1. Introduction
Lignocellulose is an abundant renewable resource, and its conversion into biofuels and high-value biological products is crucial for promoting resource recycling and reducing carbon emissions [
1,
2]. Chinese distillers’ grains (CDGs), byproducts of the Baijiu industry, possess distinctive flavor profiles and valuable nutritional components, including trace elements and proteins, which makes them a potential feed ingredient for livestock. However, strong-flavor CDGs contain a significant proportion of rice husks (one-third on a dry basis), where the high lignocellulose content in rice husks severely limits the efficient utilization of CDGs [
3]. The structural complexity of lignocellulose requires the synergistic action of cellulase, hemicellulase, lignin-degrading enzymes, and various accessory proteins for complete hydrolysis. However, low enzymatic hydrolysis efficiency and the high cost of enzymes remain major bottlenecks that hinder the effective saccharification of lignocellulose [
4].
White-rot fungi (WRFs) are a group of filamentous fungi capable of completely decomposing plant-derived lignocellulose, leaving behind a characteristic white residue [
5]. Their high degradation efficiency and environmental compatibility have made them a key focus in lignocellulose degradation research [
6]. The lignocellulose degradation mechanism of WRFs mainly relies on their abundant extracellular ligninolytic and cellulolytic enzymes, which break down lignin and cellulose biopolymers, respectively. The primary lignin-degrading enzymes include laccase (Lac), lignin peroxidase (LiP), and manganese peroxidase (MnP) [
7]. WRFs are major producers of hemicellulases, and they secrete a variety of enzymes, including main-chain and side-chain enzymes such as xylanase, mannanase, galactosidase, and arabinofuranosidase [
8]. While WRFs also produce cellulolytic enzymes, including cellobiohydrolase (CBH), endoglucanase (EG), and β-glucosidase (βG) [
7], their cellulose hydrolysis mainly generates low-molecular-weight carbohydrates rather than fully converting cellulose into monosaccharides. This limitation arises from the absence of critical enzymatic components in their cellulase system that are necessary for complete cellulose depolymerization into monosaccharides [
9].
Insufficient βG activity leads to the accumulation of cellobiose, which in turn causes the severe product feedback inhibition of EG and CBH [
10]. Moreover, the limited presence of hemicellulolytic enzymes in the
Trichoderma reesei secretome significantly hinders the efficient breakdown of lignocellulosic biomass [
11]. To enhance the conversion efficiency of lignocellulosic biomass, enzyme cocktails capable of effectively degrading all components are developed [
12]. An effective strategy for optimizing enzyme cocktails and improving hydrolysis efficiency involves supplementing specific enzymes based on the composition of the native enzyme cocktail, thereby enhancing synergistic interactions among different enzymes. For example, supplementing βG and EG in
Trichoderma reesei enzyme cocktails has been shown to enhance cellulose conversion rates in sugarcane bagasse [
13]. A comprehensive understanding of the secretomes of relevant microorganisms will aid in the development of highly efficient and customized in vitro lignocellulose-hydrolyzing enzyme cocktails [
14].
To elucidate the protein composition of extracellular lignocellulose-degrading enzyme cocktails in WRFs, optimize the enzyme cocktail composition, and enhance the lignocellulose hydrolysis efficiency, this study focuses on Coriolopsis trogii (C. trogii) Mafic-2001, a WRF strain isolated in our laboratory. Mafic-2001 simultaneously secretes high levels of laccase, cellulases, and hemicellulases. However, the composition of its secretome and its degradation efficiency for agricultural waste remain uncharacterized. This study investigates the secretome of C. trogii Mafic-2001 under lignocellulosic biomass induction. Additionally, EG and βG were heterologously expressed and supplemented into the native enzyme cocktails of Mafic-2001 for the hydrolysis of CDGs. This study lays the foundation for elucidating the mechanistic roles of lignocellulose-degrading secretomes in WRFs and for developing high-efficiency enzyme cocktails. Furthermore, this study provides strategic insights for improving the utilization efficiency of agricultural waste.
3. Discussion
WRFs secrete an enzyme cocktail mainly composed of ligninolytic enzymes, cellulases, and hemicellulases to depolymerize lignocellulose into fermentable sugars, which serve as nutrients for fungal growth [
1]. Agricultural waste, such as crop straws and CDGs, represents a critical biomass resource owing to its high lignocellulosic content. The biological conversion of these residues into value-added products, including biofeed, biochemicals, and biofuels [
20], not only mitigates environmental pollution caused by agricultural waste but also alleviates resource scarcity [
21]. However, the bottleneck in producing such bioproducts lies in the low saccharification efficiency and high costs of lignocellulose-degrading enzymes [
4]. Consequently, identifying highly efficient enzyme cocktails for lignocellulose degradation is crucial to overcoming these challenges and advancing sustainable biorefinery processes.
The use of lignocellulosic biomass as cultivation substrates not only induces fungi to secrete highly active lignocellulose-degrading enzymes but also reduces enzyme production costs. For example, Li et al. [
11] reported that using sugarcane bagasse as a substrate for
T. harzianum K223452 resulted in an enzyme cocktail with a filter paper activity of 1.62 U/mL. Similarly,
P. ostreatus grown on CDGs produced lignin-degrading enzymes with notable activities (Lac 152.68 U/g, Lip 15.56 U/g, MnP 0.34 U/g) and xylanase (10.98 U/g) [
22]. Lignocellulose-degrading enzymes are inducible, and their activity and variety exhibit substrate-dependent variations. In this study,
C. trogii Mafic-2001 cultivated on rice straw exhibited the highest laccase, cellulase, and xylanase activities compared to other biomass substrates, including oat straw, corn stover, corn cob, Napier grass, wheat bran, and rice straw. The observed variability in enzyme production across different biomass substrates might be attributed to differences in the compositional and structural characteristics of the biomass, particularly the lignocellulose content and complexity [
23]. This substrate-dependent induction pattern aligned with previous studies. For example, Zhang et al. [
14] reported that corn stover induced
T. harzianum EM0925 to secrete more cellulase and xylanase compared with wheat bran, corn cob, sugarcane bagasse, and miscanthus. Similarly, steam-exploded wood residue significantly enhanced the xylanase, endoglucanase, and β-glucanase activities in
T. reesei cultures compared with untreated wood residue [
24]. Notably, structurally complex substrates might induce fungi to secrete enzyme cocktails with superior lignocellulose degradation efficiency [
25]. In this study, the maximum cellulase and xylanase activities of
C. trogii Mafic-2001 reached 3.29 U/mL and 25.83 U/mL, respectively, which surpassed those reported for other WRFs, including
Phanerochaete chrysosporium (0.61 and 3.47 U/mL) [
26],
Trametes versicolor CTB863A (1.50 and 8.30 U/mL) [
27], and
Trametes versicolor (0.49 and 0.55 U/mL) [
28]. These findings highlighted the potential of
C. trogii Mafic-2001 as a robust producer of lignocellulose-degrading enzymes and made it a promising candidate for industrial biocatalytic applications.
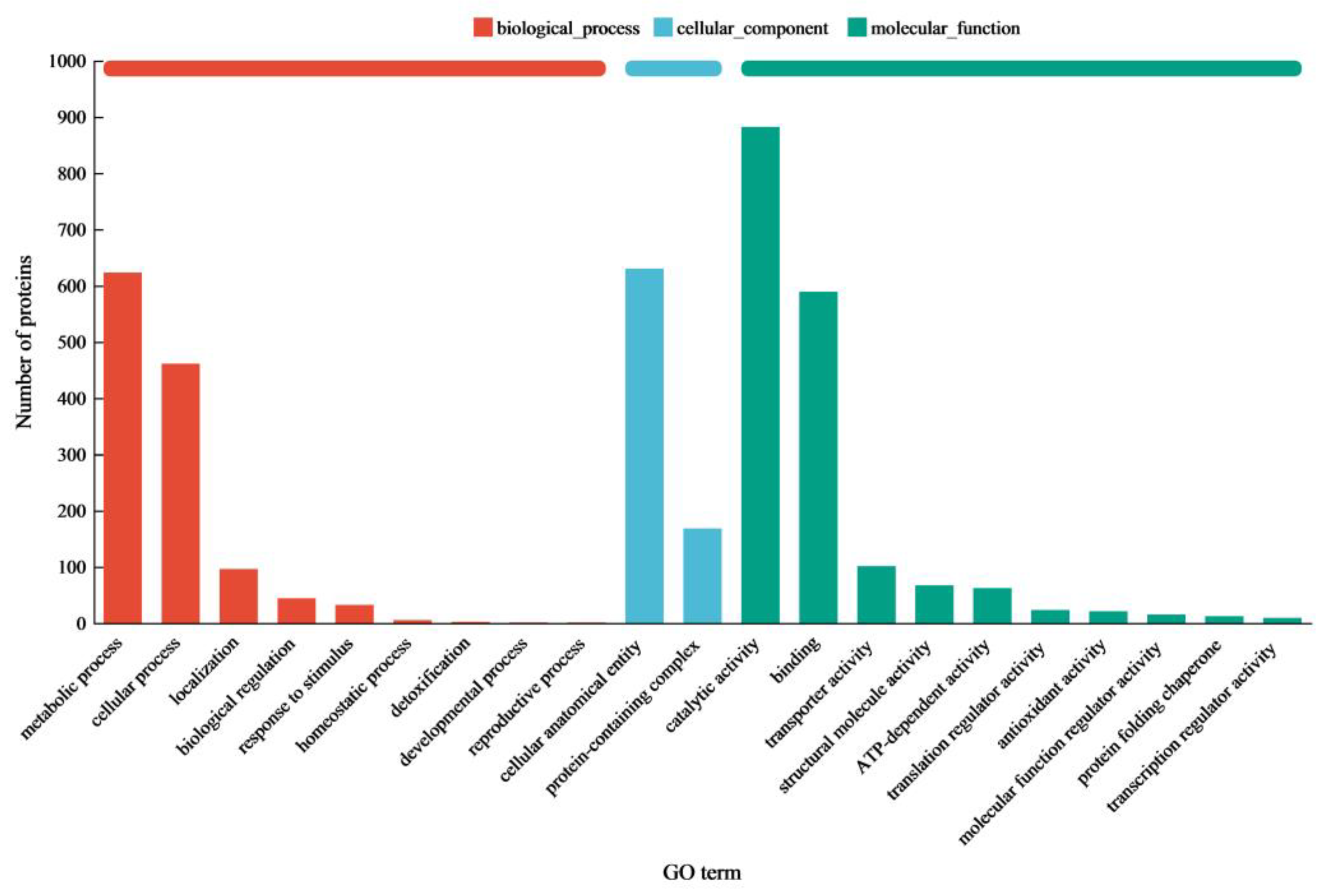
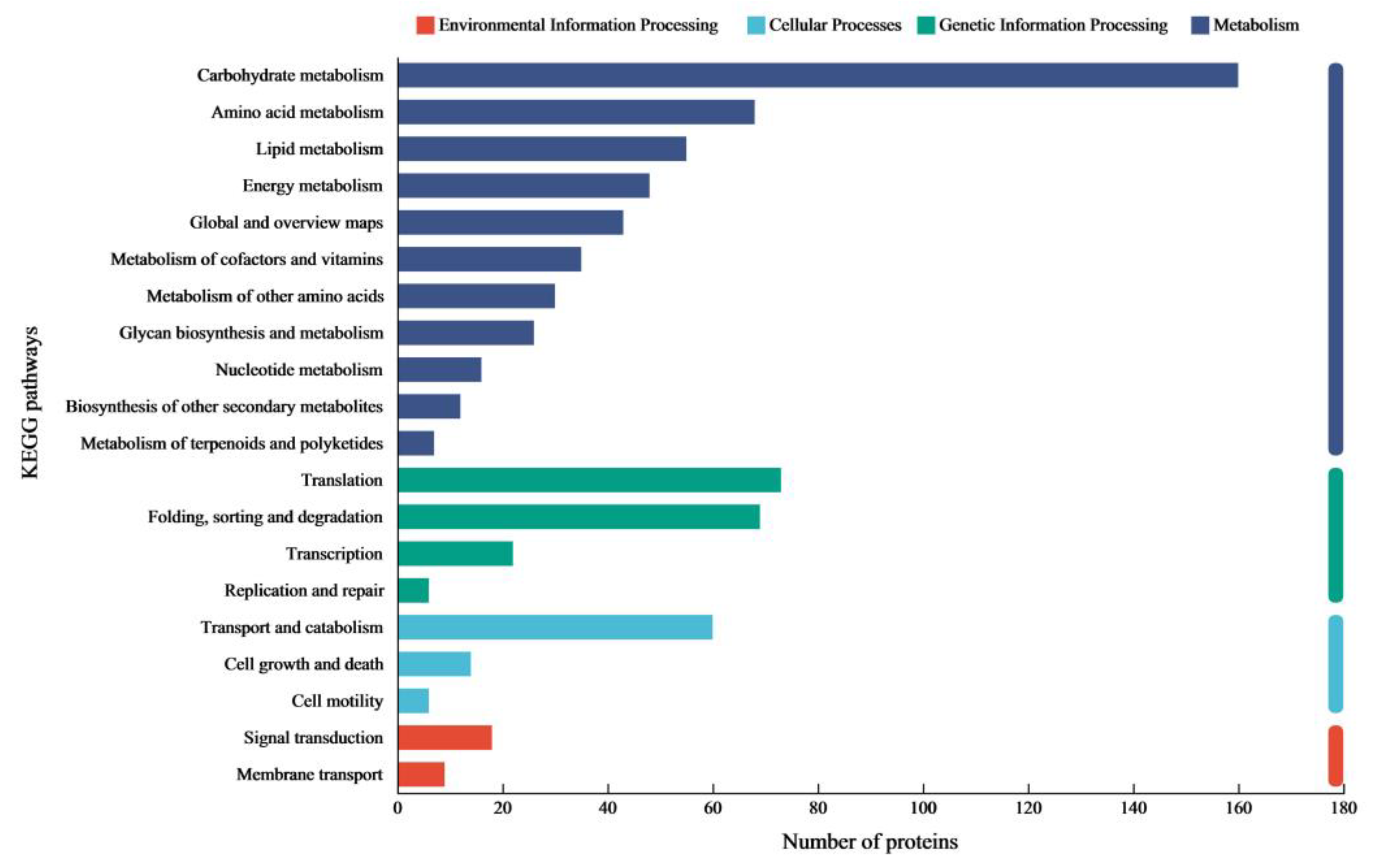
The proteomic analysis of
C. trogii Mafic-2001 induced by rice straw revealed that significant proportions of extracellular proteins were annotated to carbohydrate metabolic processes through GO and KEGG enrichment, which underscored its substantial potential for lignocellulose degradation. The complete deconstruction of lignocellulose requires the synergistic interplay of multiple enzymes [
29]. In this study, the secretome of Mafic-2001 contained 287 CAZymes, which included 147 GHs, AAs, CEs, and PLs, which together formed the enzymatic foundation for efficient lignocellulose breakdown [
8]. Notably, CBHs accounted for 7.57% of the secretome, and they hydrolyzed cellulose by progressively cleaving β-1,4-glycosidic bonds along cellulose chains to release cellobiose for further degradation by EGs and βGs [
30,
31,
32]. This finding was consistent with previous reports, such as
Phanerochaete chrysosporium secreting high-activity CBHs during tobacco straw and corn stover fermentation, which critically enhanced biomass degradation [
33,
34].
Furthermore, the Mafic-2001 secretome displayed a diverse array of hemicellulose-degrading enzymes, including backbone-cleaving enzymes (xylanases, xylosidases, and mannanases) and side-chain-targeting enzymes (galactosidases and α-L-arabinofuranosidases). Owing to the structural complexity of hemicellulose and its cross-linking with other plant cell wall components, the cooperative action of both backbone- and side-chain-degrading enzymes was crucial for efficient hydrolysis [
5]. Additionally, Mafic-2001 secreted three carboxylesterases, which facilitated the degradation of cellulose and hemicellulose by hydrolyzing ester bonds and were recognized as crucial auxiliary enzymes [
35]. In contrast to
Pycnoporus sanguineus, which was reported to secrete only two carboxylesterases [
36], Mafic-2001 exhibited a higher capacity for secreting these enzymes.
Laccases, a hallmark ligninolytic enzyme of WRFs [
37], were prominently represented in the secretome, with five laccases identified (5.45% relative abundance). Compared with other WRFs, such as
Trametes versicolor (three laccases) [
38],
Stereum hirsutum (one laccase) [
38],
Peniophora lycii (five laccases) [
37], and
Trametes hirsuta (two laccases) [
37], Mafic-2001 ranks among the most prolific laccase producers reported to date. Intriguingly, lytic polysaccharide monooxygenases (LPMOs) of the AA9 family were also detected in the secretome of Mafic-2001. LPMOs are non-hydrolytic oxidative enzymes that have recently emerged as critical players in lignocellulose degradation [
39,
40]. LPMOs can degrade insoluble polysaccharides, such as crystalline cellulose and chitin [
40], and can also co-degrade xyloglucan with glycoside hydrolases, thereby enhancing biomass conversion efficiency [
5]. The presence of LPMOs, combined with the diverse enzymatic portfolio of Mafic-2001, further highlighted the strain’s superior hydrolytic performance.
The complete degradation of cellulose relies on the synergistic action of EGs, CBHs, and βGs [
41]. Abundant EGs increase the frequency of random cleavage along cellulose backbones and generate additional sites for hydrolysis [
42]. Meanwhile, βGs further hydrolyze cellobiose produced by CBHs into glucose and complete cellulose depolymerization [
43]. The proteomic analysis of
C. trogii Mafic-2001 revealed an imbalance in its cellulase system, with insufficient EG and βG activities relative to the high abundance of CBHs. This disparity might lead to cellobiose accumulation, which could trigger the product feedback inhibition of CBHs and ultimately reduce hydrolysis efficiency [
10]. Enzymes derived from the same strain exhibit high synergies, mainly due to their co-evolution mechanism with substrate structure and similar physicochemical properties (e.g., optimal pH and temperature). The targeted supplementation of deficient enzymes, particularly those derived from the same fungal strain to maximize synergistic compatibility, has been proposed as a strategy to address such limitations [
44,
45]. In this study, the heterologous expression of
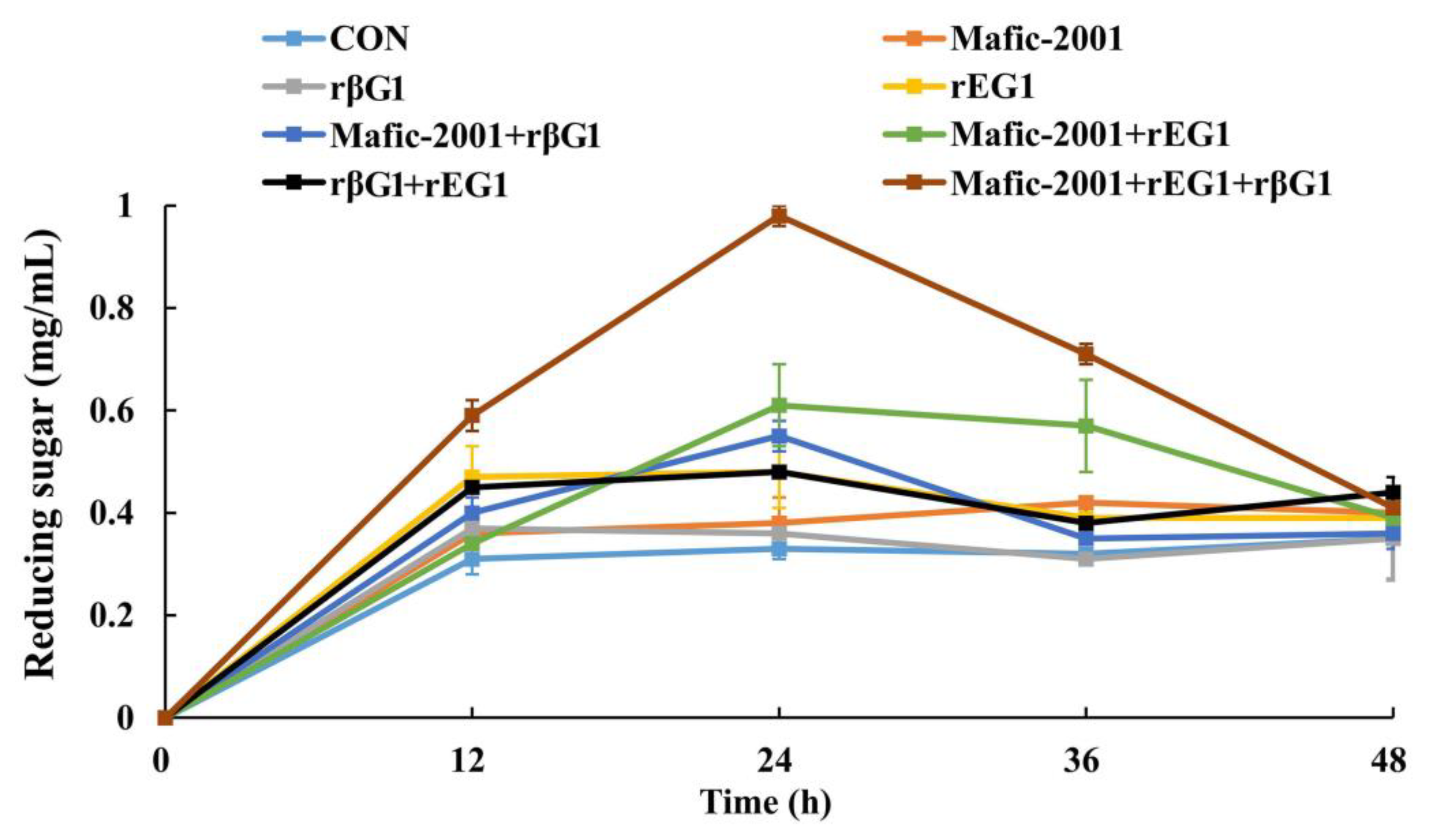
C. trogii Mafic-2001-derived EGs and βGs in
P. pastoris yielded recombinant enzymes that, when added to the native enzyme cocktail, enhanced the saccharification efficiency of CDGs by 172% compared with the effect obtained with the native enzyme cocktail alone. This enhancement aligned with prior strategies, such as those by Valadares et al. [
13], who improved sugarcane bagasse cellulose degradation from 75% to 87% by augmenting
T. reesei enzymes with white-rot fungal EGs and βGs. Similarly, the overexpression of EGs and βGs in
T. reesei significantly improved cellulolytic efficiency [
46,
47]. These findings highlighted the critical role of balanced enzyme ratios in optimizing lignocellulose conversion and validated the effectiveness of tailored enzyme cocktails for industrial biomass processing. The current study employed a strategy of heterologously expressing target enzyme components and supplementing them into the native enzyme cocktail of
C. trogii Mafic-2001 to enhance hydrolysis efficiency. While this approach demonstrated significant efficacy, the operational complexity associated with
P. pastoris engineered strain fermentation and enzyme cocktail formulation may lead to increased costs. This research lays a crucial foundation for the preparation of enzyme cocktails, as subsequent studies could build upon these findings to explore more practical optimization strategies for improving enzymatic efficiency.
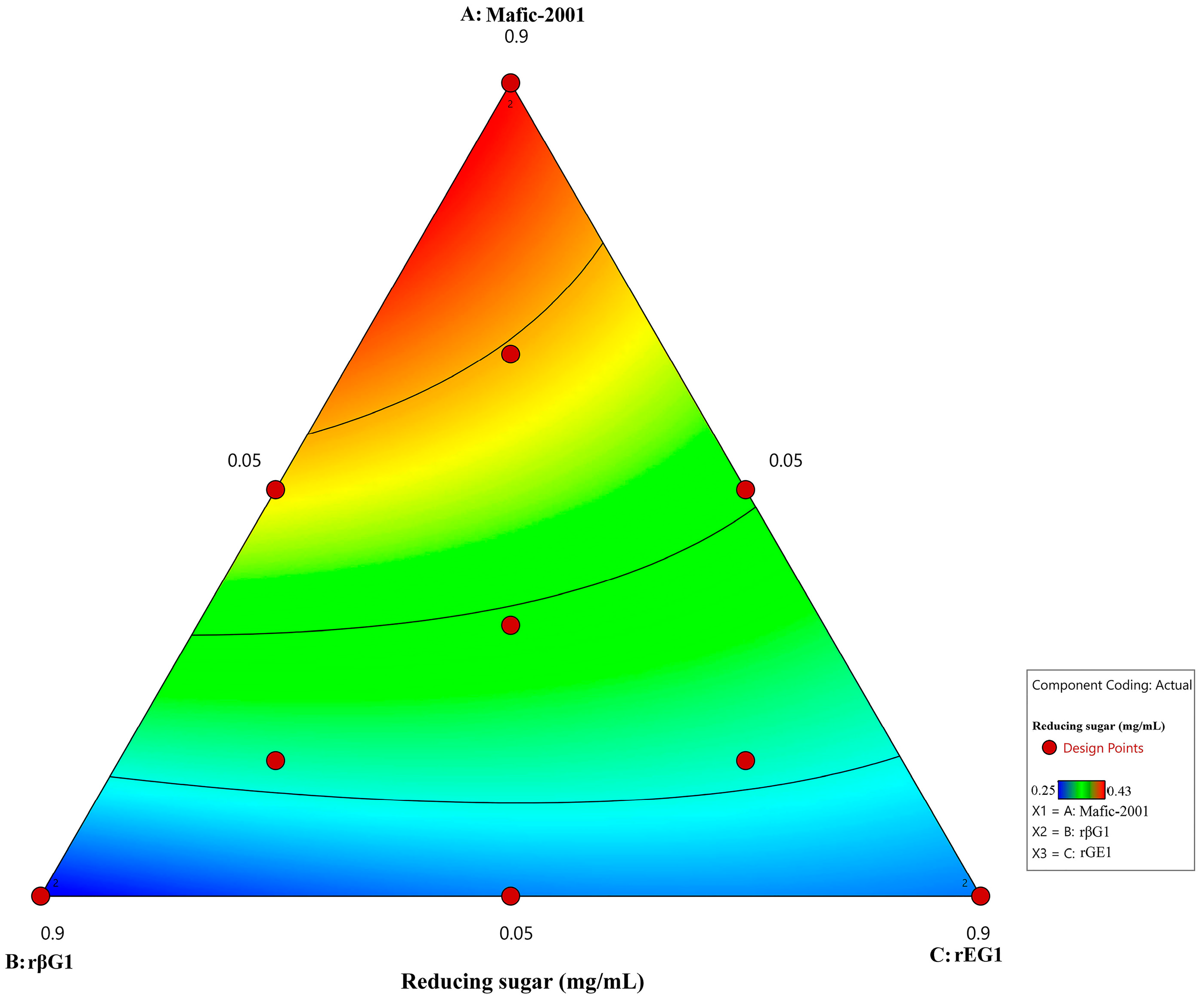
The appropriate ratio of hydrolytic enzymes within enzyme cocktails is crucial for achieving efficient hydrolysis [
48]. In this study, the proportions of recombinant enzymes (rEG1 and rβG1) and the native enzyme cocktail Mafic-2001 were optimized using a mixture design. Multivariate regression analysis revealed that the native enzyme cocktail Mafic-2001 had the highest coefficient in the model, which highlighted its dominant contribution to hydrolysis efficiency. This finding was consistent with the analysis results of the Mafic-2001 secretome and previously reported characteristics of white-rot fungal enzyme cocktails, which were enriched with diverse CAZymes capable of synergistic lignocellulose degradation [
49,
50]. Notably, while the trace supplementation of rEG1 and rβG1 significantly enhanced hydrolysis efficiency, exceeding the threshold ratio (>0.1) resulted in a decline in performance. This phenomenon might arise from competitive enzyme–substrate binding or the saturation of the available substrate surface sites [
51,
52], which was similar to the inhibitory effects of enzyme excess reported by Tanaka and Du et al. in cellulase synergy studies [
53,
54]. This phenomenon underscored the importance of the precise regulation of enzyme ratios in industrial biocatalytic processes.
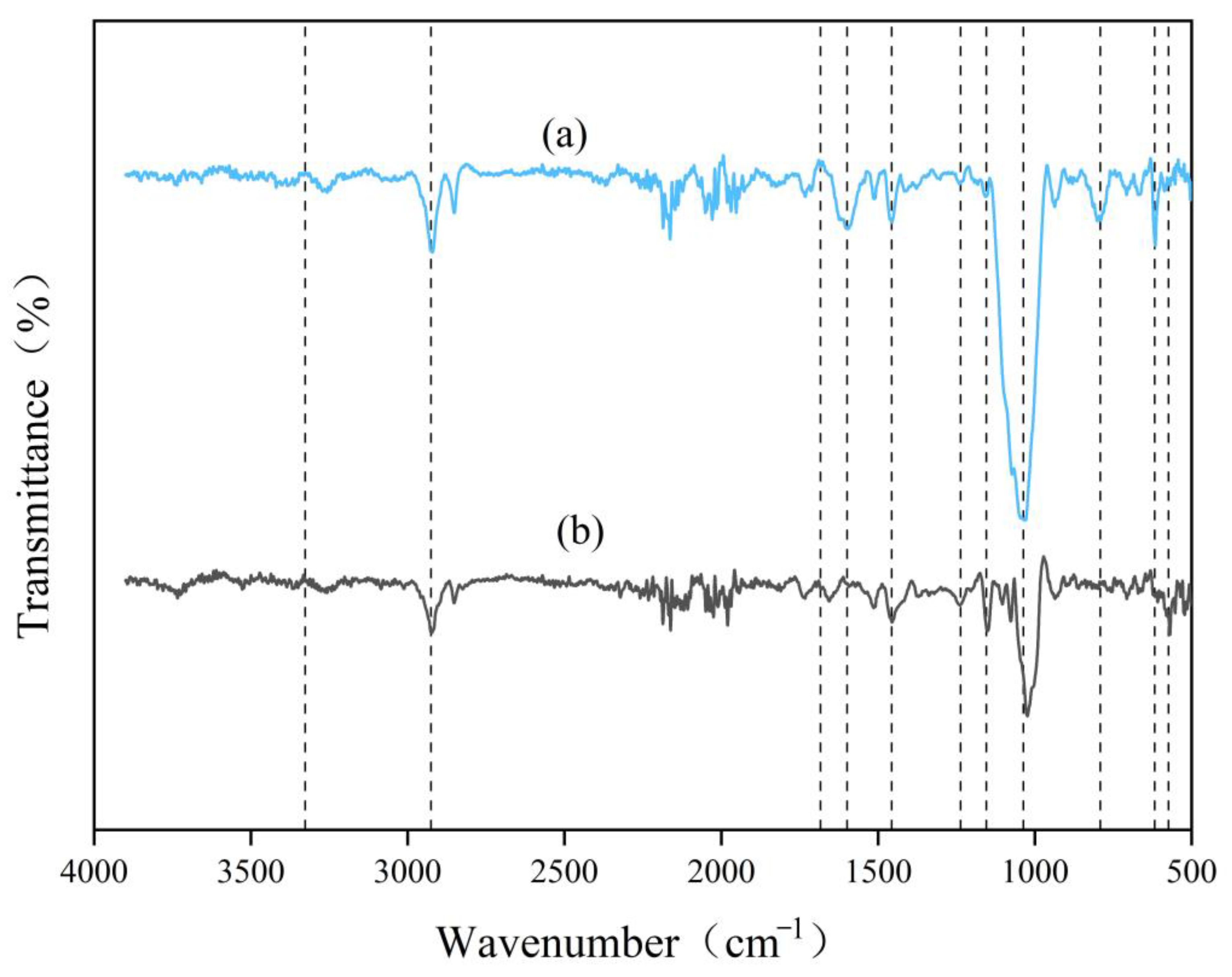
Further optimization revealed that at an enzyme loading of 320 mg/g, the reducing sugar yield reached 1.23 mg/mL, representing a significant improvement compared to pre-optimization levels. Through the holistic consideration of hydrolytic efficiency and enzyme cost, 320 mg/g was identified as the optimal enzyme loading [
55]. Maximum saccharification efficiency was observed at pH 5.0 and 40 °C, which was consistent with the acidic optima typical of most white-rot fungal lignocellulose-degrading enzymes [
56,
57]. The structural transformation of CDGs after treatment with the optimized enzyme cocktail was further investigated through SEM and FTIR analysis. SEM observations revealed that enzymatic treatment caused the fibrous architecture to shift from a compact and ordered arrangement to a loose and porous morphology. Concurrent FTIR analysis demonstrated modifications in the lignocellulosic functional groups, accompanied by the partial removal of lignin, cellulose, and hemicellulose. These findings collectively suggested that the optimized enzyme cocktail effectively disrupted the lignocellulose through synergistic interactions, exposed the polysaccharide components, and enhanced their accessibility for hydrolysis [
58]. Namnuch et al. investigated the structural modifications in sugarcane bagasse induced by enzymes derived from A. flavus KUB2 using SEM [
59]. Similarly, Zeng et al. employed FTIR to analyze functional group alterations in wheat straw and specifically evaluated lignin methylation reactions [
60]. Furthermore, Ye et al. characterized microstructural changes in treated paulownia wood through integrated FTIR and SEM analyses [
61]. The application of these multimodal analytical approaches systematically assessed the enzymatic degradation efficiency of CDGs at the microscale and provided robust evidence for the synergistic degradation mechanisms targeting the lignin–carbohydrate complex in CDGs.
4. Materials and Methods
4.1. Strains, Plasmids, Reagents, and Lignocellulosic Biomass
The C. trogii Mafic-2001, P. pastoris X-33, and pPICZαA plasmid were kindly provided by Prof. Yunhe Cao of China Agricultural University. Escherichia coli Top 10 competent cells were purchased from Tiangen Biotech Co., Ltd. (Beijing, China). Sac I, EcoRI, and Xba I were obtained from TaKaRa (Kyoto, Japan). The substrate 2, 2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) was purchased from Beyotime (Shanghai, China). Sodium carboxymethyl cellulose (CMC) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Beechwood xylan was purchased from Megazyme (Bray, Ireland). All other chemicals used in this study were of analytical grade.
The lignocellulosic biomass samples, including oat straw, corn stover, corn cob, Napier grass, wheat bran, and rice straw, were collected from Rongchang, Chongqing, China. CDGs were obtained from Wuliangye Group Co., Ltd. (Yibin, China).
4.2. Cultivation of C. trogii Mafic-2001 and Enzyme Activity Assay
Different carbon sources were used to induce enzyme production in C. trogii Mafic-2001. The fungus was aseptically transferred into an enzyme production medium containing 0.1% peptone, 0.14% ammonium sulfate, 0.2% KH2PO4, 0.03% CaCl2, 0.03% MgSO4·7H2O, 0.0005% FeSO4·7H2O, 0.00015% MnSO4·H2O, 0.00015% ZnSO4·7H2O, and 40 g of lignocellulosic biomass (including oat straw, corn stover, corn cob, Napier grass, wheat bran, or rice straw). The fermentation was conducted at 30 °C under aerobic agitation (220 rpm). Lignocellulosic enzyme activities in the culture solution were measured from day 2 to day 8 post inoculation. The optimal incubation time was determined based on the activities of cellulase, xylanase, and laccase.
Cellulase activity was measured using CMC as the substrate. A 1% (
w/
v) CMC solution was prepared in citrate–phosphate buffer (pH 5.5). One unit of cellulase activity was defined as the amount of enzyme required to release 1 μmol of reducing sugar per minute at 37 °C [
62].
Xylanase activity was measured using the DNS method, as previously reported. A 1% (
w/
v) Beechwood solution was prepared in citrate–phosphate buffer (pH 5.5). One unit of xylanase activity was defined as the amount of enzyme needed to release 1 μmol of reducing sugar per minute at 37 °C [
63].
Laccase activity was measured by using ABTS as the substrate (1 mM, pH 3.0). The culture supernatant (50 μL) was mixed with 950 μL of citrate–phosphate buffer (pH 3.0); 1 mL of ABTS solution (1 mM) was placed into another centrifuge tube and preheated at 30 °C for 3 min. The two solutions were then combined in a single centrifuge tube. The mixture was kept at 30 °C for 10 min, and the change in its absorbance at 420 nm was measured with a microplate reader. One unit of laccase activity was defined as the amount of enzyme that oxidized 1 μmol of ABTS per minute [
64].
4.3. Preparation of Enzyme Cocktail Mafic-2001 and Extracellular Quantitative Proteome Analysis
C. trogii Mafic-2001 was cultured for 6 days in a medium containing rice straw as the enzyme-inducing substrate. The culture broth was then centrifuged at 10,000× g for 15 min at 4 °C, and the supernatant was collected. Ammonium sulfate precipitation was performed to prepare the enzyme cocktail, with a final concentration of 80% (w/v). After incubation at 4 °C for 2 h, the protein precipitates were pelleted by centrifugation under the same conditions (10,000× g, 4 °C, 15 min) and then resolubilized in sodium acetate buffer (50 mM, pH 5.0).
The protein solution was dialyzed using a dialysis membrane (3500 MW, Thermo Scientific™, Waltham, MA, USA) in specific phosphate-buffered saline (PBS, pH 7.4) at 4 °C. The PBS was replaced every 4 h until a total dialysis duration of 24 h was completed. The dialyzed protein solution was then subjected to freeze-drying to obtain the enzyme cocktail. The protein concentration was determined using the BCA Protein Assay Kit (Thermo Scientific™, Waltham, MA, USA), and the enzyme cocktail was stored at −20 °C for subsequent enzymatic analyses.
After the tryptic digestion of the Mafic-2001 enzyme cocktail, peptide desalting was performed using hydrophilic–lipophilic balanced solid-phase extraction cartridges. Peptide quantification was performed using a NanoDrop One spectrophotometer (Thermo Scientific™, Waltham, MA, USA). Peptides were analyzed via a liquid chromatography–tandem mass spectrometry (LC-MS/MS) system in DIA mode. Mass spectrometry data were processed using Spectronaut software (version 18, Biognosys AG, Zurich, Switzerland) against a customized fungal proteomic database, which integrated de novo genome-predicted fungal proteins and UniProt reference entries. The functional annotation of extracellular enzymes was then performed through GO term enrichment, KEGG pathway mapping, and carbohydrate-active enzyme (CAZy) classification, to systematically characterize enzymatic functionalities and metabolic roles.
4.4. Heterologous Expression of Cellulases from C. trogii Mafic-2001 in P. pastoris
4.4.1. Cloning of Cellulase Gene from Mafic-2001 and Construction of Expressing Strains
Two pairs of primers were designed to amplify the EG1 and βG1 genes from Mafic-2001 based on the total DNA of C. trogii Mafic-2001. The primers were as follows: EG1-F 5′-ATGCCATCTTTCGCTGAGC-3′ and EG1-R 5′-TTATTTGCGCGCATTCGG-3′; βG1-F 5′-ATGTCGCGCGACTTCCTC-3′ and βG1-R 5′-TCACACCCCGTTCCATGTG-3′. The PCR products were ligated into the pGM-T vector and transformed into E. coli Top 10. Recombinant E. coli Top 10 was cultured on Luria–Bertani (LB) plates (0.5% NaCl, 0.5% yeast extract, 1% tryptone, and 1.5% agar) containing ampicillin (100 μg/mL). Recombinant plasmids (pGM-EG1 and pGM-βG1) were extracted from individual colonies and verified by Sanger sequencing using universal primers.
The EG1 and βG1 gene sequences were optimized based on the codon preference of P. pastoris. The optimized EG1 and βG1 sequences were synthesized by Tsingke Corp. (Beijing, China) and subjected to double digestion with EcoRI and XbaI. The resulting fragments were then ligated into the pPICZαA vector and transformed into E. coli Top 10. Transformed colonies were selected on LB plates supplemented with 100 µg/mL Zeocin™ and incubated at 37 °C until single colonies appeared. Individual colonies were inoculated into 50 mL flasks containing 10 mL LB liquid medium (100 µg/mL Zeocin™) and cultured overnight at 37 °C with shaking at 220 rpm. Recombinant plasmids (pPIC-EG1-opt and pPIC-βG1-opt) were then extracted using a plasmid extraction kit (Omega Bio-Tek, Norcross, GA, USA).
The plasmids pPIC-EG1-opt and pPIC-βG1-opt were linearized with SacI and subsequently electroporated into P. pastoris X-33 (2000 V, 200 Ω, 5 ms). Transformed cells were plated on YPDS agar medium (1% yeast extract, 2% peptone, 2% glucose, 1 M sorbitol, 1.5% agar, and 100 µg/mL Zeocin™) for selection. Positive colonies harboring the recombinant constructs were isolated and designated as engineered strains expressing rEG1 and rβG1.
β-glucosidase activity was measured using p-nitrophenyl-β-D-glucopyranoside (pNPG) (Shanghai Yuanye Bio-Technology Co., Ltd., Shanghai, China) as the substrate. A 5 mM pNPG solution was prepared in citrate–phosphate buffer (pH 5.0). One unit of β-glucosidase activity was defined as the amount of enzyme required to release 1 μmol of p-nitrophenol per minute.
4.4.2. Preparation of Recombinant rEG1 and rβG1 Samples
Positive clones of X-33/EG1 and X-33/βG1 were selected and cultured in 50 mL of buffered glycerol-complex (BMGY) medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 4 × 10
−5% biotin, 1.00% glycerol) at 28 °C and 220 rpm for 24 h. The cells were collected by centrifugation at 5000 rpm for 5 min at 4 °C. The cell pellet was re-suspended in 50 mL of buffered methanol-complex (BMMY) medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 4 × 10
−5% biotin, 0.5% methanol) and cultured at 28 °C with 220 rpm agitation for 96 h. Methanol (0.5%,
v/
v) was added to the culture every 12 h to induce protein expression. The detailed operation procedure was reported by Bao et al. [
64]. After fermentation, the supernatant was collected, and a small portion was concentrated using a Vivacon
® 500 (SARTORIUS, Gottingen, Germany) ultrafiltration tube. rEG1 and rβG1 were analyzed by SDS-PAGE. The rEG1 and rβG1 samples were obtained by freeze-drying most of the supernatant. The protein concentration was determined using a BCA protein detection kit, and the proteins were stored at −20 °C for later use.
4.5. Formulation of the Enzyme Cocktail Mafic-2001 and Optimization of Enzymatic Hydrolysis Conditions for CDGs
CDGs were dried at 80 °C for 24 h, pulverized, and sieved through a 40-mesh sieve. Subsequently, 200 mg of the processed CDGs was dissolved in 10 mL of Na2HPO4–citrate buffer (pH 4.0) at a solid-to-liquid ratio of 1:50 (w/v). The mixture was transferred to a 50 mL centrifuge tube containing the enzyme and incubated in a rotary shaker at 200 rpm for enzymatic hydrolysis. After centrifugation to separate the supernatant and solid residue, the reducing sugar content in the hydrolyzed supernatant was quantified using the 3,5-dinitrosalicylic acid (DNS) method. A reaction mixture containing 80 μL of distilled water, 80 μL of sample, and 200 μL of DNS was prepared, boiled at 100 °C for 5 min, and rapidly cooled to room temperature. The total volume was adjusted to 1 mL by adding 640 μL of distilled water. Absorbance was measured at 540 nm using a microplate reader, and concentrations were calculated against a glucose-derived standard curve.
The sample was mixed with DNS reagent (1:1 v/v), boiled at 100 °C for 5 min, and cooled to room temperature. Absorbance was measured at 540 nm. The reducing sugar concentration was determined using a glucose standard curve.
First, the hydrolysis efficiency of CDGs was systematically evaluated using different enzyme combinations. The experimental groups included the native enzyme cocktail Mafic-2001, rEG1, rβG1, Mafic-2001–rEG1 (1:1, w/w), Mafic-2001–rβG1 (1:1, w/w), rEG1–rβG1 (1:1, w/w), and a ternary combination of Mafic-2001–rEG1–rβG1 (1:0.5:0.5, w/w). Enzyme loading was set at 30 mg enzyme protein per gram of substrate (30 mg/g, w/w). Reactions were conducted in triplicate by incubating the mixtures in a shaking incubator at 200 rpm and 40 °C for 24 h. Samples were collected at 12 h intervals for reducing sugar content analysis.
According to the aforementioned results, the optimal enzyme combination was selected for further optimization. A simplex-lattice mixture design comprising 13 experimental runs was implemented using Design-Expert 12.0 software (Stat-Ease, Minneapolis, MN, USA). The relative proportions of the three enzymes in the combinatorial system were defined as independent variables, with reducing sugar yields from the hydrolysis of CDGs serving as the response variable. The minimum value of the three enzymes was set to 0.05, and the sum of the three enzymes was 1.0. A response surface methodology was employed to fit the experimental data and identify the best-performing regression model for predicting the optimal enzyme ratios. The detailed procedure was reported by Du et al. [
65]. Following the determination of the optimal ratio of the three enzymes through regression modeling, an enzyme cocktail formulated at this optimized ratio was employed to hydrolyze Chinese distillers’ grains (CDGs), and validation experiments were subsequently conducted.
According to the identification of the optimal enzyme combination and ratio, the concentration-dependent hydrolysis efficacy of the formulated enzyme cocktail on CDGs was investigated. Enzyme loadings were tested at various concentrations (80, 120, 160, 200, 240, 280, 320, and 360 mg/g). Hydrolysis reactions were performed in a shaker at 200 rpm for 24 h. The concentration of reducing sugars in the supernatant was quantified to evaluate enzymatic performance.
Finally, under the optimal ratio and dosage of the optimized enzyme cocktail, the optimal pH and temperature for degrading CDGs were investigated. To determine the optimal pH, the enzyme cocktail and CDGs were incubated in buffer solutions with pH values of 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, and 8.0 while maintaining other reaction parameters constant. The reducing sugar content in the hydrolysate was then analyzed. For temperature optimization, enzymatic reactions were conducted in a shaker at 200 rpm, with temperatures set at 30 °C, 35 °C, 40 °C, 45 °C, 50 °C, 55 °C, 60 °C, and 65 °C. The reducing sugar content in the supernatant was quantified to evaluate enzymatic performance.
4.6. Characterization of CDGs Before and After Enzymatic Hydrolysis
The untreated and treated CDGs were dehydrated using an air-dry oven (Shanghai Yiheng Scientific Instrument Co., Ltd., Shanghai, China). A scanning electron microscope (SU8600, Hitachi, Japan) was employed to characterize the changes in the surface morphology of the untreated and enzyme-treated CDGs. The CDGs were sputter-coated with gold and photographed at a magnification of 500×.
An FTIR spectrometer (Nicolet iN10, Thermo Scientific™, Waltham, MA, USA) and KBr pellet preparation were used to investigate the chemical structure changes in the untreated and enzyme-treated CDGs in the range of 500–4000 cm−1.


 and
and 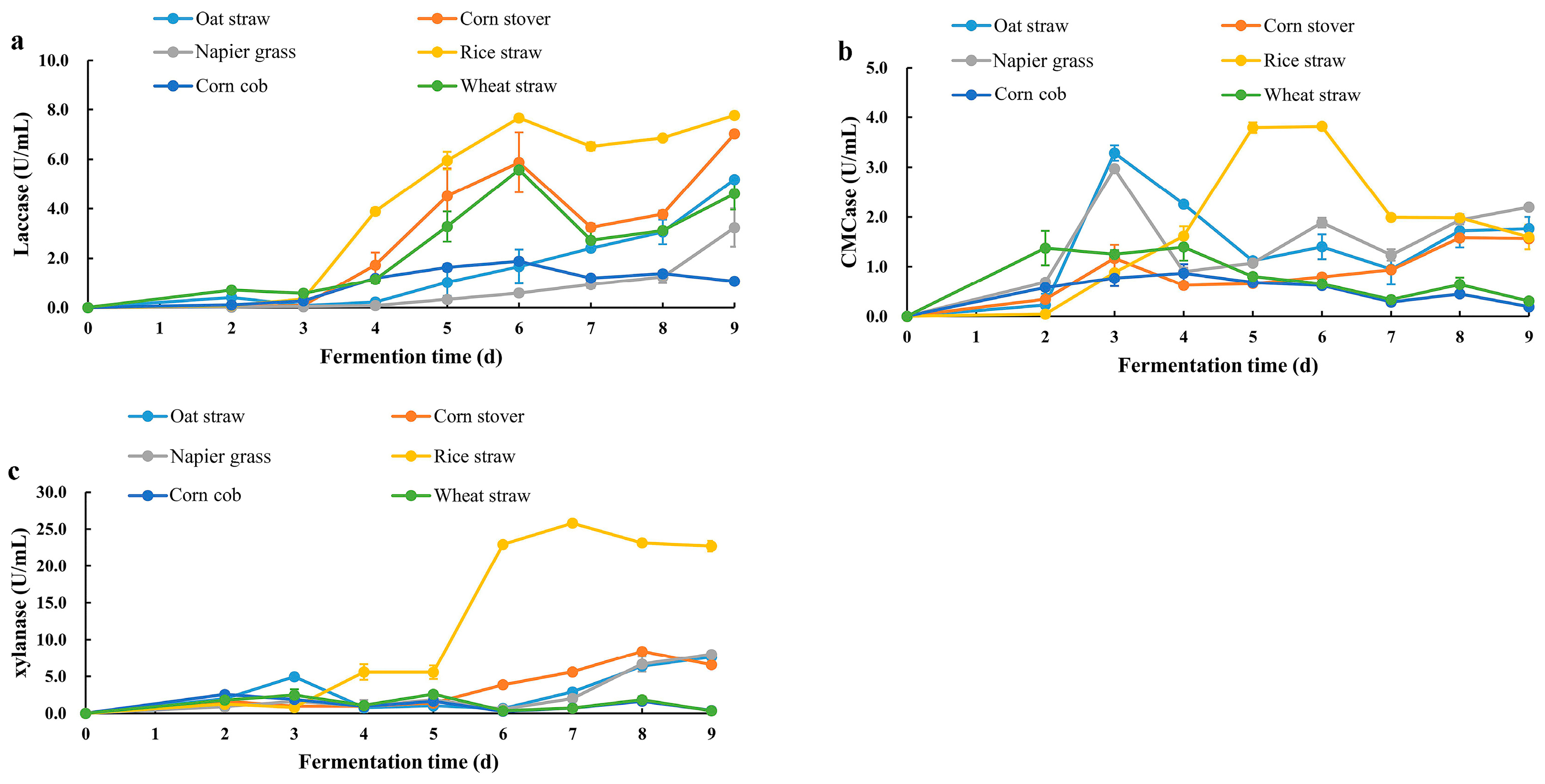





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}