
Biomarkers of Progression Independent of Relapse Activity—Can We Actually Measure It Yet?



Abstract
1. Introduction
2. Clinical Biomarkers
2.1. Expanded Disability Status Scale
2.2. Multiple Sclerosis Functional Composite
2.3. Symbol Digit Modalities Test
3. Imaging Biomarkers
3.1. MRI Based Biomarkers
3.1.1. Both Brain and Spinal Cord Volume Loss
3.1.2. Focal Neuroinflammation
3.1.3. Diffuse Neuroinflammation
3.2. Optical Coherence Tomography
3.2.1. Inner Retinal Layer Thickness
3.2.2. Inner Retinal Layer Thinning
4. Body Fluid Biomarkers: Neurodegeneration vs. Inflammation
4.1. Neurofilament Light Chain
4.2. Glial Fibrillary Acidic Protein
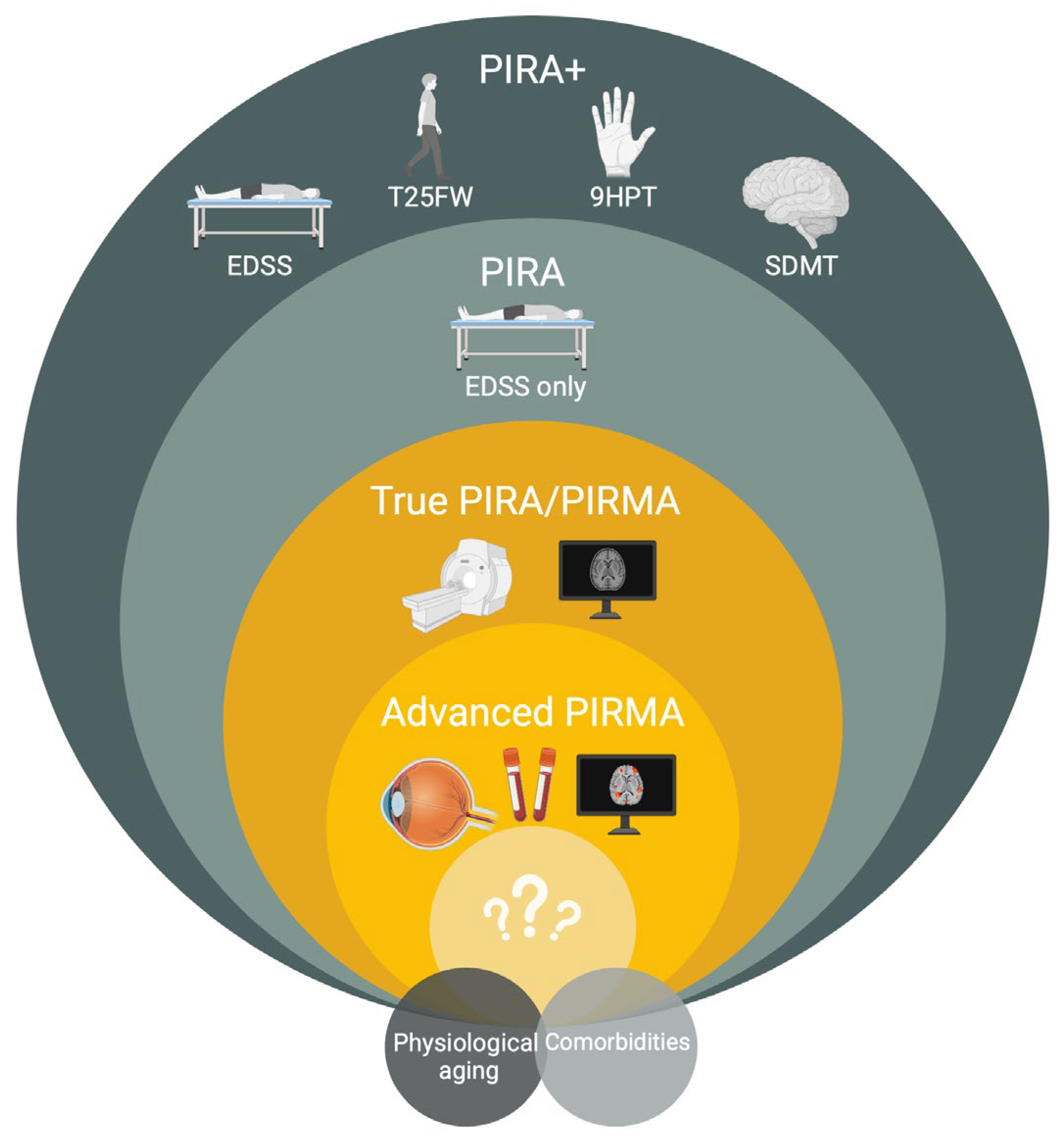
5. Conceptual Challenges in Defining and Quantifying PIRA—What Are We Measuring?
6. Future Directions for Biomarker Research
6.1. Multi-Omics and Systems Biology Approaches
6.2. AI and Predictive Models
6.3. Clarifying the Definition of PIRA
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jakimovski, D.; Bittner, S.; Zivadinov, R.; Morrow, S.A.; Benedict, R.H.; Zipp, F.; Weinstock-Guttman, B. Multiple Sclerosis. Lancet 2024, 403, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the Clinical Course of Multiple Sclerosis: The 2013 Revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Wolinsky, J.S.; Giovannoni, G.; Arnold, D.L.; Wang, Q.; Bernasconi, C.; Model, F.; Koendgen, H.; Manfrini, M.; Belachew, S.; et al. Contribution of Relapse-Independent Progression vs Relapse-Associated Worsening to Overall Confirmed Disability Accumulation in Typical Relapsing Multiple Sclerosis in a Pooled Analysis of 2 Randomized Clinical Trials. JAMA Neurol. 2020, 77, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Häring, D.A.; Ganjgahi, H.; Ocampo, A.; Hatami, F.; Čuklina, J.; Aarden, P.; Dahlke, F.; Arnold, D.L.; Wiendl, H.; et al. How Patients with Multiple Sclerosis Acquire Disability. Brain 2022, 145, 3147–3161. [Google Scholar] [CrossRef]
- Müller, J.; Cagol, A.; Lorscheider, J.; Tsagkas, C.; Benkert, P.; Yaldizli, Ö.; Kuhle, J.; Derfuss, T.; Sormani, M.P.; Thompson, A.; et al. Harmonizing Definitions for Progression Independent of Relapse Activity in Multiple Sclerosis: A Systematic Review. JAMA Neurol. 2023, 80, 1232–1245. [Google Scholar] [CrossRef]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms Underlying Progression in Multiple Sclerosis. Curr. Opin. Neurol. 2020, 33, 277–285. [Google Scholar] [CrossRef]
- Kurtzke, J.F. Rating Neurologic Impairment in Multiple Sclerosis An Expanded Disability Status Scale (EDSS). Neurology 1983, 33, 1444. [Google Scholar] [CrossRef]
- Kappos, L.; D’Souza, M.; Lechner-Scott, J.; Lienert, C. On the Origin of Neurostatus. Mult. Scler. Relat. Disord. 2015, 4, 182–185. [Google Scholar] [CrossRef]
- Koch, M.W.; Mostert, J.; Repovic, P.; Bowen, J.D.; Uitdehaag, B.; Cutter, G. Reliability of Outcome Measures in Clinical Trials in Secondary Progressive Multiple Sclerosis. Neurology 2020, 96, e111–e120. [Google Scholar] [CrossRef]
- Koch, M.W.; Mostert, J.P.; Wolinsky, J.S.; Lublin, F.D.; Uitdehaag, B.; Cutter, G.R. Comparison of the EDSS, Timed 25-Foot Walk, and the 9-Hole Peg Test as Clinical Trial Outcomes in Relapsing-Remitting Multiple Sclerosis. Neurology 2021, 97, e1560–e1570. [Google Scholar] [CrossRef]
- Portaccio, E.; Bellinvia, A.; Fonderico, M.; Pastò, L.; Razzolini, L.; Totaro, R.; Spitaleri, D.; Lugaresi, A.; Cocco, E.; Onofrj, M.; et al. Progression Is Independent of Relapse Activity in Early Multiple Sclerosis: A Real-Life Cohort Study. Brain 2022, 145, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Motamedi, S.; Asseyer, S.; Chien, C.; Saidha, S.; Calabresi, P.A.; Fitzgerald, K.C.; Samadzadeh, S.; Villoslada, P.; Llufriu, S.; et al. Individual Prognostication of Disease Activity and Disability Worsening in Multiple Sclerosis with Retinal Layer Thickness z Scores. Neurol. Neuroimmunol. Neuroinflammation 2024, 11, e200269. [Google Scholar] [CrossRef] [PubMed]
- Amato, M.P.; Fratiglioni, L.; Groppi, C.; Siracusa, G.; Amaducci, L. Interrater Reliability in Assessing Functional Systems and Disability on the Kurtzke Scale in Multiple Sclerosis. Arch. Neurol. 1988, 45, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Noseworthy, J.H.; Vandervoort, M.K.; Wong, C.J.; Ebers, G.C. Interrater Variability with the Expanded Disability Status Scale (EDSS) and Functional Systems (FS) in a Multiple Sclerosis Clinical Trial. Neurology 1990, 40, 971. [Google Scholar] [CrossRef]
- Hyland, M.; Rudick, R.A. Challenges to Clinical Trials in Multiple Sclerosis: Outcome Measures in the Era of Disease-Modifying Drugs. Curr. Opin. Neurol. 2011, 24, 255–261. [Google Scholar] [CrossRef]
- Kalincik, T.; Cutter, G.; Spelman, T.; Jokubaitis, V.; Havrdova, E.; Horakova, D.; Trojano, M.; Izquierdo, G.; Girard, M.; Duquette, P.; et al. Defining Reliable Disability Outcomes in Multiple Sclerosis. Brain 2015, 138, 3287–3298. [Google Scholar] [CrossRef]
- Kappos, L.; Butzkueven, H.; Wiendl, H.; Spelman, T.; Pellegrini, F.; Chen, Y.; Dong, Q.; Koendgen, H.; Belachew, S.; Trojano, M.; et al. Greater Sensitivity to Multiple Sclerosis Disability Worsening and Progression Events Using a Roving versus a Fixed Reference Value in a Prospective Cohort Study. Mult. Scler. J. 2017, 24, 963–973. [Google Scholar] [CrossRef]
- Bsteh, G.; Marti, S.; Krajnc, N.; Traxler, G.; Salmen, A.; Hammer, H.; Leutmezer, F.; Rommer, P.; Pauli, F.D.; Chan, A.; et al. Disability Progression Is a Question of Definition—A Methodological Reappraisal by Example of Primary Progressive Multiple Sclerosis. Mult. Scler. Relat. Disord. 2024, 93, 106215. [Google Scholar] [CrossRef]
- Tur, C.; Carbonell-Mirabent, P.; Cobo-Calvo, Á.; Otero-Romero, S.; Arrambide, G.; Midaglia, L.; Castilló, J.; Vidal-Jordana, Á.; Rodríguez-Acevedo, B.; Zabalza, A.; et al. Association of Early Progression Independent of Relapse Activity with Long-Term Disability After a First Demyelinating Event in Multiple Sclerosis. JAMA Neurol. 2023, 80, 151–160. [Google Scholar] [CrossRef]
- Sawad, A.B.; Seoane-Vazquez, E.; Rodriguez-Monguio, R.; Turkistani, F. Evaluation of the Expanded Disability Status Scale and the Multiple Sclerosis Functional Composite as Clinical Endpoints in Multiple Sclerosis Clinical Trials: Quantitative Meta-Analyses. Curr. Med. Res. Opin. 2016, 32, 1969–1974. [Google Scholar] [CrossRef]
- Kragt, J.J.; van der Linden, F.A.; Nielsen, J.M.; Uitdehaag, B.M.; Polman, C.H. Clinical Impact of 20% Worsening on Timed 25-Foot Walk and 9-Hole Peg Test in Multiple Sclerosis. Mult. Scler. J. 2006, 12, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Bosma, L.; Kragt, J.; Brieva, L.; Khaleeli, Z.; Montalban, X.; Polman, C.; Thompson, A.; Tintoré, M.; Uitdehaag, B. Progression on the Multiple Sclerosis Functional Composite in Multiple Sclerosis: What Is the Optimal Cut-off for the Three Components? Mult. Scler. J. 2010, 16, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Rudick, R.A.; Cutter, G.; Reingold, S. The Multiple Sclerosis Functional Composite: A New Clinical Outcome Measure for Multiple Sclerosis Trials. Mult. Scler. J. 2002, 8, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, D.; Cohen, J.A.; Freedman, M.S.; Goldman, M.D.; Hartung, H.-P.; Havrdova, E.; Jeffery, D.; Kapoor, R.; Miller, A.; Sellebjerg, F.; et al. The EDSS-Plus, an Improved Endpoint for Disability Progression in Secondary Progressive Multiple Sclerosis. Mult. Scler. J. 2016, 23, 94–105. [Google Scholar] [CrossRef]
- Strijbis, E.M.M.; Mostert, J.; Comtois, J.; Salter, A.; Repovic, P.; Bowen, J.D.; Uitdehaag, B.M.J.; Cutter, G.R.; Koch, M.W. Utility of Progression Independent of Relapse Activity as a Trial Outcome in Relapsing-Remitting Multiple Sclerosis. Neurology 2024, 104, e210153. [Google Scholar] [CrossRef]
- Bsteh, G.; Krajnc, N.; Altmann, P.; Hendin, B.; Bharadia, T.; Jaruszowic, S.; Lublin, F.; Oh, J.; Parow, D.; Ribbens, A.; et al. Treating to Target in Multiple Sclerosis: Do We Know How to Measure Whether We Hit It? Eur. J. Neurol. 2024, 31, e16526. [Google Scholar] [CrossRef]
- Kalinowski, A.; Cutter, G.; Bozinov, N.; Hinman, J.A.; Hittle, M.; Motl, R.; Odden, M.; Nelson, L.M. The Timed 25-Foot Walk in a Large Cohort of Multiple Sclerosis Patients. Mult. Scler. J. 2022, 28, 289–299. [Google Scholar] [CrossRef]
- Chow, H.H.; Schreiber, K.; Magyari, M.; Ammitzbøll, C.; Börnsen, L.; Christensen, J.R.; Ratzer, R.; Sørensen, P.S.; Sellebjerg, F. Progressive Multiple Sclerosis, Cognitive Function, and Quality of Life. Brain Behav. 2018, 8, e00875. [Google Scholar] [CrossRef]
- Oreja-Guevara, C.; Blanco, T.A.; Ruiz, L.B.; Pérez, M.Á.H.; Meca-Lallana, V.; Ramió-Torrentà, L. Cognitive Dysfunctions and Assessments in Multiple Sclerosis. Front. Neurol. 2019, 10, 581. [Google Scholar] [CrossRef]
- López-Góngora, M.; Querol, L.; Escartín, A. A One-Year Follow-up Study of the Symbol Digit Modalities Test (SDMT) and the Paced Auditory Serial Addition Test (PASAT) in Relapsing-Remitting Multiple Sclerosis: An Appraisal of Comparative Longitudinal Sensitivity. BMC Neurol. 2015, 15, 40. [Google Scholar] [CrossRef]
- Strober, L.; DeLuca, J.; Benedict, R.H.; Jacobs, A.; Cohen, J.A.; Chiaravalloti, N.; Hudson, L.D.; Rudick, R.A.; LaRocca, N.G. Symbol Digit Modalities Test: A Valid Clinical Trial Endpoint for Measuring Cognition in Multiple Sclerosis. Mult. Scler. J. 2018, 25, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Benedict, R.H.; DeLuca, J.; Phillips, G.; LaRocca, N.; Hudson, L.D.; Rudick, R.; Consortium, M.S.O.A. Validity of the Symbol Digit Modalities Test as a Cognition Performance Outcome Measure for Multiple Sclerosis. Mult. Scler. J. 2017, 27, 721–733. [Google Scholar] [CrossRef]
- Goldman, M.D.; LaRocca, N.G.; Rudick, R.A.; Hudson, L.D.; Chin, P.S.; Francis, G.S.; Jacobs, A.; Kapoor, R.; Matthews, P.M.; Mowry, E.M.; et al. Evaluation of Multiple Sclerosis Disability Outcome Measures Using Pooled Clinical Trial Data. Neurology 2019, 93, e1921–e1931. [Google Scholar] [CrossRef]
- Ciubotaru, A.; Grosu, C.; Alexa, D.; Covali, R.; Maștaleru, A.; Leon, M.M.; Schreiner, T.G.; Ghiciuc, C.M.; Roman, E.M.; Azoicăi, D.; et al. The Faces of “Too Late”—A Surprisingly Progressive Cohort of “Stable” Relapsing Remitting Multiple Sclerosis Patients. Medicina 2024, 60, 1401. [Google Scholar] [CrossRef]
- Biasi, M.M.; Manni, A.; Pepe, I.; Abbatantuono, C.; Gasparre, D.; Iaffaldano, P.; Simone, M.; Caro, M.F.D.; Trojano, M.; Taurisano, P.; et al. Impact of Depression on the Perception of Fatigue and Information Processing Speed in a Cohort of Multiple Sclerosis Patients. BMC Psychol. 2023, 11, 208. [Google Scholar] [CrossRef]
- Gopalakrishnan, N.; Cadden, M.; Barker, L.; Healy, B.C.; Chitnis, T.; Weiner, H.L.; Glanz, B.I. Baseline Predictors of Cross-Sectional and Longitudinal Performance on the Symbol Digit Modalities Test in Individuals with Multiple Sclerosis. J. Neurol. Sci. 2025, 469, 123384. [Google Scholar] [CrossRef]
- Pereira, D.R.; Costa, P.; Cerqueira, J.J. Repeated Assessment and Practice Effects of the Written Symbol Digit Modalities Test Using a Short Inter-Test Interval. Arch. Clin. Neuropsychol. 2015, 30, 424–434. [Google Scholar] [CrossRef]
- Roar, M.; Illes, Z.; Sejbaek, T. Practice Effect in Symbol Digit Modalities Test in Multiple Sclerosis Patients Treated with Natalizumab. Mult. Scler. Relat. Disord. 2016, 10, 116–122. [Google Scholar] [CrossRef]
- Ruggieri, S.; Petracca, M.; Giglio, L.D.; Luca, F.D.; Giannì, C.; Gurreri, F.; Petsas, N.; Tommasin, S.; Pozzilli, C.; Pantano, P. A Matter of Atrophy: Differential Impact of Brain and Spine Damage on Disability Worsening in Multiple Sclerosis. J. Neurol. 2021, 268, 4698–4706. [Google Scholar] [CrossRef]
- Eshaghi, A.; Prados, F.; Brownlee, W.J.; Altmann, D.R.; Tur, C.; Cardoso, M.J.; Angelis, F.D.; van de Pavert, S.H.; Cawley, N.; Stefano, N.D.; et al. Deep Gray Matter Volume Loss Drives Disability Worsening in Multiple Sclerosis. Ann. Neurol. 2018, 83, 210–222. [Google Scholar] [CrossRef]
- Cagol, A.; Schaedelin, S.; Barakovic, M.; Benkert, P.; Todea, R.-A.; Rahmanzadeh, R.; Galbusera, R.; Lu, P.-J.; Weigel, M.; Melie-Garcia, L.; et al. Association of Brain Atrophy with Disease Progression Independent of Relapse Activity in Patients with Relapsing Multiple Sclerosis. JAMA Neurol. 2022, 79, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Cagol, A.; Benkert, P.; Melie-Garcia, L.; Schaedelin, S.A.; Leber, S.; Tsagkas, C.; Barakovic, M.; Galbusera, R.; Lu, P.-J.; Weigel, M.; et al. Association of Spinal Cord Atrophy and Brain Paramagnetic Rim Lesions with Progression Independent of Relapse Activity in People with MS. Neurology 2024, 102, e207768. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.J.; Cen, S.Y.; Khadka, S.; Liu, S.; Kornak, J.; Shi, Y.; Zheng, L.; Hauser, S.L.; Pelletier, D. Thalamic Atrophy in Multiple Sclerosis: A Magnetic Resonance Imaging Marker of Neurodegeneration throughout Disease. Ann. Neurol. 2018, 83, 223–234. [Google Scholar] [CrossRef]
- Mahajan, K.R.; Nakamura, K.; Cohen, J.A.; Trapp, B.D.; Ontaneda, D. Intrinsic and Extrinsic Mechanisms of Thalamic Pathology in Multiple Sclerosis. Ann. Neurol. 2020, 88, 81–92. [Google Scholar] [CrossRef]
- Rocca, M.A.; Mesaros, S.; Pagani, E.; Sormani, M.P.; Comi, G.; Filippi, M. Thalamic Damage and Long-Term Progression of Disability in Multiple Sclerosis. Radiology 2010, 257, 463–469. [Google Scholar] [CrossRef]
- Biberacher, V.; Schmidt, P.; Keshavan, A.; Boucard, C.C.; Righart, R.; Sämann, P.; Preibisch, C.; Fröbel, D.; Aly, L.; Hemmer, B.; et al. Intra- and Interscanner Variability of Magnetic Resonance Imaging Based Volumetry in Multiple Sclerosis. Neuroimage 2016, 142, 188–197. [Google Scholar] [CrossRef]
- Zivadinov, R.; Jakimovski, D.; Gandhi, S.; Ahmed, R.; Dwyer, M.G.; Horakova, D.; Weinstock-Guttman, B.; Benedict, R.R.H.; Vaneckova, M.; Barnett, M.; et al. Clinical Relevance of Brain Atrophy Assessment in Multiple Sclerosis. Implications for Its Use in a Clinical Routine. Expert Rev. Neurother. 2016, 16, 777–793. [Google Scholar] [CrossRef]
- Storelli, L.; Rocca, M.A.; Pagani, E.; Hecke, W.V.; Horsfield, M.A.; Stefano, N.D.; Rovira, A.; Sastre-Garriga, J.; Palace, J.; Sima, D.; et al. Measurement of Whole-Brain and Gray Matter Atrophy in Multiple Sclerosis: Assessment with MR Imaging. Radiology 2018, 288, 554–564. [Google Scholar] [CrossRef]
- Zivadinov, R.; Tranquille, A.; Reeves, J.A.; Dwyer, M.G.; Bergsland, N. Brain Atrophy Assessment in Multiple Sclerosis: Technical– and Subject-Related Barriers for Translation to Real-World Application in Individual Subjects. Expert Rev. Neurother. 2024, 24, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Stefano, N.D.; Stromillo, M.L.; Giorgio, A.; Bartolozzi, M.L.; Battaglini, M.; Baldini, M.; Portaccio, E.; Amato, M.P.; Sormani, M.P. Establishing Pathological Cut-Offs of Brain Atrophy Rates in Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 93–99. [Google Scholar] [CrossRef]
- Sastre-Garriga, J.; Pareto, D.; Battaglini, M.; Rocca, M.A.; Ciccarelli, O.; Enzinger, C.; Wuerfel, J.; Sormani, M.P.; Barkhof, F.; Yousry, T.A.; et al. MAGNIMS Consensus Recommendations on the Use of Brain and Spinal Cord Atrophy Measures in Clinical Practice. Nat. Rev. Neurol. 2020, 16, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Sormani, M.P.; Rovaris, M.; Valsasina, P.; Wolinsky, J.S.; Comi, G.; Filippi, M. Measurement Error of Two Different Techniques for Brain Atrophy Assessment in Multiple Sclerosis. Neurology 2004, 62, 1432–1434. [Google Scholar] [CrossRef]
- Pontillo, G.; Prados, F.; Colman, J.; Kanber, B.; Abdel-Mannan, O.; Al-Araji, S.; Bellenberg, B.; Bianchi, A.; Bisecco, A.; Brownlee, W.J.; et al. Disentangling Neurodegeneration from Aging in Multiple Sclerosis Using Deep Learning: The Brain-Predicted Disease Duration Gap. Neurology 2024, 103, e209976. [Google Scholar] [CrossRef] [PubMed]
- Krajnc, N.; Schmidbauer, V.; Leinkauf, J.; Haider, L.; Bsteh, G.; Kasprian, G.; Leutmezer, F.; Kornek, B.; Rommer, P.S.; Berger, T.; et al. Paramagnetic Rim Lesions Lead to Pronounced Diffuse Periplaque White Matter Damage in Multiple Sclerosis. Mult. Scler. J. 2023, 29, 1406–1417. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Kornek, B.; Kasprian, G.; Berger, T.; Leutmezer, F.; Rommer, P.S.; Trattnig, S.; et al. Long-Term Evolution of Multiple Sclerosis Iron Rim Lesions in 7 T MRI. Brain 2021, 144, 833–847. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Oh, J.; Sati, P.; Absinta, M. Chronic Active Lesions in Multiple Sclerosis: Classification, Terminology, and Clinical Significance. Ther. Adv. Neurol. Disord. 2024, 17, 17562864241306684. [Google Scholar] [CrossRef]
- Liu, C.; Li, W.; Tong, K.A.; Yeom, K.W.; Kuzminski, S. Susceptibility-weighted Imaging and Quantitative Susceptibility Mapping in the Brain. J. Magn. Reson. Imaging 2015, 42, 23–41. [Google Scholar] [CrossRef]
- Reeves, J.A.; Bartnik, A.; Jakimovski, D.; Mohebbi, M.; Bergsland, N.; Salman, F.; Schweser, F.; Wilding, G.; Weinstock-Guttman, B.; Dwyer, M.G.; et al. Associations Between Paramagnetic Rim Lesion Evolution and Clinical and Radiologic Disease Progression in Persons with Multiple Sclerosis. Neurology 2024, 103, e210004. [Google Scholar] [CrossRef]
- Absinta, M.; Sati, P.; Masuzzo, F.; Nair, G.; Sethi, V.; Kolb, H.; Ohayon, J.; Wu, T.; Cortese, I.C.M.; Reich, D.S. Association of Chronic Active Multiple Sclerosis Lesions with Disability In Vivo. JAMA Neurol. 2019, 76, 1474–1483. [Google Scholar] [CrossRef]
- Reeves, J.A.; Mohebbi, M.; Wicks, T.; Salman, F.; Bartnik, A.; Jakimovski, D.; Bergsland, N.; Schweser, F.; Weinstock-Guttman, B.; Dwyer, M.G.; et al. Paramagnetic Rim Lesions Predict Greater Long-Term Relapse Rates and Clinical Progression over 10 Years. Mult. Scler. J. 2024, 30, 535–545. [Google Scholar] [CrossRef]
- Elliott, C.; Wolinsky, J.S.; Hauser, S.L.; Kappos, L.; Barkhof, F.; Bernasconi, C.; Wei, W.; Belachew, S.; Arnold, D.L. Slowly Expanding/Evolving Lesions as a Magnetic Resonance Imaging Marker of Chronic Active Multiple Sclerosis Lesions. Mult. Scler. J. 2018, 25, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Andorra, M.; Nakamura, K.; Lampert, E.J.; Pulido-Valdeolivas, I.; Zubizarreta, I.; Llufriu, S.; Martinez-Heras, E.; Sola-Valls, N.; Sepulveda, M.; Tercero-Uribe, A.; et al. Assessing Biological and Methodological Aspects of Brain Volume Loss in Multiple Sclerosis. JAMA Neurol. 2018, 75, 1246. [Google Scholar] [CrossRef] [PubMed]
- Calvi, A.; Carrasco, F.P.; Tur, C.; Chard, D.T.; Stutters, J.; Angelis, F.D.; John, N.; Williams, T.; Doshi, A.; Samson, R.S.; et al. Association of Slowly Expanding Lesions on MRI with Disability in People with Secondary Progressive Multiple Sclerosis. Neurology 2022, 98, e1783–e1793. [Google Scholar] [CrossRef]
- Calvi, A.; Clarke, M.A.; Prados, F.; Chard, D.; Ciccarelli, O.; Alberich, M.; Pareto, D.; Barranco, M.R.; Sastre-Garriga, J.; Tur, C.; et al. Relationship between Paramagnetic Rim Lesions and Slowly Expanding Lesions in Multiple Sclerosis. Mult. Scler. J. 2023, 29, 352–362. [Google Scholar] [CrossRef]
- Maggi, P.; Kuhle, J.; Schädelin, S.; van der Meer, F.; Weigel, M.; Galbusera, R.; Mathias, A.; Lu, P.-J.; Rahmanzadeh, R.; Benkert, P.; et al. Chronic White Matter Inflammation and Serum Neurofilament Levels in Multiple Sclerosis. Neurology 2021, 97, e543–e553. [Google Scholar] [CrossRef]
- Rosenstein, I.; Nordin, A.; Sabir, H.; Malmeström, C.; Blennow, K.; Axelsson, M.; Novakova, L. Association of Serum Glial Fibrillary Acidic Protein with Progression Independent of Relapse Activity in Multiple Sclerosis. J. Neurol. 2024, 271, 4412–4422. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Belachew, S.; Wolinsky, J.S.; Hauser, S.L.; Kappos, L.; Barkhof, F.; Bernasconi, C.; Fecker, J.; Model, F.; Wei, W.; et al. Chronic White Matter Lesion Activity Predicts Clinical Progression in Primary Progressive Multiple Sclerosis. Brain 2019, 142, 2787–2799. [Google Scholar] [CrossRef]
- Ciccarelli, O.; Barkhof, F.; Calabrese, M.; Stefano, N.D.; Eshaghi, A.; Filippi, M.; Gasperini, C.; Granziera, C.; Kappos, L.; Rocca, M.A.; et al. Using the Progression Independent of Relapse Activity Framework to Unveil the Pathobiological Foundations of Multiple Sclerosis. Neurology 2024, 103, e209444. [Google Scholar] [CrossRef]
- Clark, K.A.; Manning, A.R.; Chen, L.; Liu, F.; Cao, Q.; Bar-Or, A.; Shinohara, R.T.; Sweeney, E.; Schindler, M.K. Early Magnetic Resonance Imaging Features of New Paramagnetic Rim Lesions in Multiple Sclerosis. Ann. Neurol. 2023, 94, 736–744. [Google Scholar] [CrossRef]
- Kwong, K.C.N.K.; Mollison, D.; Meijboom, R.; York, E.N.; Kampaite, A.; Thrippleton, M.J.; Chandran, S.; Waldman, A.D. The Prevalence of Paramagnetic Rim Lesions in Multiple Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2021, 16, e0256845. [Google Scholar] [CrossRef]
- Bonacchi, R.; Pagani, E.; Meani, A.; Cacciaguerra, L.; Preziosa, P.; Meo, E.D.; Filippi, M.; Rocca, M.A. Clinical Relevance of Multiparametric MRI Assessment of Cervical Cord Damage in Multiple Sclerosis. Radiology 2020, 296, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Lauerer, M.; Wiltgen, T.; Brückner, C.; Engl, C.; Giglhuber, K.; Lambrecht, S.; Pongratz, V.; Berthele, A.; Gasperi, C.; Kirschke, J.S.; et al. Predictors of Early Disability Accumulation in Newly Diagnosed Multiple Sclerosis: Clinical, Imaging and Cerebrospinal Fluid Measures. J. Neurol. Neurosurg. Psychiatry 2025. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-Cell Follicles in Secondary Progressive Multiple Sclerosis Associate with Early Onset of Disease and Severe Cortical Pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef]
- Treaba, C.A.; Granberg, T.E.; Sormani, M.P.; Herranz, E.; Ouellette, R.A.; Louapre, C.; Sloane, J.A.; Kinkel, R.P.; Mainero, C. Longitudinal Characterization of Cortical Lesion Development and Evolution in Multiple Sclerosis with 7.0-T MRI. Radiology 2019, 291, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. Multiple Sclerosis: Role of Meningeal Lymphoid Aggregates in Progression Independent of Relapse Activity. Trends Immunol. 2023, 44, 266–275. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.W.; Calabrese, M.; Reynolds, R. Meningeal Inflammation as a Driver of Cortical Grey Matter Pathology and Clinical Progression in Multiple Sclerosis. Nat. Rev. Neurol. 2023, 19, 461–476. [Google Scholar] [CrossRef]
- Kilsdonk, I.D.; Jonkman, L.E.; Klaver, R.; van Veluw, S.J.; Zwanenburg, J.J.M.; Kuijer, J.P.A.; Pouwels, P.J.W.; Twisk, J.W.R.; Wattjes, M.P.; Luijten, P.R.; et al. Increased Cortical Grey Matter Lesion Detection in Multiple Sclerosis with 7 T MRI: A Post-Mortem Verification Study. Brain 2016, 139, 1472–1481. [Google Scholar] [CrossRef]
- Sethi, V.; Yousry, T.A.; Muhlert, N.; Ron, M.; Golay, X.; Wheeler-Kingshott, C.; Miller, D.H.; Chard, D.T. Improved Detection of Cortical MS Lesions with Phase-Sensitive Inversion Recovery MRI. J. Neurol. Neurosurg. Psychiatry 2012, 83, 877. [Google Scholar] [CrossRef]
- Favaretto, A.; Poggiali, D.; Lazzarotto, A.; Rolma, G.; Causin, F.; Gallo, P. The Parallel Analysis of Phase Sensitive Inversion Recovery (PSIR) and Double Inversion Recovery (DIR) Images Significantly Improves the Detection of Cortical Lesions in Multiple Sclerosis (MS) since Clinical Onset. PLoS ONE 2015, 10, e0127805. [Google Scholar] [CrossRef]
- Beck, E.S.; Sati, P.; Sethi, V.; Kober, T.; Dewey, B.; Bhargava, P.; Nair, G.; Cortese, I.C.; Reich, D.S. Improved Visualization of Cortical Lesions in Multiple Sclerosis Using 7T MP2RAGE. Am. J. Neuroradiol. 2018, 39, 459–466. [Google Scholar] [CrossRef]
- Metere, R.; Kober, T.; Möller, H.E.; Schäfer, A. Simultaneous Quantitative MRI Mapping of T1, T2* and Magnetic Susceptibility with Multi-Echo MP2RAGE. PLoS ONE 2017, 12, e0169265. [Google Scholar] [CrossRef] [PubMed]
- Pracht, E.D.; Feiweier, T.; Ehses, P.; Brenner, D.; Roebroeck, A.; Weber, B.; Stöcker, T. SAR and Scan-time Optimized 3D Whole-brain Double Inversion Recovery Imaging at 7T. Magn. Reson. Med. 2018, 79, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Mougin, O.; Abdel-Fahim, R.; Dineen, R.; Pitiot, A.; Evangelou, N.; Gowland, P. Imaging Gray Matter with Concomitant Null Point Imaging from the Phase Sensitive Inversion Recovery Sequence. Magn. Reson. Med. 2016, 76, 1512–1516. [Google Scholar] [CrossRef]
- Harrison, D.M.; Allette, Y.M.; Zeng, Y.; Cohen, A.; Dahal, S.; Choi, S.; Zhuo, J.; Hua, J. Meningeal Contrast Enhancement in Multiple Sclerosis: Assessment of Field Strength, Acquisition Delay, and Clinical Relevance. PLoS ONE 2024, 19, e0300298. [Google Scholar] [CrossRef]
- Okar, S.V.; Dieckhaus, H.; Beck, E.S.; Gaitán, M.I.; Norato, G.; Pham, D.L.; Absinta, M.; Cortese, I.C.; Fletcher, A.; Jacobson, S.; et al. Highly Sensitive 3-Tesla Real Inversion Recovery MRI Detects Leptomeningeal Contrast Enhancement in Chronic Active Multiple Sclerosis. Investig. Radiol. 2024, 59, 243–251. [Google Scholar] [CrossRef]
- Ahn, S.J.; Taoka, T.; Moon, W.; Naganawa, S. Contrast-Enhanced Fluid-Attenuated Inversion Recovery in Neuroimaging: A Narrative Review on Clinical Applications and Technical Advances. J. Magn. Reson. Imaging 2022, 56, 341–353. [Google Scholar] [CrossRef]
- Zwanenburg, J.J.M.; Hendrikse, J.; Visser, F.; Takahara, T.; Luijten, P.R. Fluid Attenuated Inversion Recovery (FLAIR) MRI at 7.0 Tesla: Comparison with 1.5 and 3.0 Tesla. Eur. Radiol. 2010, 20, 915–922. [Google Scholar] [CrossRef]
- Saranathan, M.; Tourdias, T.; Kerr, A.B.; Bernstein, J.D.; Kerchner, G.A.; Han, M.H.; Rutt, B.K. Optimization of Magnetization-Prepared 3-Dimensional Fluid Attenuated Inversion Recovery Imaging for Lesion Detection at 7 T. Investig. Radiol. 2014, 49, 290–298. [Google Scholar] [CrossRef]
- Giannetti, P.; Politis, M.; Su, P.; Turkheimer, F.E.; Malik, O.; Keihaninejad, S.; Wu, K.; Waldman, A.; Reynolds, R.; Nicholas, R.; et al. Increased PK11195-PET Binding in Normal-Appearing White Matter in Clinically Isolated Syndrome. Brain 2015, 138, 110–119. [Google Scholar] [CrossRef]
- Sucksdorff, M.; Matilainen, M.; Tuisku, J.; Polvinen, E.; Vuorimaa, A.; Rokka, J.; Nylund, M.; Rissanen, E.; Airas, L. Brain TSPO-PET Predicts Later Disease Progression Independent of Relapses in Multiple Sclerosis. Brain 2020, 143, 3318–3330. [Google Scholar] [CrossRef]
- Bodini, B.; Tonietto, M.; Airas, L.; Stankoff, B. Positron Emission Tomography in Multiple Sclerosis—Straight to the Target. Nat. Rev. Neurol. 2021, 17, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Högel, H.; Rissanen, E.; Vuorimaa, A.; Airas, L. Positron Emission Tomography Imaging in Evaluation of MS Pathology in Vivo. Mult. Scler. J. 2018, 24, 1399–1412. [Google Scholar] [CrossRef]
- Dadar, M.; Mahmoud, S.; Narayanan, S.; Collins, D.L.; Arnold, D.L.; Maranzano, J. Diffusely Abnormal White Matter Converts to T2 Lesion Volume in the Absence of MRI-Detectable Acute Inflammation. Brain 2021, 145, 2008–2017. [Google Scholar] [CrossRef]
- Hamzaoui, M.; Garcia, J.; Boffa, G.; Lazzarotto, A.; Absinta, M.; Ricigliano, V.A.G.; Soulier, T.; Tonietto, M.; Gervais, P.; Bissery, A.; et al. Positron Emission Tomography with [18F]-DPA-714 Unveils a Smoldering Component in Most Multiple Sclerosis Lesions Which Drives Disease Progression. Ann. Neurol. 2023, 94, 366–383. [Google Scholar] [CrossRef]
- Tozlu, C.; Jamison, K.; Kang, Y.; Rua, S.H.; Kaunzner, U.W.; Nguyen, T.; Kuceyeski, A.; Gauthier, S.A. TSPO-PET Reveals Higher Inflammation in White Matter Disrupted by Paramagnetic Rim Lesions in Multiple Sclerosis. bioRxiv 2025. [Google Scholar] [CrossRef]
- Nutma, E.; Stephenson, J.A.; Gorter, R.P.; de Bruin, J.; Boucherie, D.M.; Donat, C.K.; Breur, M.; van der Valk, P.; Matthews, P.M.; Owen, D.R.; et al. A Quantitative Neuropathological Assessment of Translocator Protein Expression in Multiple Sclerosis. Brain 2019, 142, 3440–3455. [Google Scholar] [CrossRef]
- Nylund, M.; Sucksdorff, M.; Matilainen, M.; Polvinen, E.; Tuisku, J.; Airas, L. Phenotyping of Multiple Sclerosis Lesions According to Innate Immune Cell Activation Using 18 KDa Translocator Protein-PET. Brain Commun. 2021, 4, fcab301. [Google Scholar] [CrossRef]
- Horti, A.G.; Naik, R.; Foss, C.A.; Minn, I.; Misheneva, V.; Du, Y.; Wang, Y.; Mathews, W.B.; Wu, Y.; Hall, A.; et al. PET Imaging of Microglia by Targeting Macrophage Colony-Stimulating Factor 1 Receptor (CSF1R). Proc. Natl. Acad. Sci. USA 2019, 116, 1686–1691. [Google Scholar] [CrossRef]
- Airas, L.; Yong, V.W. Microglia in Multiple Sclerosis—Pathogenesis and Imaging. Curr. Opin. Neurol. 2022, 35, 299–306. [Google Scholar] [CrossRef]
- Obert, D.; Helms, G.; Sättler, M.B.; Jung, K.; Kretzschmar, B.; Bähr, M.; Dechent, P.; Diem, R.; Hein, K. Brain Metabolite Changes in Patients with Relapsing-Remitting and Secondary Progressive Multiple Sclerosis: A Two-Year Follow-Up Study. PLoS ONE 2016, 11, e0162583. [Google Scholar] [CrossRef]
- Heckova, E.; Dal-Bianco, A.; Strasser, B.; Hangel, G.J.; Lipka, A.; Motyka, S.; Hingerl, L.; Rommer, P.S.; Berger, T.; Hnilicová, P.; et al. Extensive Brain Pathologic Alterations Detected with 7.0-T MR Spectroscopic Imaging Associated with Disability in Multiple Sclerosis. Radiology 2022, 303, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Niess, E.; Dal-Bianco, A.; Strasser, B.; Niess, F.; Hingerl, L.; Bachrata, B.; Motyka, S.; Rommer, P.; Trattnig, S.; Bogner, W. Topographical Mapping of Metabolic Abnormalities in Multiple Sclerosis Using Rapid Echo-Less 3D-MR Spectroscopic Imaging at 7T. NeuroImage 2025, 308, 121043. [Google Scholar] [CrossRef]
- Hannoun, S.; Bagory, M.; Durand-Dubief, F.; Ibarrola, D.; Comte, J.-C.; Confavreux, C.; Cotton, F.; Sappey-Marinier, D. Correlation of Diffusion and Metabolic Alterations in Different Clinical Forms of Multiple Sclerosis. PLoS ONE 2012, 7, e32525. [Google Scholar] [CrossRef]
- Caranova, M.; Soares, J.F.; Batista, S.; Castelo-Branco, M.; Duarte, J.V. A Systematic Review of Microstructural Abnormalities in Multiple Sclerosis Detected with NODDI and DTI Models of Diffusion-Weighted Magnetic Resonance Imaging. Magn. Reson. Imaging 2023, 104, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Collorone, S.; Prados, F.; Kanber, B.; Cawley, N.M.; Tur, C.; Grussu, F.; Solanky, B.S.; Yiannakas, M.; Davagnanam, I.; Wheeler-Kingshott, C.A.M.G.; et al. Brain Microstructural and Metabolic Alterations Detected in Vivo at Onset of the First Demyelinating Event. Brain 2021, 144, 1409–1421. [Google Scholar] [CrossRef]
- Seyedmirzaei, H.; Nabizadeh, F.; Aarabi, M.H.; Pini, L. Neurite Orientation Dispersion and Density Imaging in Multiple Sclerosis: A Systematic Review. J. Magn. Reson. Imaging 2023, 58, 1011–1029. [Google Scholar] [CrossRef] [PubMed]
- Rahmanzadeh, R.; Lu, P.-J.; Barakovic, M.; Weigel, M.; Maggi, P.; Nguyen, T.D.; Schiavi, S.; Daducci, A.; Rosa, F.L.; Schaedelin, S.; et al. Myelin and Axon Pathology in Multiple Sclerosis Assessed by Myelin Water and Multi-Shell Diffusion Imaging. Brain 2021, 144, 1684–1696. [Google Scholar] [CrossRef]
- Moccia, M.; van de Pavert, S.; Eshaghi, A.; Haider, L.; Pichat, J.; Yiannakas, M.; Ourselin, S.; Wang, Y.; Wheeler-Kingshott, C.; Thompson, A.; et al. Pathologic Correlates of the Magnetization Transfer Ratio in Multiple Sclerosis. Neurology 2020, 95, e2965–e2976. [Google Scholar] [CrossRef]
- Moll, N.M.; Rietsch, A.M.; Thomas, S.; Ransohoff, A.J.; Lee, J.; Fox, R.; Chang, A.; Ransohoff, R.M.; Fisher, E. Multiple Sclerosis Normal-appearing White Matter: Pathology–Imaging Correlations. Ann. Neurol. 2011, 70, 764–773. [Google Scholar] [CrossRef]
- Shin, H.-G.; Oh, S.-H.; Fukunaga, M.; Nam, Y.; Lee, D.; Jung, W.; Jo, M.; Ji, S.; Choi, J.Y.; Lee, J. Advances in Gradient Echo Myelin Water Imaging at 3T and 7T. NeuroImage 2019, 188, 835–844. [Google Scholar] [CrossRef]
- Vavasour, I.M.; Sun, P.; Graf, C.; Yik, J.T.; Kolind, S.H.; Li, D.K.; Tam, R.; Sayao, A.-L.; Schabas, A.; Devonshire, V.; et al. Characterization of Multiple Sclerosis Neuroinflammation and Neurodegeneration with Relaxation and Diffusion Basis Spectrum Imaging. Mult. Scler. J. 2021, 28, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Rúa, S.M.H.; Kaunzner, U.W.; Pandya, S.; Sweeney, E.; Tozlu, C.; Kuceyeski, A.; Nguyen, T.D.; Gauthier, S.A. Lesion Features on Magnetic Resonance Imaging Discriminate Multiple Sclerosis Patients. Eur. J. Neurol. 2022, 29, 237–246. [Google Scholar] [CrossRef]
- Chawla, S.; Kister, I.; Sinnecker, T.; Wuerfel, J.; Brisset, J.-C.; Paul, F.; Ge, Y. Longitudinal Study of Multiple Sclerosis Lesions Using Ultra-High Field (7T) Multiparametric MR Imaging. PLoS ONE 2018, 13, e0202918. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, T.; Takahashi, T.; Fujihara, K.; Misu, T.; Mugikura, S.; Abe, M.; Ishii, T.; Aoki, M.; Nakashima, I. Number of MRI T1-Hypointensity Corrected by T2/FLAIR Lesion Volume Indicates Clinical Severity in Patients with Multiple Sclerosis. PLoS ONE 2020, 15, e0231225. [Google Scholar] [CrossRef] [PubMed]
- Thaler, C.; Faizy, T.D.; Sedlacik, J.; Bester, M.; Stellmann, J.-P.; Heesen, C.; Fiehler, J.; Siemonsen, S. The Use of Multiparametric Quantitative Magnetic Resonance Imaging for Evaluating Visually Assigned Lesion Groups in Patients with Multiple Sclerosis. J. Neurol. 2018, 265, 127–133. [Google Scholar] [CrossRef]
- Essel, R.R.; Krieger, B.; Bellenberg, B.; Müller, D.; Ladopoulos, T.; Gold, R.; Schneider, R.; Lukas, C. Lesion Assessment in Multiple Sclerosis: A Comparison between Synthetic and Conventional Fluid-Attenuated Inversion Recovery Imaging. Front. Neurol. 2025, 16, 1537465. [Google Scholar] [CrossRef]
- Tazza, F.; Boffa, G.; Schiavi, S.; Lapucci, C.; Piredda, G.F.; Cipriano, E.; Zacà, D.; Roccatagliata, L.; Hilbert, T.; Kober, T.; et al. Multiparametric Characterization and Spatial Distribution of Different MS Lesion Phenotypes. Am. J. Neuroradiol. 2024, 45, 1166–1174. [Google Scholar] [CrossRef]
- Granziera, C.; Wuerfel, J.; Barkhof, F.; Calabrese, M.; Stefano, N.D.; Enzinger, C.; Evangelou, N.; Filippi, M.; Geurts, J.J.G.; Reich, D.S.; et al. Quantitative Magnetic Resonance Imaging towards Clinical Application in Multiple Sclerosis. Brain 2021, 144, 1296–1311. [Google Scholar] [CrossRef]
- Petzold, A. Optical Coherence Tomography to Assess Neurodegeneration in Multiple Sclerosis. Methods Mol. Biol. 2014, 1304, 131–141. [Google Scholar] [CrossRef]
- Saidha, S.; Sotirchos, E.S.; Oh, J. Relationships between Retinal Axonal and Neuronal Measures and Global Central Nervous System Pathology in Multiple Sclerosis. JAMA Neurol. 2013, 70, 34–43. [Google Scholar] [CrossRef]
- Pemp, B.; Kardon, R.H.; Kircher, K.; Pernicka, E.; Schmidt-Erfurth, U.; Reitner, A. Effectiveness of Averaging Strategies to Reduce Variance in Retinal Nerve Fibre Layer Thickness Measurements Using Spectral-Domain Optical Coherence Tomography. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Seigo, M.A.; Sotirchos, E.S.; Newsome, S.; Babiarz, A.; Eckstein, C.; Ford, E.; Oakley, J.D.; Syc, S.B.; Frohman, T.C.; Ratchford, J.N.; et al. In Vivo Assessment of Retinal Neuronal Layers in Multiple Sclerosis with Manual and Automated Optical Coherence Tomography Segmentation Techniques. J. Neurol. 2012, 259, 2119–2130. [Google Scholar] [CrossRef]
- Petzold, A.; Balcer, L.J.; Calabresi, P.A.; Costello, F.; Frohman, T.C.; Frohman, E.M.; Martínez-Lapiscina, E.H.; Green, A.J.; Kardon, R.; Outteryck, O.; et al. Retinal Layer Segmentation in Multiple Sclerosis: A Systematic Review and Meta-Analysis. Lancet Neurol. 2017, 16, 797–812. [Google Scholar] [CrossRef]
- Wadhwani, M.; Bali, S.J.; Satyapal, R.; Angmo, D.; Sharma, R.; Pandey, V.; Dada, T. Test-Retest Variability of Retinal Nerve Fiber Layer Thickness and Macular Ganglion Cell-Inner Plexiform Layer Thickness Measurements Using Spectral-Domain Optical Coherence Tomography. J. Glaucoma 2015, 24, e109–e115. [Google Scholar] [CrossRef]
- Martínez-Lapiscina, E.H.; Arnow, S.; Wilson, J.A.; Saidha, S.; Preiningerova, J.L.; Oberwahrenbrock, T.; Brandt, A.U.; Pablo, L.E.; Guerrieri, S.; Gonzalez, I.; et al. Retinal Thickness Measured with Optical Coherence Tomography and Risk of Disability Worsening in Multiple Sclerosis: A Cohort Study. Lancet Neurol. 2016, 15, 574–584. [Google Scholar] [CrossRef]
- Bsteh, G.; Hegen, H.; Teuchner, B.; Berek, K.; Wurth, S.; Auer, M.; Pauli, F.D.; Deisenhammer, F.; Berger, T. Peripapillary Retinal Nerve Fibre Layer Thinning Rate as a Biomarker Discriminating Stable and Progressing Relapsing–Remitting Multiple Sclerosis. Eur. J. Neurol. 2019, 26, 865–871. [Google Scholar] [CrossRef]
- Bsteh, G.; Berek, K.; Hegen, H.; Altmann, P.; Wurth, S.; Auer, M.; Zinganell, A.; Pauli, F.D.; Rommer, P.; Leutmezer, F.; et al. Macular Ganglion Cell–Inner Plexiform Layer Thinning as a Biomarker of Disability Progression in Relapsing Multiple Sclerosis. Mult. Scler. J. 2020, 27, 684–694. [Google Scholar] [CrossRef]
- Lambe, J.; Fitzgerald, K.C.; Murphy, O.C.; Filippatou, A.G.; Sotirchos, E.S.; Kalaitzidis, G.; Vasileiou, E.; Pellegrini, N.; Ogbuokiri, E.; Toliver, B.; et al. Association of Spectral-Domain OCT with Long-Term Disability Worsening in Multiple Sclerosis. Neurology 2021, 96, e2058–e2069. [Google Scholar] [CrossRef]
- Knier, B.; Leppenetier, G.; Wetzlmair, C.; Aly, L.; Hoshi, M.-M.; Pernpeintner, V.; Biberacher, V.; Berthele, A.; Mühlau, M.; Zimmer, C.; et al. Association of Retinal Architecture, Intrathecal Immunity, and Clinical Course in Multiple Sclerosis. JAMA Neurol. 2017, 74, 847. [Google Scholar] [CrossRef] [PubMed]
- Ratchford, J.N.; Saidha, S.; Sotirchos, E.S.; Oh, J.A.; Seigo, M.A.; Eckstein, C.; Durbin, M.K.; Oakley, J.D.; Meyer, S.A.; Conger, A.; et al. Active MS Is Associated with Accelerated Retinal Ganglion Cell/Inner Plexiform Layer Thinning. Neurology 2013, 80, 47–54. [Google Scholar] [CrossRef]
- Balk, L.J.; Cruz-Herranz, A.; Albrecht, P.; Arnow, S.; Gelfand, J.M.; Tewarie, P.; Killestein, J.; Uitdehaag, B.M.J.; Petzold, A.; Green, A.J. Timing of Retinal Neuronal and Axonal Loss in MS: A Longitudinal OCT Study. J. Neurol. 2016, 263, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.C.; You, Y.; Yiannikas, C.; Garrick, R.; Parratt, J.; Barnett, M.H.; Klistorner, A. Progressive Loss of Retinal Ganglion Cells and Axons in Nonoptic Neuritis Eyes in Multiple Sclerosis: A Longitudinal Optical Coherence Tomography Study. Investig. Ophth Vis. Sci. 2016, 57, 2311–2317. [Google Scholar] [CrossRef]
- Bsteh, G.; Hegen, H.; Altmann, P.; Auer, M.; Berek, K.; Pauli, F.D.; Wurth, S.; Zinganell, A.; Rommer, P.; Deisenhammer, F.; et al. Retinal Layer Thinning Is Reflecting Disability Progression Independent of Relapse Activity in Multiple Sclerosis. Mult. Scler. J.-Exp. Transl. Clin. 2020, 6, 205521732096634. [Google Scholar] [CrossRef]
- Bsteh, G.; Berek, K.; Hegen, H.; Teuchner, B.; Buchmann, A.; Voortman, M.M.; Auer, M.; Wurth, S.; Zinganell, A.; Pauli, F.D.; et al. Serum Neurofilament Levels Correlate with Retinal Nerve Fiber Layer Thinning in Multiple Sclerosis. Mult. Scler. J. 2019, 26, 1682–1690. [Google Scholar] [CrossRef]
- Jenkins, T.M.; Toosy, A.T. Optical Coherence Tomography Should Be Part of the Routine Monitoring of Patients with Multiple Sclerosis: No. Mult. Scler. J. 2014, 20, 1299–1301. [Google Scholar] [CrossRef]
- Ringelstein, M.; Albrecht, P.; Südmeyer, M.; Harmel, J.; Müller, A.; Keser, N.; Finis, D.; Ferrea, S.; Guthoff, R.; Schnitzler, A.; et al. Subtle Retinal Pathology in Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2014, 1, 290–297. [Google Scholar] [CrossRef]
- Paul, F.; Calabresi, P.A.; Barkhof, F.; Green, A.J.; Kardon, R.; Sastre-Garriga, J.; Schippling, S.; Vermersch, P.; Saidha, S.; Gerendas, B.S.; et al. Optical Coherence Tomography in Multiple Sclerosis: A 3-year Prospective Multicenter Study. Ann. Clin. Transl. Neurol. 2021, 8, 2235–2251. [Google Scholar] [CrossRef]
- Bittner, S.; Oh, J.; Havrdová, E.K.; Tintoré, M.; Zipp, F. The Potential of Serum Neurofilament as Biomarker for Multiple Sclerosis. Brain 2021, 144, 2954–2963. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as Biomarkers in Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Disanto, G.; Barro, C.; Benkert, P.; Naegelin, Y.; Schädelin, S.; Giardiello, A.; Zecca, C.; Blennow, K.; Zetterberg, H.; Leppert, D.; et al. Serum Neurofilament Light: A Biomarker of Neuronal Damage in Multiple Sclerosis. Ann. Neurol. 2017, 81, 857–870. [Google Scholar] [CrossRef]
- Benkert, P.; Meier, S.; Schaedelin, S.; Manouchehrinia, A.; Yaldizli, Ö.; Maceski, A.; Oechtering, J.; Achtnichts, L.; Conen, D.; Derfuss, T.; et al. Serum Neurofilament Light Chain for Individual Prognostication of Disease Activity in People with Multiple Sclerosis: A Retrospective Modelling and Validation Study. Lancet Neurol. 2022, 21, 246–257. [Google Scholar] [CrossRef]
- Novakova, L.; Zetterberg, H.; Sundström, P.; Axelsson, M.; Khademi, M.; Gunnarsson, M.; Malmeström, C.; Svenningsson, A.; Olsson, T.; Piehl, F.; et al. Monitoring Disease Activity in Multiple Sclerosis Using Serum Neurofilament Light Protein. Neurology 2017, 89, 2230–2237. [Google Scholar] [CrossRef]
- Gafson, A.R.; Jiang, X.; Shen, C.; Kapoor, R.; Zetterberg, H.; Fox, R.J.; Belachew, S. Serum Neurofilament Light and Multiple Sclerosis Progression Independent of Acute Inflammation. JAMA Netw. Open 2022, 5, e2147588. [Google Scholar] [CrossRef]
- Cantó, E.; Barro, C.; Zhao, C.; Caillier, S.J.; Michalak, Z.; Bove, R.; Tomic, D.; Santaniello, A.; Häring, D.A.; Hollenbach, J.; et al. Association Between Serum Neurofilament Light Chain Levels and Long-Term Disease Course Among Patients with Multiple Sclerosis Followed up for 12 Years. JAMA Neurol. 2019, 76, 1359–1366. [Google Scholar] [CrossRef]
- Häring, D.A.; Kropshofer, H.; Kappos, L.; Cohen, J.A.; Shah, A.; Meinert, R.; Leppert, D.; Tomic, D.; Kuhle, J. Long-Term Prognostic Value of Longitudinal Measurements of Blood Neurofilament Levels. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e856. [Google Scholar] [CrossRef]
- Leppert, D.; Kropshofer, H.; Häring, D.A.; Dahlke, F.; Patil, A.; Meinert, R.; Tomic, D.; Kappos, L.; Kuhle, J. Blood Neurofilament Light in Progressive Multiple Sclerosis: Post Hoc Analysis of 2 Randomized Controlled Trials. Neurology 2021, 98, e2120–e2131. [Google Scholar] [CrossRef]
- Benkert, P.; Maceski, A.M.; Schaedelin, S.; Oechtering, J.; Zadic, A.; Gomez, J.F.V.; Melie-Garcia, L.; Cagol, A.; Galbusera, R.; Subramaniam, S.; et al. Serum Glial Fibrillary Acidic Protein and Neurofilament Light Chain Levels Reflect Different Mechanisms of Disease Progression under B-Cell Depleting Treatment in Multiple Sclerosis. Ann. Neurol. 2025, 97, 104–115. [Google Scholar] [CrossRef]
- Khalil, M.; Pirpamer, L.; Hofer, E.; Voortman, M.M.; Barro, C.; Leppert, D.; Benkert, P.; Ropele, S.; Enzinger, C.; Fazekas, F.; et al. Serum Neurofilament Light Levels in Normal Aging and Their Association with Morphologic Brain Changes. Nat. Commun. 2020, 11, 812. [Google Scholar] [CrossRef]
- Chitnis, T.; Gonzalez, C.; Healy, B.C.; Saxena, S.; Rosso, M.; Barro, C.; Michalak, Z.; Paul, A.; Kivisakk, P.; Cruz, C.D.; et al. Neurofilament Light Chain Serum Levels Correlate with 10-year MRIoutcomes in Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 1478–1491. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K.W. Glial Fibrillary Acidic Protein: From Intermediate Filament Assembly and Gliosis to Neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef]
- Meier, S.; Willemse, E.A.J.; Schaedelin, S.; Oechtering, J.; Lorscheider, J.; Melie-Garcia, L.; Cagol, A.; Barakovic, M.; Galbusera, R.; Subramaniam, S.; et al. Serum Glial Fibrillary Acidic Protein Compared with Neurofilament Light Chain as a Biomarker for Disease Progression in Multiple Sclerosis. JAMA Neurol. 2023, 80, 287–297. [Google Scholar] [CrossRef]
- Madill, E.; Healy, B.C.; Molazadeh, N.; Polgar-Turcsanyi, M.; Glanz, B.I.; Weiner, H.L.; Chitnis, T. Serum Glial Fibrillary Acidic Protein Predicts Disease Progression in Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2024, 11, 2719–2730. [Google Scholar] [CrossRef]
- Thebault, S.; Fereshtehnejad, S.-M.; Bergman, H.P.; Breville, G.; Abdoli, M.; Booth, R.A.; Fadda, G.; Freedman, M.S.; Bose, G. The Combination of CSF Neurofilament Light Chain and Glial Fibrillary Acidic Protein Improves the Prediction of Long-Term Confirmed Disability Worsening in Multiple Sclerosis. Sci. Rep. 2024, 14, 29135. [Google Scholar] [CrossRef]
- Norgren, N.; Sundström, P.; Svenningsson, A.; Rosengren, L.; Stigbrand, T.; Gunnarsson, M. Neurofilament and Glial Fibrillary Acidic Protein in Multiple Sclerosis. Neurology 2004, 63, 1586–1590. [Google Scholar] [CrossRef]
- Cross, A.H.; Gelfand, J.M.; Thebault, S.; Bennett, J.L.; von Büdingen, H.C.; Cameron, B.; Carruthers, R.; Edwards, K.; Fallis, R.; Gerstein, R.; et al. Emerging Cerebrospinal Fluid Biomarkers of Disease Activity and Progression in Multiple Sclerosis. JAMA Neurol. 2024, 81, 373–383. [Google Scholar] [CrossRef]
- Cruciani, A.; Toosy, A.T. Progression Independent of Relapse Activity in Multiple Sclerosis: More, or Less, Than Meets the Eye? Neurology 2025, 104, e210296. [Google Scholar] [CrossRef]
- Goyne, C.E.; Fair, A.E.; Sumowski, P.E.; Graves, J.S. The Impact of Aging on Multiple Sclerosis. Curr. Neurol. Neurosci. Rep. 2024, 24, 83–93. [Google Scholar] [CrossRef]
- Scalfari, A.; Traboulsee, A.; Oh, J.; Airas, L.; Bittner, S.; Calabrese, M.; Dominguez, J.M.G.; Granziera, C.; Greenberg, B.; Hellwig, K.; et al. Smouldering-Associated Worsening in Multiple Sclerosis: An International Consensus Statement on Definition, Biology, Clinical Implications, and Future Directions. Ann. Neurol. 2024, 96, 826–845. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Moccia, M.; Coetzee, T.; Cohen, J.A.; Correale, J.; Graves, J.; Marrie, R.A.; Montalban, X.; Yong, V.W.; Thompson, A.J.; et al. Multiple Sclerosis Progression: Time for a New Mechanism-Driven Framework. Lancet Neurol. 2023, 22, 78–88. [Google Scholar] [CrossRef]
- di Sturmeck, T.G.; Malimpensa, L.; Ferrazzano, G.; Belvisi, D.; Leodori, G.; Lembo, F.; Brandi, R.; Pascale, E.; Cattaneo, A.; Salvetti, M.; et al. Exploring MiRNAs’ Based Modeling Approach for Predicting PIRA in Multiple Sclerosis: A Comprehensive Analysis. Int. J. Mol. Sci. 2024, 25, 6342. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, E.D.; Becker, T.; Werthen-Brabants, L.; Dewulf, P.; Iliadis, D.; Dekeyser, C.; Laureys, G.; Wijmeersch, B.V.; Popescu, V.; Dhaene, T.; et al. Machine-Learning-Based Prediction of Disability Progression in Multiple Sclerosis: An Observational, International, Multi-Center Study. PLoS Digit. Health 2024, 3, e0000533. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Modality | Measure | Marker of Ongoing PIRA | Predictor of Future PIRA |
|---|---|---|---|
| Clinical | EDSS | yes | Limited value |
| Clinical | T25FW | yes—within PIRA plus | Limited value |
| Clinical | 9HPT | Marginal value within PIRA plus | Limited value |
| Clinical | SDMT | yes—within PIRA plus | Limited value |
| MRI | T2L | Negative marker within PIRMA | Limited value as negative predictor |
| MRI | Brain/spinal cord atrophy | yes | Potential predictor with limited evidence |
| MRI | PRL | yes | Potential predictor with limited evidence |
| MRI | SEL | yes | Potential predictor with limited evidence |
| MRI | CL/meningeal inflammation | Positive association | Potential predictor with limited evidence |
| PET-TSPO | Microglial activation | Positive association | Potential predictor with limited evidence |
| OCT | pRNFL | Negative association | Potential predictor with limited evidence |
| OCT | GCIPL | Negative association | Potential predictor with limited evidence |
| Fluid biomarkers | sNfL | Positive association but lacks specificity | Potential predictor but lacks specificity |
| Fluid biomarkers | GFAP | Positive association | Potential predictor with limited evidence |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bsteh, G.; Dal-Bianco, A.; Krajnc, N.; Berger, T. Biomarkers of Progression Independent of Relapse Activity—Can We Actually Measure It Yet? Int. J. Mol. Sci. 2025, 26, 4704. https://doi.org/10.3390/ijms26104704
Bsteh G, Dal-Bianco A, Krajnc N, Berger T. Biomarkers of Progression Independent of Relapse Activity—Can We Actually Measure It Yet? International Journal of Molecular Sciences. 2025; 26(10):4704. https://doi.org/10.3390/ijms26104704
Chicago/Turabian StyleBsteh, Gabriel, Assunta Dal-Bianco, Nik Krajnc, and Thomas Berger. 2025. "Biomarkers of Progression Independent of Relapse Activity—Can We Actually Measure It Yet?" International Journal of Molecular Sciences 26, no. 10: 4704. https://doi.org/10.3390/ijms26104704
APA StyleBsteh, G., Dal-Bianco, A., Krajnc, N., & Berger, T. (2025). Biomarkers of Progression Independent of Relapse Activity—Can We Actually Measure It Yet? International Journal of Molecular Sciences, 26(10), 4704. https://doi.org/10.3390/ijms26104704