; Gaster, M.; Juel, C.; Wojtaszewski, J.F.P.; Dela, F. Strength Training Increases Insulin-Mediated Glucose Uptake, GLUT4 Content, and Insulin Signaling in Skeletal Muscle in Patients with Type 2 Diabetes. Diabetes 2004, 53, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Hirshman, M.F.; Wallberg-Henriksson, H.; Wardzala, L.J.; Horton, E.D.; Horton, E.S. Acute Exercise Increases the Number of Plasma Membrane Glucose Transporters in Rat Skeletal Muscle. FEBS Lett. 1988, 238, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Yaspelkis, B.B.; Singh, M.K.; Trevino, B.; Krisan, A.D.; Collins, D.E. Resistance Training Increases Glucose Uptake and Transport in Rat Skeletal Muscle. Acta Physiol. Scand. 2002, 175, 315–323. [Google Scholar] [CrossRef]
- Amor, M.; Itariu, B.K.; Moreno-Viedma, V.; Keindl, M.; Jürets, A.; Prager, G.; Langer, F.; Grablowitz, V.; Zeyda, M.; Stulnig, T.M. Serum Myostatin Is Upregulated in Obesity and Correlates with Insulin Resistance in Humans. Exp. Clin. Endocrinol. Diabetes 2019, 127, 550–556. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, L.; Ye, J.; Zhu, D. Upregulation of Myostatin Gene Expression in Streptozotocin-Induced Type 1 Diabetes Mice is Attenuated by Insulin. Biochem. Biophys. Res. Commun. 2009, 388, 112–116. [Google Scholar] [CrossRef]
- Lenk, K.; Erbs, S.; Höllriegel, R.; Beck, E.; Linke, A.; Gielen, S.; Winkler, S.M.; Sandri, M.; Hambrecht, R.; Schuler, G.; et al. Exercise Training Leads to a Reduction of Elevated Myostatin Levels in Patients with Chronic Heart Failure. Eur. J. Prev. Cardiol. 2012, 19, 404–411. [Google Scholar] [CrossRef]
- Hittel, D.S.; Axelson, M.; Sarna, N.; Shearer, J.; Huffman, K.M.; Kraus, W.E. Myostatin Decreases with Aerobic Exercise and Associates with Insulin Resistance. Med. Sci. Sports Exerc. 2010, 42, 2023–2029. [Google Scholar] [CrossRef]
- Butcher, J.T.; Mintz, J.D.; Larion, S.; Qiu, S.; Ruan, L.; Fulton, D.J.; Stepp, D.W. Increased Muscle Mass Protects Against Hypertension and Renal Injury in Obesity. J. Am. Heart Assoc. 2018, 7, e009358. [Google Scholar] [CrossRef]
- Matsakas, A.; Foster, K.; Otto, A.; Macharia, R.; Elashry, M.I.; Feist, S.; Graham, I.; Foster, H.; Yaworsky, P.; Walsh, F.; et al. Molecular, Cellular and Physiological Investigation of Myostatin Propeptide-Mediated Muscle Growth in Adult Mice. Neuromuscul. Disord. 2009, 19, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Hadhazy, M.; Wehling, M.; Tidball, J.G.; McNally, E.M. Dominant Negative Myostatin Produces Hypertrophy without Hyperplasia in Muscle. FEBS Lett. 2000, 474, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, G.; Bonfanti, R.; Meschi, F.; Bognetti, E.; Paesano, P.L.; Gianolli, L.; Querques, M.; Maestroni, A.; Calori, G.; Del Maschio, A.; et al. Persistent Renal Hypertrophy and Faster Decline of Glomerular Filtration Rate Precede the Development of Microalbuminuria in Type 1 Diabetes. Diabetes 2006, 55, 2620–2625. [Google Scholar] [CrossRef]
- Mertens, J.; De Block, C.; Spinhoven, M.; Driessen, A.; Francque, S.M.; Kwanten, W.J. Hepatopathy Associated With Type 1 Diabetes: Distinguishing Non-Alcoholic Fatty Liver Disease From Glycogenic Hepatopathy. Front. Pharmacol. 2021, 12, 768576. [Google Scholar] [CrossRef]
- Della Pepa, G.; Lupoli, R.; Masulli, M.; Boccia, R.; De Angelis, R.; Gianfrancesco, S.; Piccolo, R.; Rainone, C.; Rivellese, A.A.; Annuzzi, G.; et al. Blood Glucose Control and Metabolic Dysfunction-Associated Steatotic Liver Disease in People with Type 1 Diabetes. J. Endocrinol. Investig. 2024, 47, 2371–2378. [Google Scholar] [CrossRef]
- Al-Hussaini, A.A.; Sulaiman, N.M.; Alzahrani, M.D.; Alenizi, A.S.; Khan, M. Prevalence of Hepatopathy in Type 1 Diabetic Children. BMC Pediatr. 2012, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic Myopathy: Impact of Diabetes Mellitus on Skeletal Muscle Progenitor Cells. Front. Physiol. 2013, 4, 379. [Google Scholar] [CrossRef]
- Hernández-Ochoa, E.O.; Vanegas, C. Diabetic Myopathy and Mechanisms of Disease. Biochem. Pharmacol. 2015, 4, e179. [Google Scholar] [CrossRef]
- Mortensen, S.P.; Damsgaard, R.; Dawson, E.A.; Secher, N.H.; González-Alonso, J. Restrictions in Systemic and Locomotor Skeletal Muscle Perfusion, Oxygen Supply and VO2 during High-Intensity Whole-Body Exercise in Humans. J. Physiol. 2008, 586, 2621–2635. [Google Scholar] [CrossRef]
- Frisbee, J.C.; Goodwill, A.G.; Butcher, J.T.; Olfert, I.M. Divergence between Arterial Perfusion and Fatigue Resistance in Skeletal Muscle in the Metabolic Syndrome. Exp. Physiol. 2011, 96, 369–383. [Google Scholar] [CrossRef]
- Schnell, O.; Cappuccio, F.; Genovese, S.; Standl, E.; Valensi, P.; Ceriello, A. Type 1 Diabetes and Cardiovascular Disease. Cardiovasc. Diabetol. 2013, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-M.; Zhao, Y.-P.; Zhao, Y.; Deng, S.-L.; Yu, K. Regulation of Myostatin on the Growth and Development of Skeletal Muscle. Front. Cell Dev. Biol. 2021, 9, 785712. [Google Scholar] [CrossRef]
- Mouisel, E.; Relizani, K.; Mille-Hamard, L.; Denis, R.; Hourdé, C.; Agbulut, O.; Patel, K.; Arandel, L.; Morales-Gonzalez, S.; Vignaud, A.; et al. Myostatin is a Key Mediator between Energy Metabolism and Endurance Capacity of Skeletal Muscle. Am. J. Physiol. 2014, 307, R444–R454. [Google Scholar] [CrossRef] [PubMed]
- Mafi, F.; Biglari, S.; Ghardashi Afousi, A.; Gaeini, A.A. Improvement in Skeletal Muscle Strength and Plasma Levels of Follistatin and Myostatin Induced by an 8-Week Resistance Training and Epicatechin Supplementation in Sarcopenic Older Adults. J. Aging Phys. Act. 2019, 27, 384–391. [Google Scholar] [CrossRef]
- Bagheri, R.; Rashidlamir, A.; Motevalli, M.S.; Elliott, B.T.; Mehrabani, J.; Wong, A. Effects of Upper-Body, Lower-Body, or Combined Resistance Training on the Ratio of Follistatin and Myostatin in Middle-Aged Men. Eur. J. Appl. Physiol. 2019, 119, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Khalafi, M.; Aria, B.; Symonds, M.E.; Rosenkranz, S.K. The Effects of Resistance Training on Myostatin and Follistatin in Adults: A Systematic Review and Meta-Analysis. Physiol. Behav. 2023, 269, 114272. [Google Scholar] [CrossRef]
- Wolosowicz, M.; Lukaszuk, B.; Chabowski, A. The Causes of Insulin Resistance in Type 1 Diabetes Mellitus: Is There a Place for Quaternary Prevention? Int. J. Environ. Res. Public. Health 2020, 17, 8651. [Google Scholar] [CrossRef]
- Nunan, E.; Wright, C.L.; Semola, O.A.; Subramanian, M.; Balasubramanian, P.; Lovern, P.C.; Fancher, I.S.; Butcher, J.T. Obesity as a Premature Aging Phenotype—Implications for Sarcopenic Obesity. GeroScience 2022, 44, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Schiphof-Godart, L.; Roelands, B.; Hettinga, F.J. Drive in Sports: How Mental Fatigue Affects Endurance Performance. Front. Psychol. 2018, 9, 1383. [Google Scholar] [CrossRef]
- Boolani, A.; Manierre, M. An Exploratory Multivariate Study Examining Correlates of Trait Mental and Physical Fatigue and Energy. Fatigue Biomed. Health Behav. 2019, 7, 29–40. [Google Scholar] [CrossRef]
- Fuller, D.T.; Smith, M.L.; Boolani, A. Trait Energy and Fatigue Modify the Effects of Caffeine on Mood, Cognitive and Fine-Motor Task Performance: A Post-Hoc Study. Nutrients 2021, 13, 412. [Google Scholar] [CrossRef] [PubMed]
- Boolani, A.; Ryan, J.; Vo, T.; Wong, B.; Banerjee, N.K.; Banerjee, S.; Fulk, G.; Smith, M.L.; Martin, R. Do Changes in Mental Energy and Fatigue Impact Functional Assessments Associated with Fall Risks? An Exploratory Study Using Machine Learning. Phys. Occup. Ther. Geriatr. 2020, 38, 283–301. [Google Scholar] [CrossRef]
- Kowalski, K.L.; Boolani, A.; Christie, A.D. State and Trait Fatigue and Energy Predictors of Postural Control and Gait. Mot. Control. 2021, 25, 519–536. [Google Scholar] [CrossRef]
- Gigliotti, H.M.; Hodgson, C.; Riley, M.; Marshall, B.; Ward-Ritacco, C.L.; Martin, J.; Boolani, A. Trait Energy and Fatigue Influence Inter-Individual Mood and Neurocognitive Responses during Work Done While Sitting, Standing, and Intermittent Walking: A Randomized-Controlled Crossover Design. Appl. Sci. 2023, 13, 4241. [Google Scholar] [CrossRef]
- Goedendorp, M.M.; Tack, C.J.; Steggink, E.; Bloot, L.; Bazelmans, E.; Knoop, H. Chronic Fatigue in Type 1 Diabetes: Highly Prevalent but not Explained by Hyperglycemia or Glucose Variability. Diabetes Care 2014, 37, 73–80. [Google Scholar] [CrossRef]
- Griggs, S.; Morris, N.S. Fatigue Among Adults with Type 1 Diabetes Mellitus and Implications for Self-Management: An Integrative Review. Diabetes Educ. 2018, 44, 325–339. [Google Scholar] [CrossRef]
- Kalra, S.; Sahay, R. Diabetes Fatigue Syndrome. Diabetes Ther. 2018, 9, 1421–1429. [Google Scholar] [CrossRef]
- Nuzzo, J.L. Sex Differences in Skeletal Muscle Fiber Types: A Meta-analysis. Clin. Anat. 2024, 37, 81–91. [Google Scholar] [CrossRef]
- Lundsgaard, A.-M.; Kiens, B. Gender Differences in Skeletal Muscle Substrate Metabolism—Molecular Mechanisms and Insulin Sensitivity. Front. Endocrinol. 2014, 5, 195. [Google Scholar] [CrossRef]
- Haizlip, K.M.; Harrison, B.C.; Leinwand, L.A. Sex-Based Differences in Skeletal Muscle Kinetics and Fiber-Type Composition. Physiology 2015, 30, 30–39. [Google Scholar] [CrossRef]
- Butcher, J.T.; Ali, M.I.; Ma, M.W.; McCarthy, C.G.; Islam, B.N.; Fox, L.G.; Mintz, J.D.; Larion, S.; Fulton, D.J.; Stepp, D.W. Effect of Myostatin Deletion on Cardiac and Microvascular Function. Physiol. Rep. 2017, 5, e13525. [Google Scholar] [CrossRef] [PubMed]
- Larion, S.; Padgett, C.A.; Mintz, J.D.; Thompson, J.A.; Butcher, J.T.; Belin de Chantemèle, E.J.; Haigh, S.; Khurana, S.; Fulton, D.J.; Stepp, D.W. NADPH Oxidase 1 Promotes Hepatic Steatosis in Obese Mice and Is Abrogated by Augmented Skeletal Muscle Mass. Am. J. 2024, 326, G264–G273. [Google Scholar] [CrossRef] [PubMed]
- Herren, D.J.; Norman, J.B.; Anderson, R.; Tremblay, M.L.; Huby, A.-C.; Belin de Chantemèle, E.J. Deletion of Protein Tyrosine Phosphatase 1B (PTP1B) Enhances Endothelial Cyclooxygenase 2 Expression and Protects Mice from Type 1 Diabetes-Induced Endothelial Dysfunction. PLoS ONE 2015, 10, e0126866. [Google Scholar] [CrossRef]
- Padgett, C.A.; Butcher, J.T.; Haigh, S.B.; Speese, A.C.; Corley, Z.L.; Rosewater, C.L.; Sellers, H.G.; Larion, S.; Mintz, J.D.; Fulton, D.J.R.; et al. Obesity Induces Disruption of Microvascular Endothelial Circadian Rhythm. Front. Physiol. 2022, 13, 887559. [Google Scholar] [CrossRef]
- Butcher, J.T.; Goodwill, A.G.; Frisbee, J.C. The Ex Vivo Isolated Skeletal Microvessel Preparation for Investigation of Vascular Reactivity. J. Vis. Exp. 2012, 62, e3674. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Body Composition | WT | KO | WT | KO |
|---|---|---|---|---|
| Baseline | T1D (Day 28) | |||
| Weight (g) | 38.7 ± 1.0 | 39.5 ± 0.8 | 33.9 ± 0.6 | 35.3 ± 0.7 |
| Weight Loss (%) | 12.1 ± 1.5 | 10.5 ± 1.5 | ||
| Fat (%) | 13.9 ± 2.4 | 3.2 ± 1.8 ^ | 12.0 ± 1.6 | 3.2 ± 1.7 * |
| Organ Weights (mg) | ||||
| Liver | 1282 ± 64.8 | 1050 ± 55.8 * | ||
| Gastroc | 187.0 ± 6.1 | 293.2 ± 15.3 * | ||
| Heart (wet wt.) | 164.4 ± 8.0 | 169.4 ± 8.0 | ||
| Kidney | 228.8 ± 6.7 | 202.9 ± 6.5 * | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunan, E.; Huff, D.R.; Gore, J.L.; Wright, C.L.; Harris, T.; Butler, L.; Padgett, C.A.; Rochowski, M.T.; Lovern, P.C.; Boolani, A.; et al. Targeting Myostatin as an Adjunct Treatment for the Preservation of Cardiometabolic and Skeletal Muscle Function in Type 1 Diabetes. Int. J. Mol. Sci. 2025, 26, 4830. https://doi.org/10.3390/ijms26104830
Nunan E, Huff DR, Gore JL, Wright CL, Harris T, Butler L, Padgett CA, Rochowski MT, Lovern PC, Boolani A, et al. Targeting Myostatin as an Adjunct Treatment for the Preservation of Cardiometabolic and Skeletal Muscle Function in Type 1 Diabetes. International Journal of Molecular Sciences. 2025; 26(10):4830. https://doi.org/10.3390/ijms26104830
Chicago/Turabian StyleNunan, Emily, Denton R. Huff, Jillian L. Gore, Carson L. Wright, Tag Harris, Landon Butler, Caleb A. Padgett, Matthew T. Rochowski, Pamela C. Lovern, Ali Boolani, and et al. 2025. "Targeting Myostatin as an Adjunct Treatment for the Preservation of Cardiometabolic and Skeletal Muscle Function in Type 1 Diabetes" International Journal of Molecular Sciences 26, no. 10: 4830. https://doi.org/10.3390/ijms26104830
APA StyleNunan, E., Huff, D. R., Gore, J. L., Wright, C. L., Harris, T., Butler, L., Padgett, C. A., Rochowski, M. T., Lovern, P. C., Boolani, A., Valdez, C., & Butcher, J. T. (2025). Targeting Myostatin as an Adjunct Treatment for the Preservation of Cardiometabolic and Skeletal Muscle Function in Type 1 Diabetes. International Journal of Molecular Sciences, 26(10), 4830. https://doi.org/10.3390/ijms26104830