1. Introduction
Erythropoiesis is a tightly regulated process involving hierarchical differentiation stages, in which hematopoietic stem and progenitor cells (HSPCs) stepwise differentiate into lineage-committed erythroid progenitors and precursors, ultimately giving rise to mature circulating red blood cells (RBCs). Approximately 200 billion erythrocytes must be produced each day to ensure efficient oxygen transport, a process achieved by coordinating erythrocyte proliferation, differentiation, and apoptosis [
1]. Dysfunction in erythropoiesis can lead to various hematological disorders. Defective erythropoiesis results in diseases like sickle cell anemia, Diamond–Blackfan anemia (DBA), and β-thalassemia [
2]. Conversely, excessive RBC production causes polycythemia vera (PV), characterized by increased hematocrit and hemoglobin levels, and is associated with an elevated risk of thrombosis [
3]. Therefore, elucidating the molecular mechanisms governing erythropoiesis is crucial for developing therapies to address hematologic disorders.
The stepwise progression of erythroid development is governed by multiple regulators, including cytokines, transcription factors, and chromatin modifiers [
4,
5]. Growing evidence also reveals the important role of translational control in erythropoiesis. Mutations in ribosomal proteins (RPs) are implicated in the majority of DBA cases [
6,
7]. The erythroid-specific RNA-binding protein RBM38 interacts with the translation initiation factor eIF4G (eukaryotic initiation factor 4G) to regulate erythropoiesis, and its inhibition impairs erythroid maturation [
8]. Moreover, during iron or heme deficiency, activation of heme-regulated eIF2α kinase (HRI) leads to eIF2α phosphorylation, which suppresses globin translation while selectively promoting ATF4 translation to mitigate oxidative stress and prevent ineffective erythropoiesis [
9,
10]. A recent study also showed that eIF4E modulates erythroid maturation, with its overexpression arresting cells at an early erythroid stage [
11]. Despite these insights, how protein synthesis is maintained at appropriate levels during erythroid differentiation under steady state and the potential physiological constraints governing this process remain largely unexplored.
Eukaryotic elongation factor 2 (eEF2) is essential for translation elongation, facilitating tRNA translocation along the ribosome [
12]. Its activity is predominantly regulated through phosphorylation at threonine 56 (Thr56), a modification that inactivates eEF2, slows down the elongation rate, and ultimately represses protein synthesis [
13]. eEF2 kinase (eEF2K), a calcium/calmodulin (CaM)-dependent enzyme, is the only known kinase in mammals that phosphorylates eEF2 at Thr56, thereby inhibiting its activity and suppressing protein synthesis [
14]. While eEF2 phosphorylation has been explored in cancer and neurological disorders [
15,
16,
17], its regulatory role in erythropoiesis remains undefined.
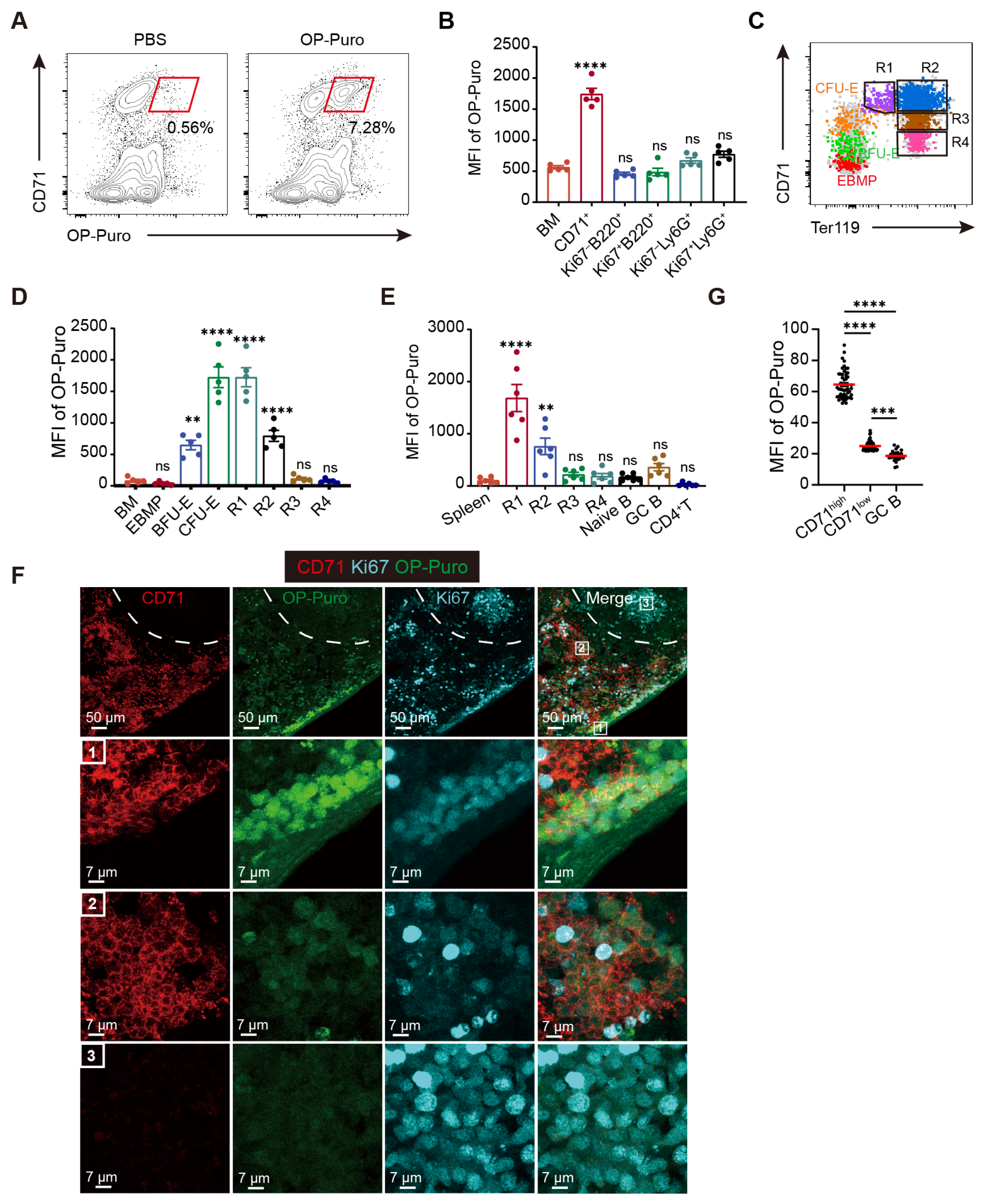
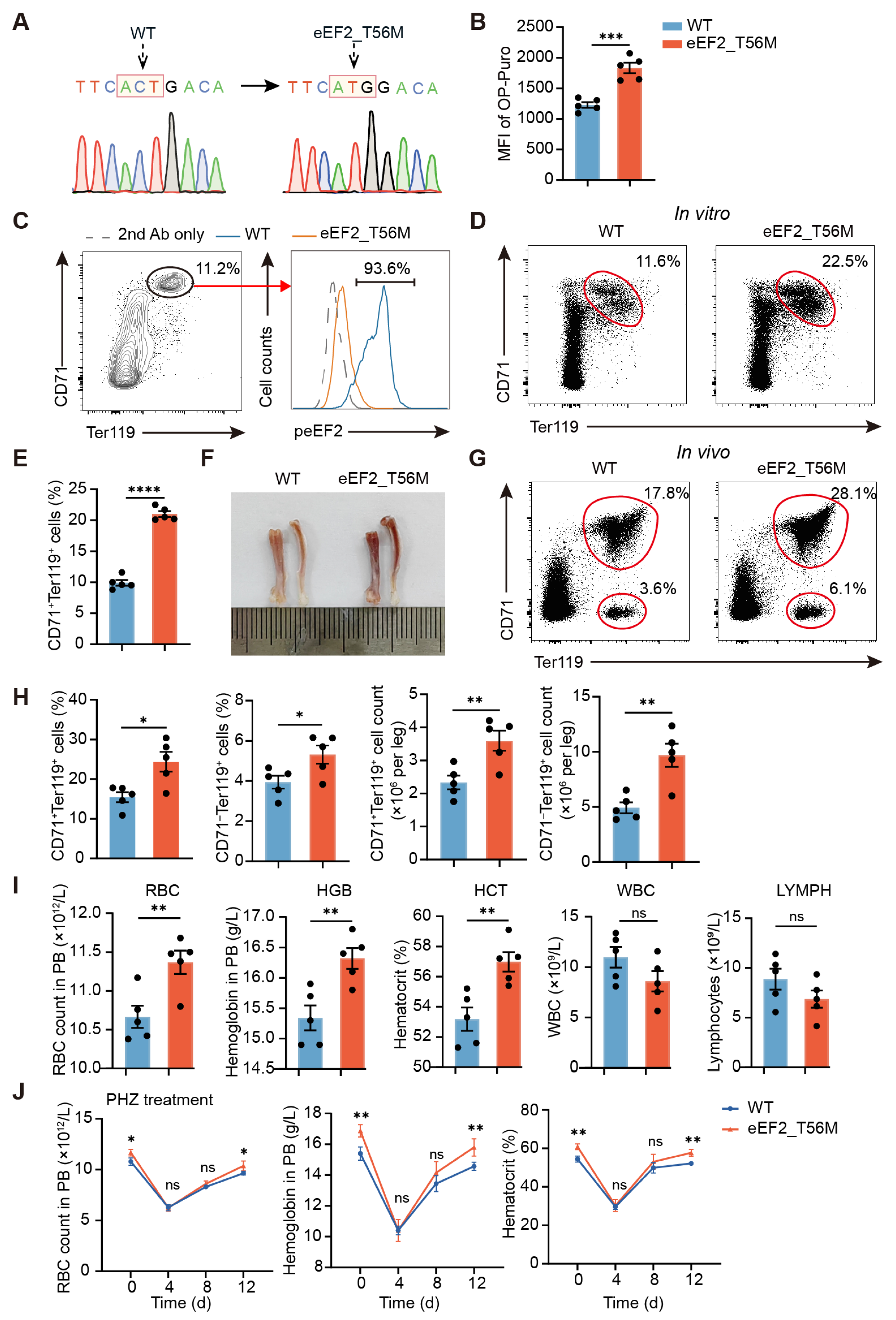
In this study, we show that erythroid cells exhibit an exceptionally high demand for protein synthesis, which far exceeds that of other cell types and is critical for their differentiation. However, negative regulation by eEF2 phosphorylation is also required to maintain protein synthesis at optimal levels. In eEF2_T56M mice, which harbor a mutation abolishing eEF2 phosphorylation, we observed increased RBC counts in peripheral blood, concomitant with a heightened risk of thrombosis and blood stasis, strongly supporting the role of this modification in preventing overactive erythropoiesis. Mechanistically, eEF2 phosphorylation modulates erythroid differentiation in part by selectively regulating NFE2 translation, a transcription factor that controls the expression of key erythroid genes, including β-globin [
18]. Overall, our findings highlight eEF2 phosphorylation as a pivotal regulator of erythropoietic homeostasis through coordinated translational control.
3. Discussion
In this study, we revealed the distinct characteristics of protein synthesis during erythroid differentiation and identified a critical protective mechanism mediated by eEF2 phosphorylation, which serves as a molecular brake to ensure optimal and balanced protein synthesis, thereby preserving erythropoietic homeostasis.
Several studies [
11,
38], including our own, have shown that protein synthesis levels in erythroid cells initially increase and subsequently decline throughout erythropoiesis. This trend aligns with the developmental trajectory of erythroid cells, which begins with an early differentiation phase characterized by rapid proliferation and transitions into a maturation phase marked by reduced proliferative capacity and cell size, ultimately yielding functional red blood cells devoid of most organelles. Strikingly, we observed that protein synthesis rates in early erythroid precursors are extraordinarily high, far surpassing not only those in rapidly proliferating neutrophil and B-cell precursors in bone marrow, but also those in activated germinal center B cells in peripheral lymphoid organs, which are generally regarded as possessing robust protein synthesis capacity for antibody production. This raises an intriguing question about the specific proteins synthesized at this stage, with proteomic analysis providing preliminary insights. Studies from Xu’s and Blanc’s laboratories have revealed that [
37,
39], in addition to central erythroid transcription factors such as GATA1 and KLF1, proteins with increased abundance during the early phase of human erythropoiesis are predominantly enriched in two categories, with one associated with hemoglobin and heme biosynthesis, which are hallmarks of erythroid differentiation, and the other related to mitochondrial biogenesis, which is consistent with the markedly elevated mitochondrial biomass and potential observed in ProEs compared to HSPCs. While changes in protein stability may contribute, it is likely that the increased abundance of those specific proteins is primarily attributed to the significantly heightened protein synthesis capacity, a hypothesis that requires further investigation with more precise approaches. Another distinct feature of translational control in erythroid cells is their extreme sensitivity to fluctuations in protein synthesis levels, as we found that even minor disruptions can lead to defective erythroid output in vitro. Supporting this, dysregulated erythropoiesis was observed in eEF2_T56M mice, whereas the development of other cell types, such as lymphocytes, remained unaffected. This hypersensitivity may also explain why mutations in over 20 RP genes are implicated in DBA [
7]. Together, these two unique characteristics underscore the importance of translational regulation in erythroid differentiation and offer promising therapeutic avenues for hematologic disorders.
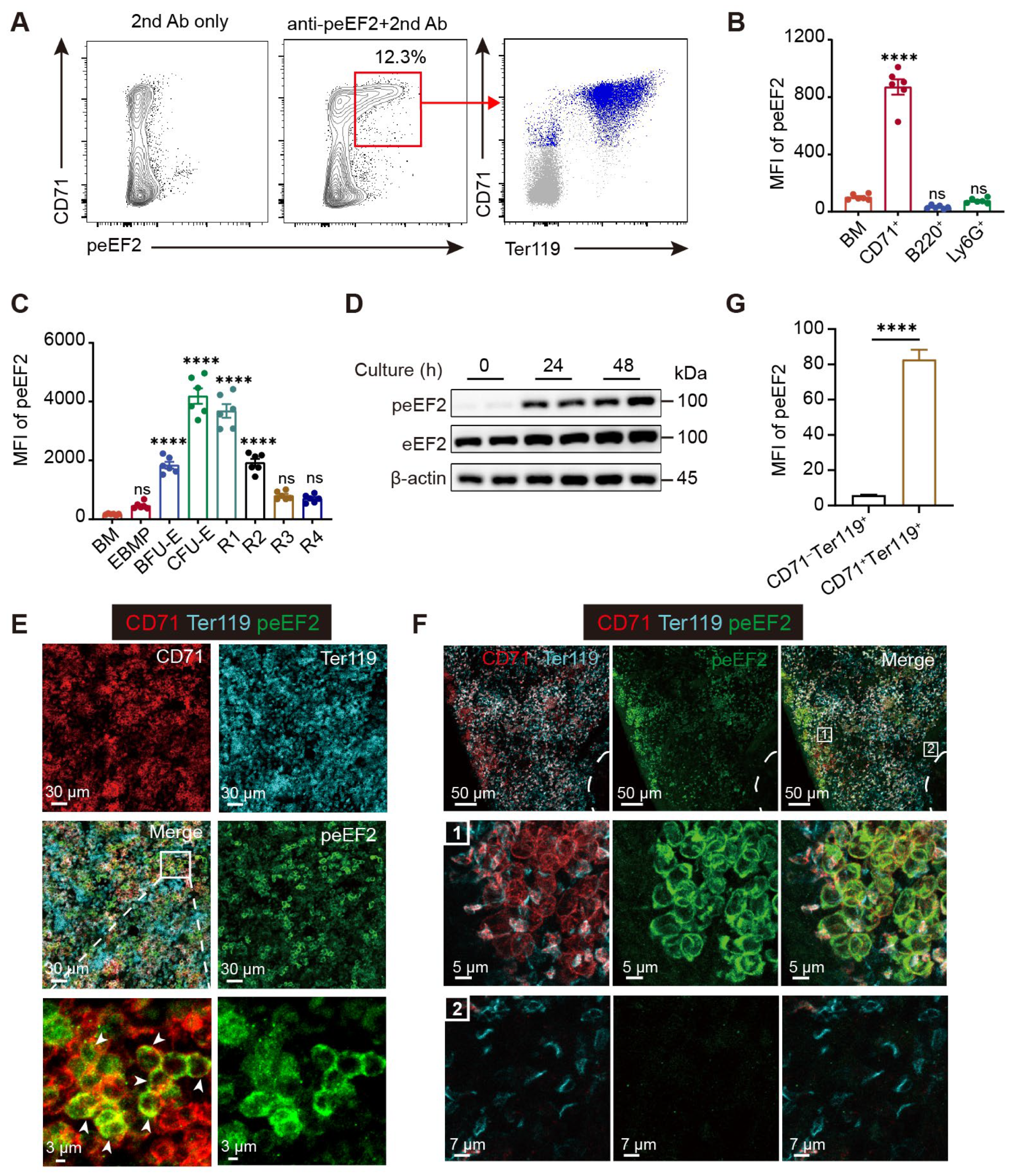
Translation initiation is widely considered the rate-limiting step in protein synthesis, and recent research on the regulation of protein synthesis during erythropoiesis has predominantly focused on initiation factors within the translation machinery, such as eIF2α, eIF4E, and eIF5A [
9,
11,
40]. In contrast, our study uncovers a previously unexplored connection between translation elongation and erythroid differentiation, mediated by eEF2 phosphorylation at Thr56. Although the molecular basis underlying its effect on eEF2 function has been extensively studied, its physiological significance remains poorly understood and is largely confined to roles in the brain and skeletal muscle. Remarkably, we observed strong eEF2 phosphorylation signals in erythroid precursors in both bone marrow and fetal liver under steady state, as well as in the spleen during stress erythropoiesis. These findings, along with data from the eEF2_T56M mouse model we developed, establish erythropoiesis as a critical physiological context for understanding eEF2-mediated translational control in vivo.
It may appear paradoxical that eEF2-mediated translational suppression occurs in erythroid cells, which concurrently require extraordinarily high levels of protein synthesis for differentiation. However, such seemingly contradictory regulatory mechanisms are often indispensable for enabling a system to respond rapidly to external stimuli while maintaining homeostasis, as observed in various contexts. For instance, bacterial infections can trigger robust cytokine production by immune cells to combat the invasion, while host-derived negative regulators are simultaneously upregulated to suppress excessive inflammatory responses and prevent tissue damage. Similarly, despite the high protein synthesis demand for effective erythropoiesis, mechanisms such as eEF2 phosphorylation likely act as safeguards to modulate this capacity and ensure optimal erythroid differentiation. The factors responsible for inducing eEF2 phosphorylation in this process remain unclear. eEF2K is currently the only recognized kinase for eEF2 phosphorylation in mammals, yet our data indicate that eEF2 phosphorylation occurs independently of eEF2K, as pharmacological inhibition of eEF2K had no significant effect on peEF2 levels or erythroid differentiation (
Figure S7A). Further validation using eEF2K knockout models is warranted to confirm these findings. Notably, the EPO-EPOR axis activates the PI3K/AKT signaling pathway, which is critical for erythroid cell survival, proliferation, and differentiation via serine/threonine phosphorylation cascades [
4,
41]. Intriguingly, treatment of cultured bone marrow cells with Afuresertib (GSK2110183), an AKT inhibitor, resulted in a marked reduction in eEF2 phosphorylation levels (
Figure S7B). This observation suggests that the kinases involved in the AKT signaling pathway may be potential regulators of eEF2 phosphorylation. Although the precise kinases targeting eEF2 remain unidentified, our findings highlight the significance of an eEF2-dependent negative feedback mechanism in ensuring well-regulated erythropoiesis.
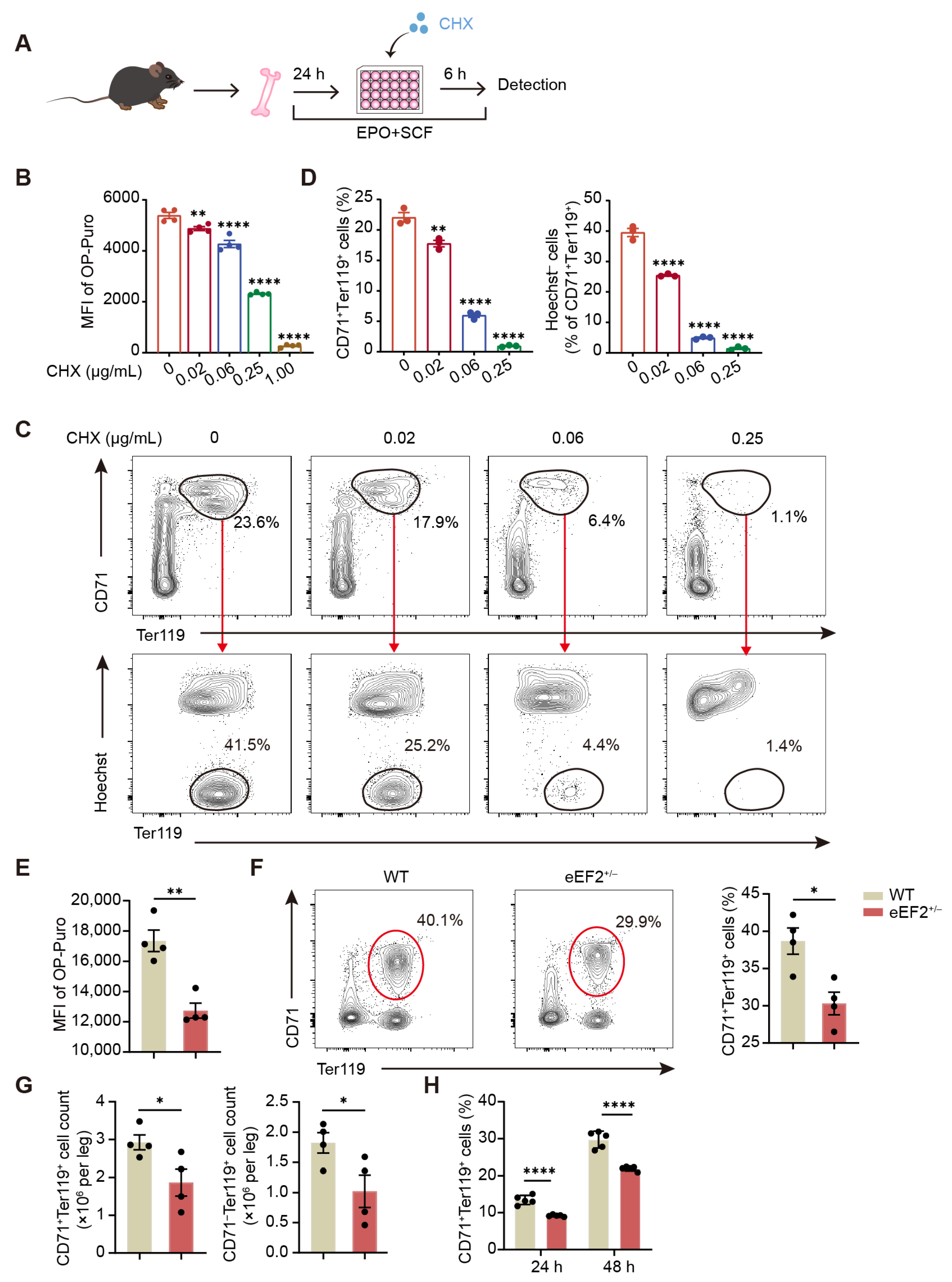
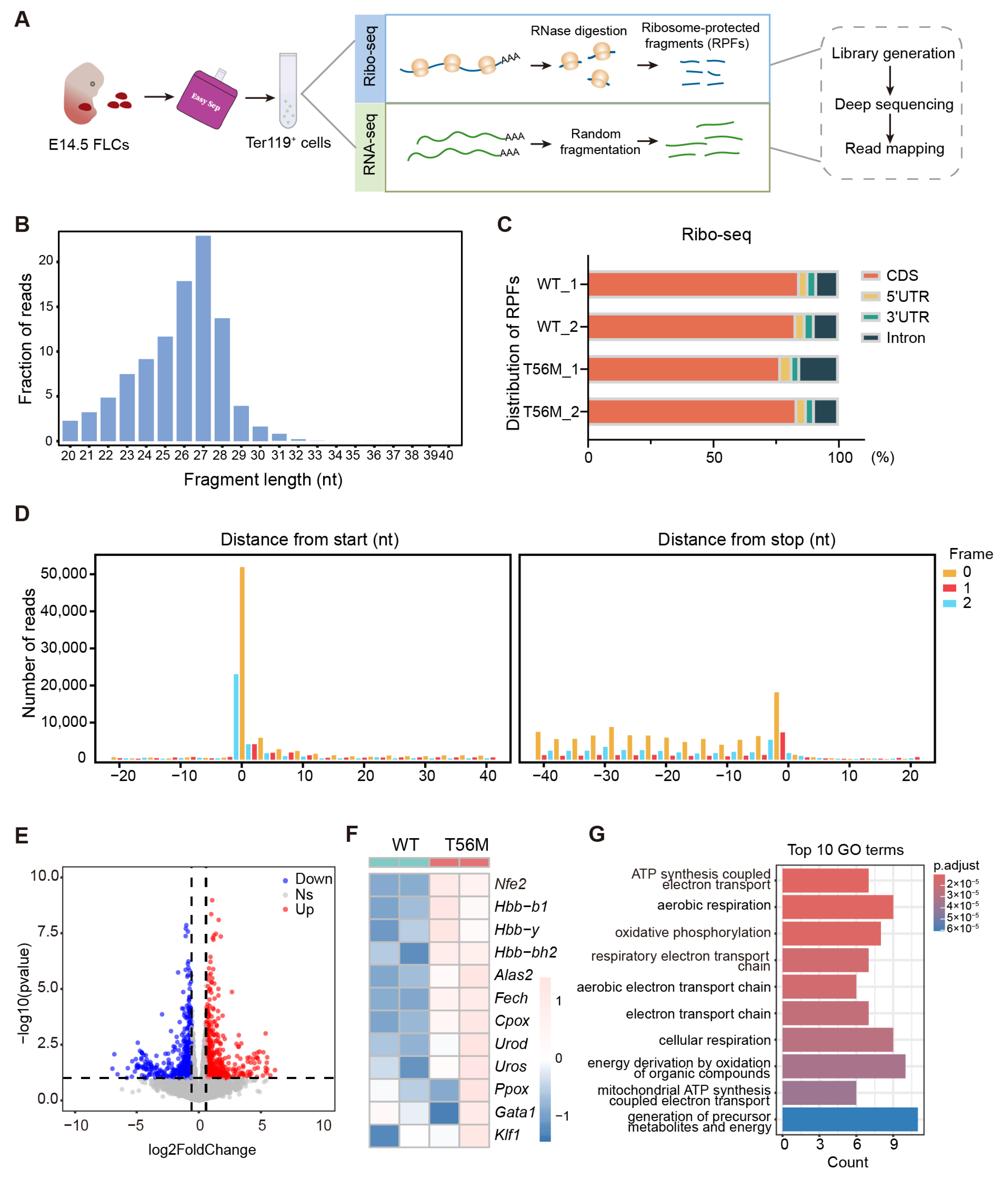
In contrast to the notion that impaired elongation universally reduces overall protein synthesis, our results reveal a specific translational control mediated by eEF2 phosphorylation during erythropoiesis, supported by proteomic profiles obtained under CHX treatment, a widely used translation elongation inhibitor targeting eEF2. Among transcripts with upregulated translation efficiency in T56M erythroblasts, a subset of genes directly involved in heme and globin biosynthesis stood out, including
Alas2,
Fech, and
Nfe2. Even subtle changes in these processes can significantly impact erythroid progression, as evidenced by reduced erythroid maturation upon NFE2 suppression in T56M cells, consistent with a previous report linking elevated NFE2 levels to polycythemia vera [
33,
34]. Another major finding is that eEF2 phosphorylation also guides the translation of mitochondrial proteins. The critical role of mitochondrial biosynthesis in erythropoiesis is underscored by evidence showing that disruptions in mitochondrial protein synthesis, such as those caused by impaired eIF5A function or depletion of mitochondrial transcription factor A (TFAM), severely hinder erythroid differentiation [
37,
40]. Overall, our studies emphasize eEF2 phosphorylation as an additional regulatory layer beyond gene transcription, adeptly coordinating erythropoiesis through a unique translational program.
It is noteworthy that, despite a concerted effort to drive basal protein synthesis, individual components of the translation machinery exhibit specialized roles in regulating distinct translational profiles. For example, unlike eEF2, eIF4E directs the selective translation of proteins implicated in early hematopoiesis, such as PTPN6 and IGF2BP1, contributing to the maintenance of the early precursor state of erythroid cells [
11]. While eIF4E preferentially translates mRNAs with conserved cytosine-rich motifs in their 5′ UTRs [
11,
42], the mechanism by which eEF2 phosphorylation confers translational specificity remains unclear. Although multiple possibilities may exist, it is tempting to hypothesize that the phosphorylation of a portion of eEF2 molecules reduces active eEF2 availability, making well-translated transcripts with abundant ribosomes more susceptible to ribosome collision and translation arrest. This assumption aligns with the proposed role of eEF2 phosphorylation as a fine-tuning mechanism for regulating massive protein synthesis during erythropoiesis. Future studies are necessary to elucidate the precise mechanisms governing translational selectivity mediated by eEF2 phosphorylation.
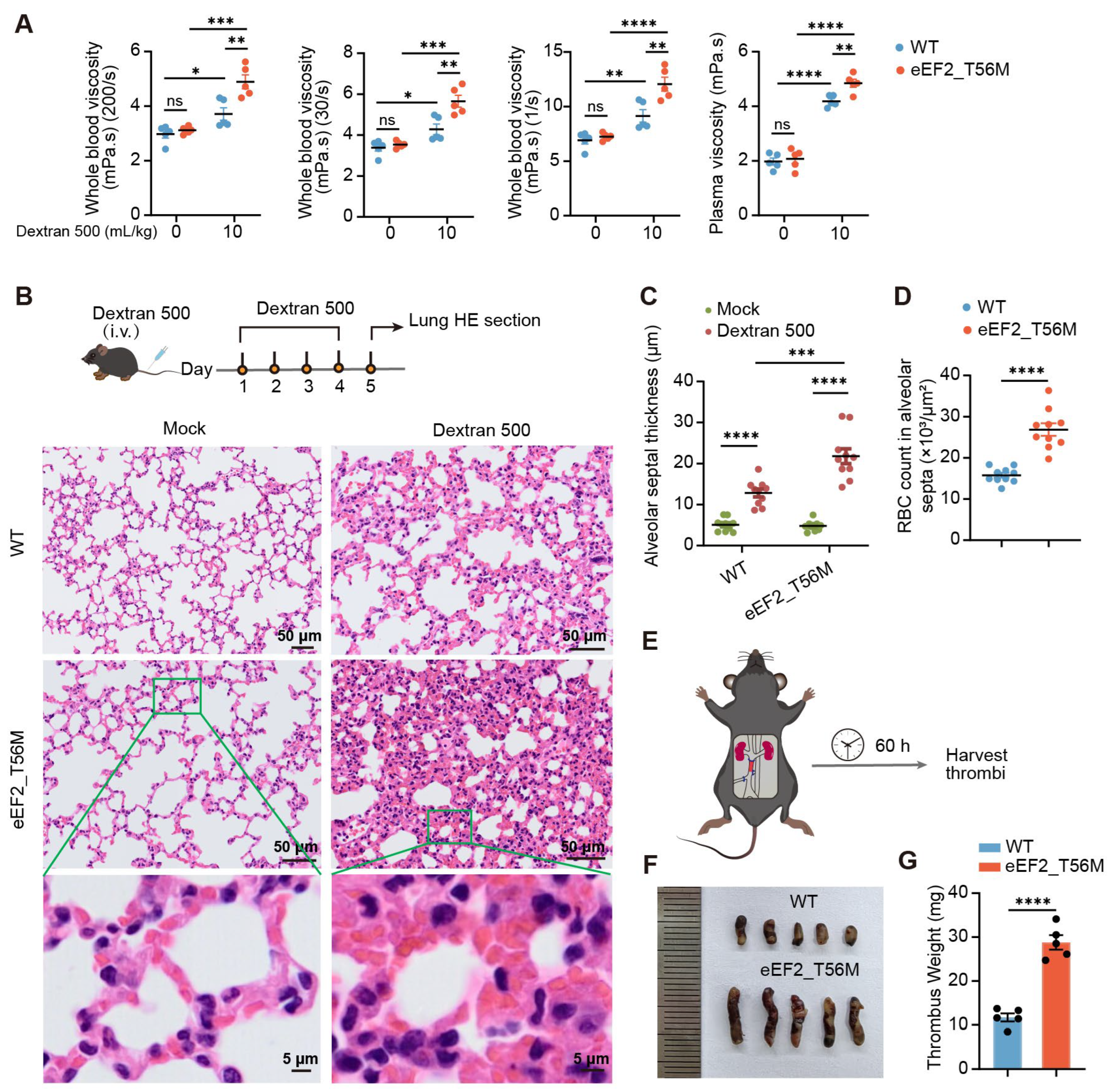
Polycythemia vera (PV) is a myeloproliferative neoplasm characterized by elevated hemoglobin and hematocrit levels, most commonly driven by constitutive activation of the JAK2 signaling pathway [
3]. Notably, in our study, T56M mutant mice exhibit increased hemoglobin and hematocrit compared to WT mice (
Figure 4), recapitulating a hallmark clinical feature of PV. Moreover, T56M mice display an enhanced propensity for thrombosis (
Figure 5), a major complication in PV patients. NFE2, a transcription factor upregulated in PV [
33,
34], is also significantly elevated in T56M mice (
Figure 6). These parallels suggest that impaired phosphorylation of eEF2 may play a previously underappreciated role in promoting erythrocytosis, independent of JAK2 mutations. Our results raise the possibility that mutations affecting eEF2 phosphorylation sites, particularly at T56, may exist in a subset of PV patients, especially those lacking the canonical JAK2V617F mutations. Screening for such mutations could help identify individuals at risk, and targeting the translational elongation machinery may represent an innovative and promising therapeutic strategy for this specific patient cohort.
Despite these findings, our study has several limitations. The precise molecular mechanisms by which eEF2 phosphorylation selectively modulates mRNA translation remain to be fully elucidated. Furthermore, identifying the upstream effectors responsible for eEF2 phosphorylation will facilitate the development of targeted strategies to regulate erythropoiesis via translational control.
In conclusion, our study highlights the importance of elaborate translational control during erythropoiesis, revealing eEF2 as a critical regulator of coordinated erythroid differentiation (
Figure S8). These findings establish a foundation for developing innovative therapies for hematologic disorders.
4. Materials and Methods
4.1. Mice
All mice used in this research were on the C57BL/6J genetic background. C57BL/6 mice were purchased from Shanghai Slaccas Company (Shanghai, China). eEF2+/− mice (strain #T027339) and eEF2_T56M mice (strain #T053825) were purchased from Jiangsu GemPharmatech Co., Ltd (Nanjing, China). eEF2_T56M homozygous mice exhibit normal body size, weight, and activity levels under standard housing conditions. For PHZ treatment, mice were intraperitoneally injected with PHZ (50 mg/kg body weight; Sigma, Burlington, MA, USA) once to induce hemolytic anemia. All mice were maintained with standard 12 h light/dark cycles under specific pathogen-free (SPF) conditions. Mice aged 8 to 16 weeks were used for experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Shanghai Jiao Tong University School of Medicine.
4.2. Cell Isolation and Culture
For bone marrow preparation, femurs and tibias were collected and placed in phosphate-buffered saline (PBS) (BasalMedia, Shanghai, China) containing 2% fetal bovine serum (FBS) (ExCell Bio, Suzhou, China). Bone marrow cells were flushed out using a 2 mL sterile syringe fitted with a 26-gauge needle and passed through a 70 µm cell strainer. After red blood cell lysis with ACK buffer, 1 × 106 bone marrow cells were seeded in a 48-well plate and cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FBS, 1% penicillin/streptomycin, 200 μg/mL holo-transferrin (Beyotime, Shanghai, China), 10 μg/mL recombinant human insulin (Beyotime), 50 ng/mL stem cell factor (SCF) (PeproTech, Cranbury, NJ, USA), 10 ng/mL erythropoietin (EPO) (R&D), and 10–4 M β-mercaptoethanol (Sigma, USA).
E14.5 fetal liver cells (FLCs) were mechanically dissociated in PBS containing 10% FBS and then labeled with FITC-conjugated anti-Ter119 antibody (clone TER119). Ter119− cells were purified using the FITC Positive Selection Kit (STEMCELL, Vancouver, BC, Canada) following the manufacturer’s instructions. Purified cells were seeded in fibronectin-coated 24-well plates at a cell density of 3 × 105/mL in IMDM medium supplemented with 15% FBS, 1% detoxified bovine serum albumin (Sigma), 2 mM l-glutamine (Gibco, Grand Island, NY, USA), 200 μg/mL holo-transferrin, 10 μg/mL recombinant human insulin, 10–4 M β-mercaptoethanol, and 10 ng/mL EPO. All cells were grown in humidified CO2 incubators at 37 °C with 5% CO2.
4.3. Flow Cytometry Analysis
Single-cell suspensions of murine bone marrow, spleen, fetal liver, or cultured cells were prepared and stained with a fixable viability dye (BD Biosciences, San Jose, CA, USA) for 10 min at 4 °C to exclude dead cells. Subsequently, cells were incubated with fluorochrome-conjugated antibodies specific for cell surface antigens in FACS buffer (PBS containing 2% FBS and 2 mM EDTA) for 30 min at 4 °C. For erythroid progenitor staining, cells were incubated with a lineage antibody cocktail as well as anti-CD55 (clone RIKO-3, Biolegend, San Diego, CA, USA), anti-CD41 (clone MWReg30, Biolegend), anti-CD105 (clone MJ7/18, Biolegend), anti-CD49f (clone GoH3, Biolegend), anti-CD117 (clone 2B8, Biolegend), anti-TER119 (clone TER119, Biolegend), and anti-CD71 (clone RI7217, Biolegend) antibodies. The lineage antibody cocktail included anti-CD11b (clone M1/70, Biolegend), anti-Ly6G (clone 1A8, Biolegend), anti-CD4 (clone GK1.5, Biolegend), anti-CD8a (clone 53-6.7, Biolegend), and anti-B220 (clone RA3-6B2, Biolegend) antibodies. For Ki67 staining, after surface staining, cells were fixed and permeabilized using the BD Fixation/Permeabilization Solution Kit (BD Biosciences) according to the manufacturer’s instructions and then incubated with the Ki67 antibody (clone SolA15, Invitrogen, Carlsbad, CA, USA). Flow cytometry assays were conducted on a BD LSRFortessa X-20 (BD Biosciences) and further analyzed using FlowJo software (version 10) (Tree Star). Detailed information about antibodies is provided in
Supplemental Table S1.
4.4. Measurement of Protein Synthesis Rate
Protein synthesis rates were measured based on O-propargyl-puromycin (OP-Puro) incorporation. For in vivo assessments, mice were injected intraperitoneally with OP-Puro (50 mg/kg) (MCE, Shanghai, China), while the vehicle group received PBS. One hour after administration, bone marrow or spleen samples were obtained for immunofluorescence or flow cytometry analysis. For ex vivo assessments, cells were incubated with OP-Puro (20 μM) for 1 h at 37 °C prior to harvesting. Following cell surface staining, cells were fixed in 1% paraformaldehyde (PFA) for 15 min at 4 °C and permeabilized with the BD Fixation/Permeabilization Solution Kit for 5 min at room temperature (RT) in the dark. Subsequently, OP-Puro-labeled polypeptides were detected using the Click-iT Plus OPP Alexa Fluor 488 Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol.
4.5. Immunofluorescence Staining
Tissues were fixed in 4% PFA and dehydrated in 30% sucrose overnight at 4 °C, then embedded in OCT compound (Sakura Finetek, Torrance, CA, USA) and frozen at −80 °C. Sections (20 µm thick) were permeabilized in pre-cooled methanol at −20 °C for 30 min, followed by incubation with blocking buffer (PBS containing 0.3% Tween 20, 1% bovine serum albumin, 1% FBS, and 0.1 M Tris-HCl) for 1 h at RT. Sections were stained with primary antibodies diluted in blocking buffer for 3 h at RT or overnight at 4 °C, and finally incubated with secondary antibodies for 3 h at RT. Fluorescence images were acquired using a Leica SP8 confocal microscope (Leica, Wetzlar, Germany) and analyzed with Imaris (version 9.7.0, Bitplane, Oxford, UK).
4.6. eEF2 Phosphorylation Detection
Mouse bone marrow, spleen, or cultured cells were collected and subjected to cell surface staining at 4 °C for 30 min. Cells were then fixed in 1% PFA for 15 min at 4 °C in the dark, followed by washing and permeabilization using the BD Fixation/Permeabilization Solution Kit. For phospho-eEF2 (Thr56) detection, cells were stained with the Phospho-eEF2 (Thr56) antibody (CST, Danvers, MA, USA) diluted in BD Perm/Wash buffer for 30 min at 4 °C. After washing twice with BD Perm/Wash buffer, cells were incubated with a goat anti-rabbit secondary antibody for 30 min at 4 °C. All stained samples were analyzed by flow cytometry.
4.7. Western Blot and Quantitative PCR Assay
For western blot, cells were washed with PBS and then lysed by RIPA lysis buffer containing Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and phosphatase inhibitor (Beyotime). Protein concentrations were determined using the BCA Protein Assay Kit (Yeasen, Shanghai, China). Equal amounts of proteins were separated by 8–12% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Merck Millipore, Darmstadt, Germany). Membranes were blocked with 5% non-fat milk in TBST (50 mM Tris, 150 mM NaCl, 1% Tween-20, pH 7.4) for 1 h at RT, followed by incubation with primary antibodies overnight at 4 °C. After washing three times with TBST, membranes were incubated with HRP-conjugated secondary antibodies (CST) diluted 1:5000 in 5% non-fat milk for 1 h at RT.
For qPCR, total RNA was extracted using TRIzol Reagent (Ambion, Austin, TX, USA) following the manufacturer’s instructions and quantified with a NanoDrop spectrophotometer. cDNA was synthesized through reverse transcription using the Hifair II 1st Strand cDNA Synthesis Kit (Yeasen). Subsequent qPCR was performed with SYBR Green Realtime PCR Master Mix (TOYOBO, Osaka, Japan) on a ViiA7 real-time PCR system (Applied Biosystems, Waltham, MA, USA). Relative gene expression levels were calculated based on the 2
–∆∆Ct method and normalized to the housekeeping genes
18s or
Actb. The primer sequences used for qPCR are listed in
Supplemental Table S2.
4.8. Proteomic Analysis
Murine bone marrow cells were subjected to cycloheximide (CHX) treatment (0.5 µg/mL; Selleck, Houston, TX, USA) during the final 6 h of a 30 h in vitro culture with EPO and SCF. Then, cells were harvested and stained with surface antibodies against CD71 and Ter119. CD71+Ter119+ erythroid cells were sorted on a BD FACS Aria III (BD Biosciences) and washed twice with cold PBS prior to being frozen in liquid nitrogen for proteomic analysis, which was performed using a mass spectrometer in data-independent acquisition (DIA) mode. Briefly, cells were lysed in 2% SDS buffer containing 50 mM DTT for 20 min at RT, and the supernatants were boiled at 100 °C for 5 min before alkylation with iodoacetamide for 1 h at RT. Proteins were precipitated with pre-cooled acetone and digested overnight at 37 °C with sequencing-grade modified trypsin (Promega, Madison, WI, USA). Tryptic peptides were obtained by centrifugation at 14,000 g for 20 min at 20 °C, purified using C18 Ziptips, and eluted with 0.1% TFA in 50–70% acetonitrile. The eluted peptides were lyophilized using a SpeedVac (ThermoSavant, Holbrook, NY, USA), and the iRT peptides (Biognosys, Schlieren, Switzerland) were spiked into the sample according to the manufacturer’s instructions prior to analysis. DIA was performed on an Orbitrap Exploris 480 coupled to a FAIMS and EASY-nanoLC 1200 system (Thermo Scientific). The peptides were reconstituted in 0.1% formic acid and separated on a 20 cm analytical column using a 120 min gradient. The DIA data were processed and analyzed by Spectronaut 18 (Biognosys AG) with default settings. The resulting sequences were analyzed using the mouse Uniprot FASTA database.
4.9. RNA Sequencing
E14.5 FLCs were purified for Ter119+ erythroblasts, followed by RNA extraction as described above. RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). mRNAs were enriched with Oligo (dT) beads, and RNA libraries were constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s instructions. Sequencing was conducted on an Illumina Novoseq 6000 by Gene Denovo Biotechnology Co. (Guangzhou, China). Low-quality reads were removed, and the filtered clean reads were mapped to the reference genome using HISAT2 2.1.0. The expression abundance and variations were quantified by calculating TPM (Transcripts per kilobase of exon model per million mapped reads) values using RSEM software (v1.3.3). Differentially expressed genes were analyzed by DESeq2.
4.10. Ribosome Profiling
E14.5 FLCs were purified for Ter119+ erythroblasts and washed twice with cold PBS containing cycloheximide (100 ug/mL) at 4 °C for 5 min to block translational elongation. Cells were then rapidly frozen in liquid nitrogen for 1 h and stored at −80 °C for ribosome profiling. In brief, cell extracts were prepared in lysis buffer, triturated through a 26-G needle, and centrifuged to collect the supernatant. Ribosome-protected fragments (RPFs) were generated by incubating lysates with RNase I (New England Biolabs) and DNase I (New England Biolabs) for 45 min at RT, followed by inactivation with SUPERase·In RNase inhibitor (Ambion). RPFs with a size larger than 17 nt were isolated using size exclusion columns and the RNA Clean and Concentrator-25 Kit (Zymo Research, Irvine, CA, USA). rRNA was removed by antisense DNA probes and RNase H digestion, with final purification achieved using magnetic beads (Vazyme, Nanjing, China). Subsequently, Ribo-seq libraries were constructed using the NEBNext® Multiple Small RNA Library Prep Set for Illumina® (E7300S, E7300L) and sequenced using Illumina NovaSeq X Plus by Gene Denovo Biotechnology Co. (Guangzhou, China). Low-quality reads were filtered by fastp, and processed RNA-seq reads were aligned to the genome using whole genome alignment by STAR with the 2-pass setting enabled. Gene expression levels were normalized using the TPM (transcripts per kilobase of exon model per million mapped reads) method.
4.11. Plasmid Construction and Retroviral Infection of FLCs
To generate retroviral particles, 293T cells were cultured in DMEM medium supplemented with 10% FBS. For overexpression and knockdown of NFE2, the plasmids Migr1-NFE2-IRES-EGFP or MSCV-miR30-shNFE2-EGFP were co-transfected with the retroviral packaging plasmid pCL-Eco at a 3:1 ratio using Lipofectamine™ 3000 (Invitrogen). Viral supernatants were harvested at 48 and 72 h post-transfection, then filtered through a 0.45 μm cell strainer. The sequences of retroviral shRNA oligonucleotides targeting mouse NFE2 are provided in
Supplemental Table S2. Retroviral infection of the purified Ter119 negative mouse FLCs was performed as previously described. Briefly, FLCs were resuspended in viral supernatants in the presence of 10 μg/mL polybrene (Sigma) and centrifuged at 800 g for 1.5 h at 32 °C. After spin-infection for 4 h, the viral supernatants were replaced with fresh medium.
4.12. Blood Stasis Syndrome Model
Via the tail vein, 10% dextran 500 (molecular weight 500,000) (Yeasen) in 0.9% saline was administered into WT and eEF2_T56M mice at a dosage of 10 mL/kg, and the control group was injected with 0.9% saline. For blood viscosity detection, abdominal aortic blood was collected 1 h after a single dextran 500 injection. For the establishment of the blood stasis mouse model, dextran 500 was injected via the tail vein once daily for four consecutive days, then lung tissues were harvested for hematoxylin and eosin (HE) staining to evaluate the aggregation of red blood cells.
4.13. Inferior Vena Cava (IVC) Stenosis Model
Mice were anesthetized with isoflurane, and a median laparotomy was performed to expose the IVC and its lateral branches. Complete ligation of all lateral branches of the IVC was achieved using a 7-0 polypropylene suture. After careful isolation of the IVC from the aorta, a partial ligature (stenosis) of the IVC was conducted by tying a 7-0 polypropylene suture over a blunted 30-gauge needle. The needle was subsequently removed to allow partial restoration of blood flow, and the abdominal cavity was gently closed. Operated mice were euthanized 60–72 h post-surgery, and the thrombi developed within the IVC were obtained for measurement and weighing.
4.14. Statistical Analyses
Data are presented as the mean ± standard error of the mean (SEM). All statistical analyses were performed using GraphPad Prism software (v8.0, San Diego, CA, USA). Statistical significance was determined using unpaired two-tailed Student’s t-test between two groups and one-way ANOVA with Tukey’s multiple comparison test between multiple groups. Assays were conducted at least three times. p-values less than 0.05 were considered significant.


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}