
AI-Driven Drug Target Screening Platform Identified Oncogene CACNA2D1 Activated by Enhancer Infestation in Epstein-Barr Virus-Associated Nasopharyngeal Carcinoma
 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
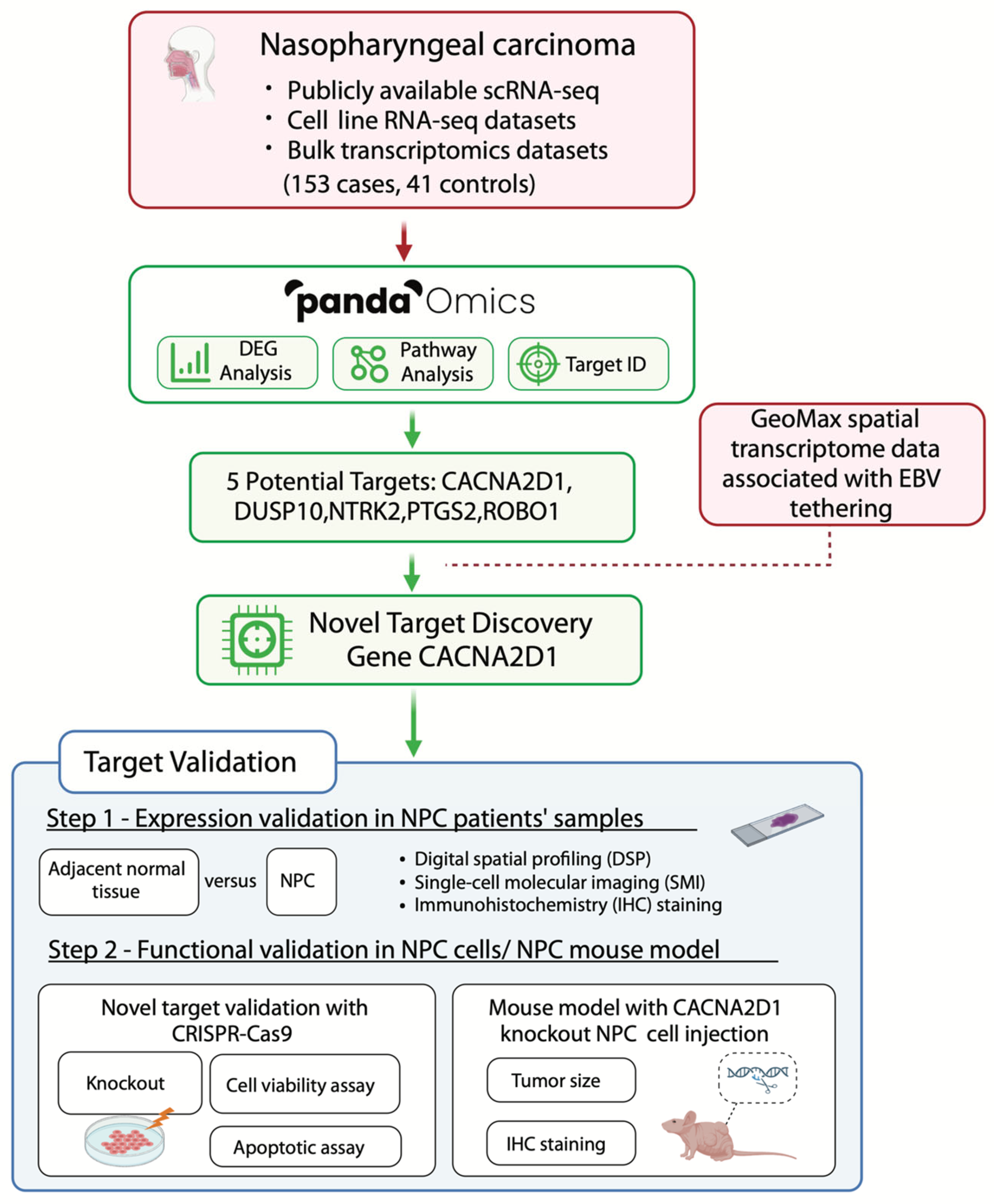
2.1. AI-Driven Platform Identifies Novel Therapeutic Targets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | NPC vs. Control | Proposed Therapy | Protein Family | Tissue Specificity (HPA) | Biological Processes | # Clinical Trials | Any Clinical Trials for Cancers | Gene–Disease Association | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Rank | Median LFC | % | % | ||||||||
| Up | p < 0.05 | ||||||||||
| Investigational target for NPC | |||||||||||
| PTGS2 | 5 | 2.47 | 100 | 75 | Antagonist | Oxidoreductase | Enhanced in the bone marrow, seminal vesicle, and urinary bladder | Biosynthesis and metabolism of fatty acid, lipid, and prostaglandin | 5104 | Yes | Involved in three clinical trials (max. P3) for NPC (not a novel target for NPC). |
| Potential novel targets for NPC | |||||||||||
| NTRK2 | 46 | 1.61 | 100 | 100 | Antagonist | Receptor kinase | Enhanced in the brain and thyroid gland | Differentiation and neurogenesis | 211 | Yes | LMP1 promotes NPC metastasis through NTRK2-mediated anoikis resistance. NTRK2 downregulation inhibits the metastasis of LMP1-positive NPC [36]. |
| CACNA2D1 | 10 | 2.5 | 100 | 100 | Antagonist | Ion channel | Enhanced in the skeletal muscle and tongue | Calcium transport, ion transport, and transport | 922 | None * | Enhanced radioresistance in cancer stem-like cells [37]; critical for HCC stemness and predictive of poor prognosis for HCC [38,39]. |
| ROBO1 | 38 | 2.13 | 100 | 100 | Antagonist | Immunoglobulin | Low tissue specificity | Chemotaxi, differentiation, and neurogenesis | 0 | None | Reported to promote anti-tumor activities upon ROBO1 knockout/knockdown. |
| DUSP10 | 171 | 0.82 | 100 | 75 | Antagonist | Esterase | Enhanced in the liver | N/A | 0 | None | DUSP10 inhibition in esophagus cancer cells increases apoptosis after irradiation [40]. DUSP10 is a regulator of YAP1 activity, promoting cell proliferation and colorectal cancer progression [41]. |
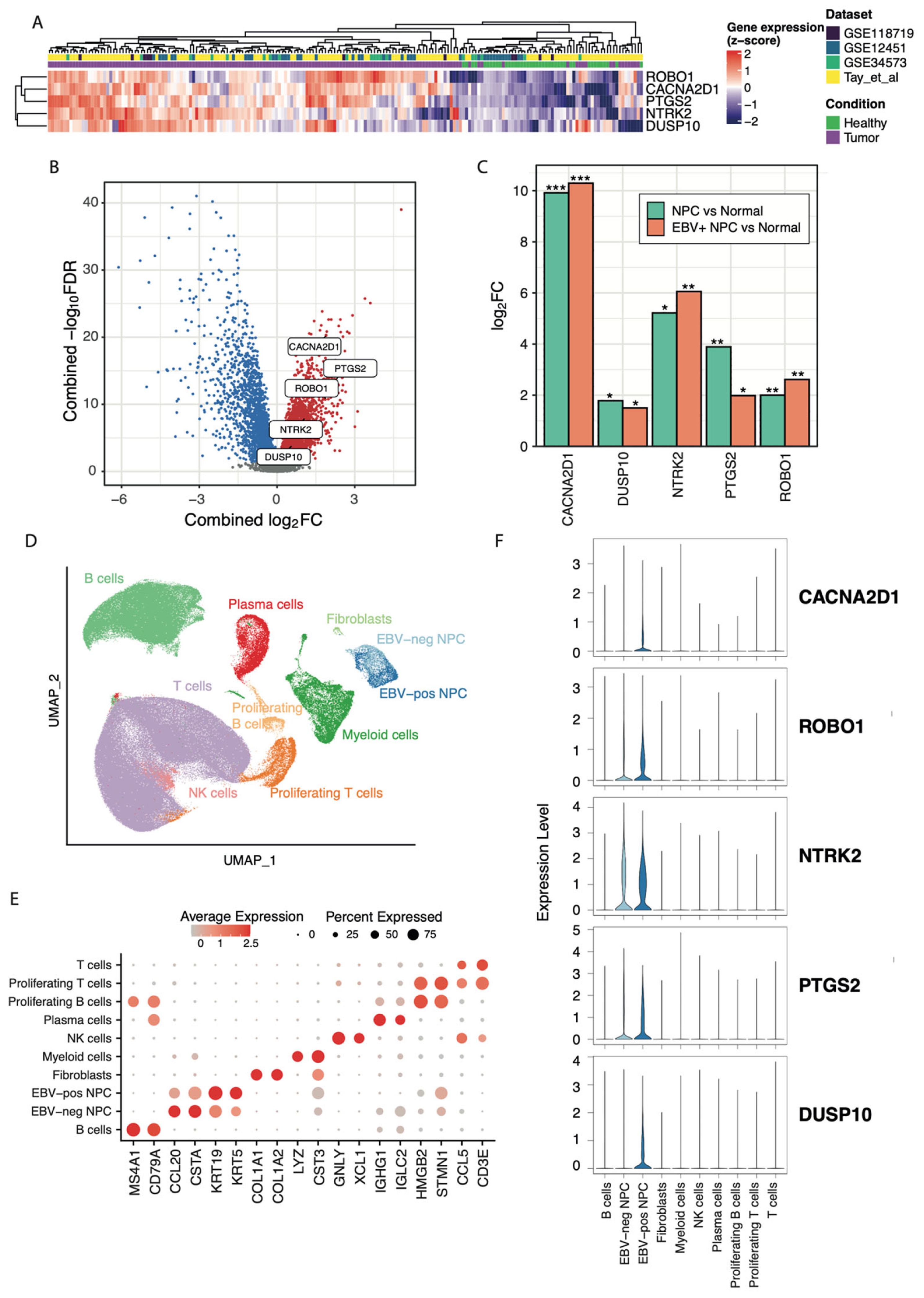
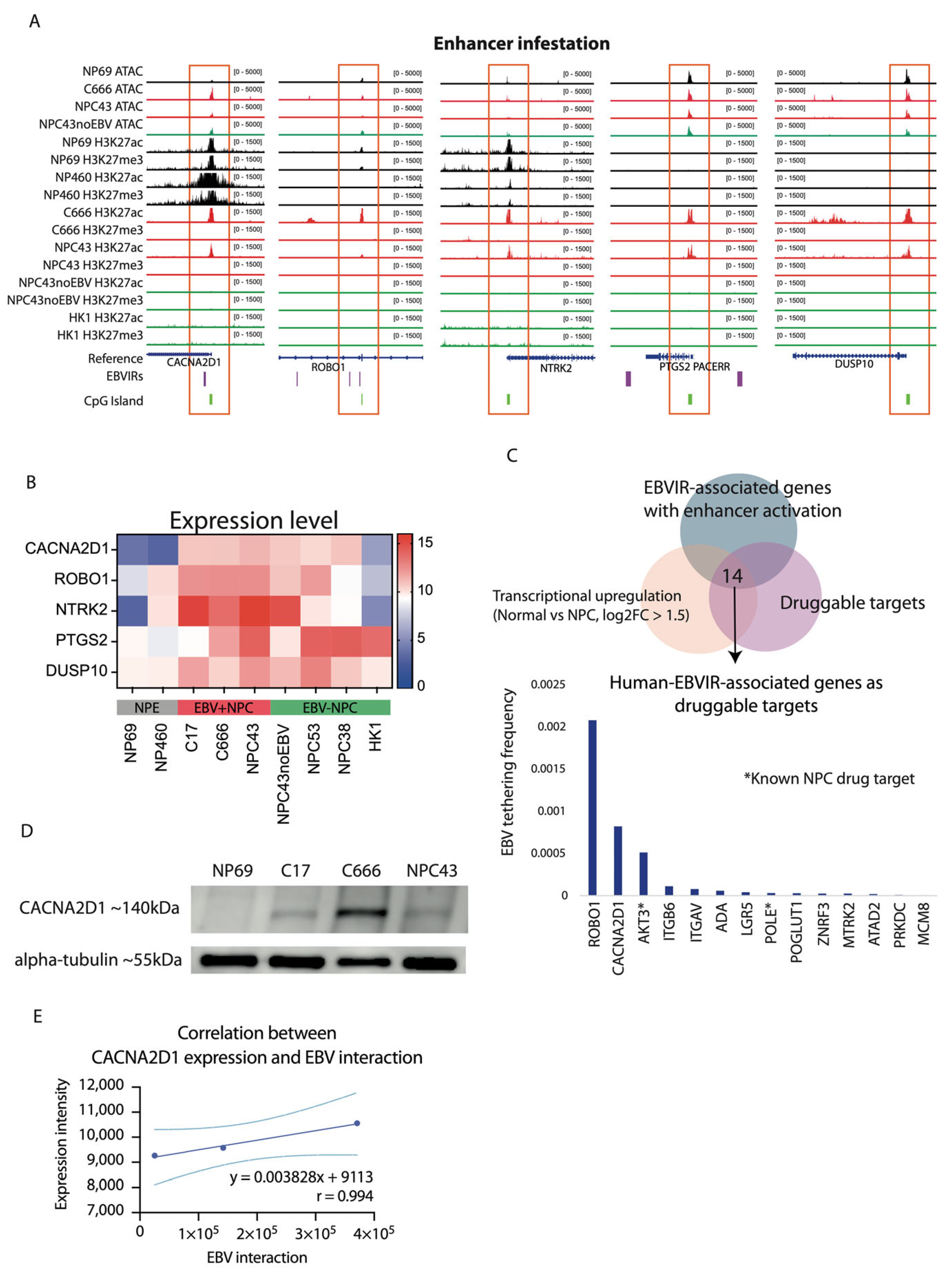
2.2. Enhancer Infestation at CACNA2D1 and ROBO1
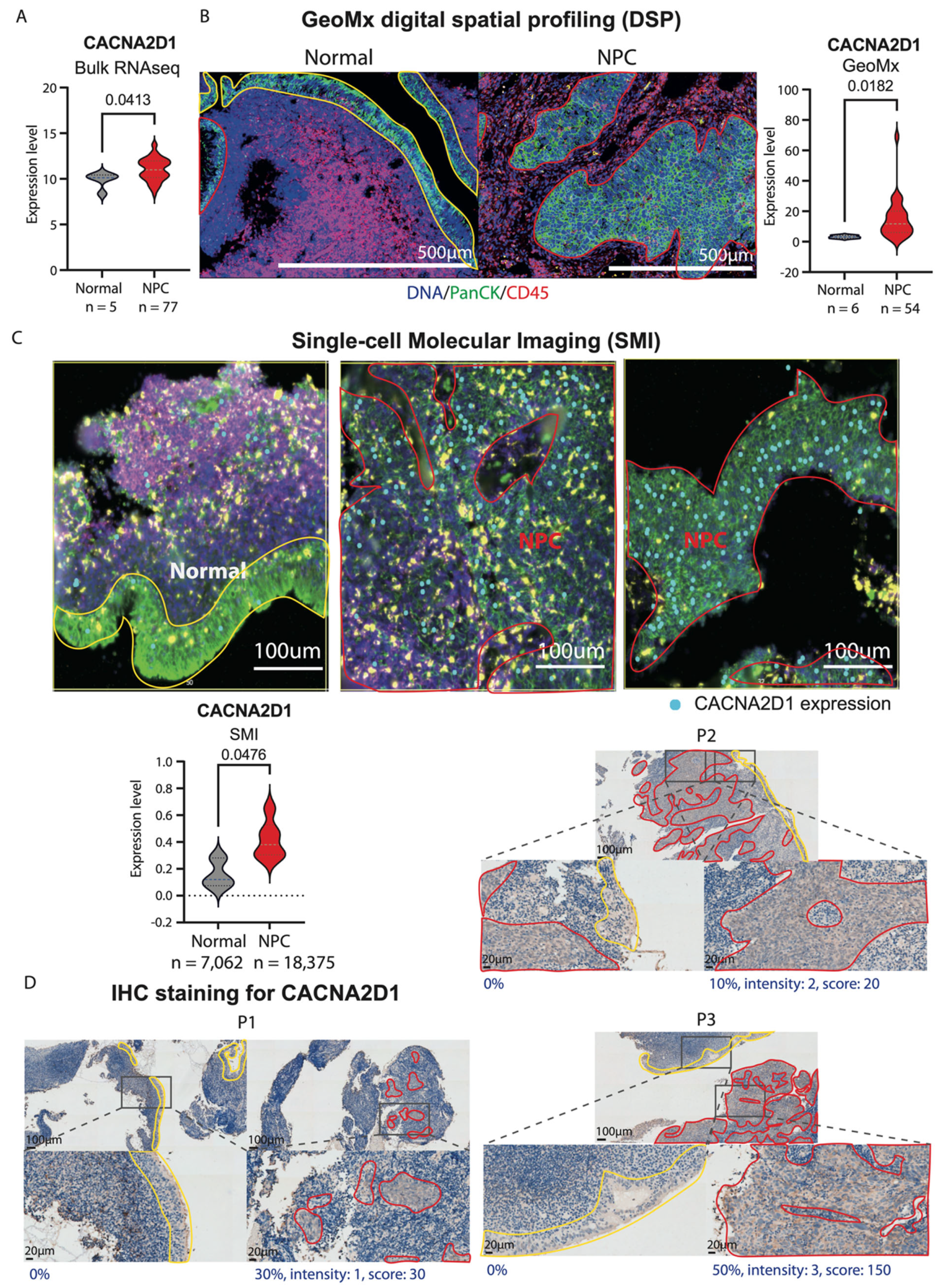
2.3. Aberrant Upregulation of CACNA2D1 in NPC
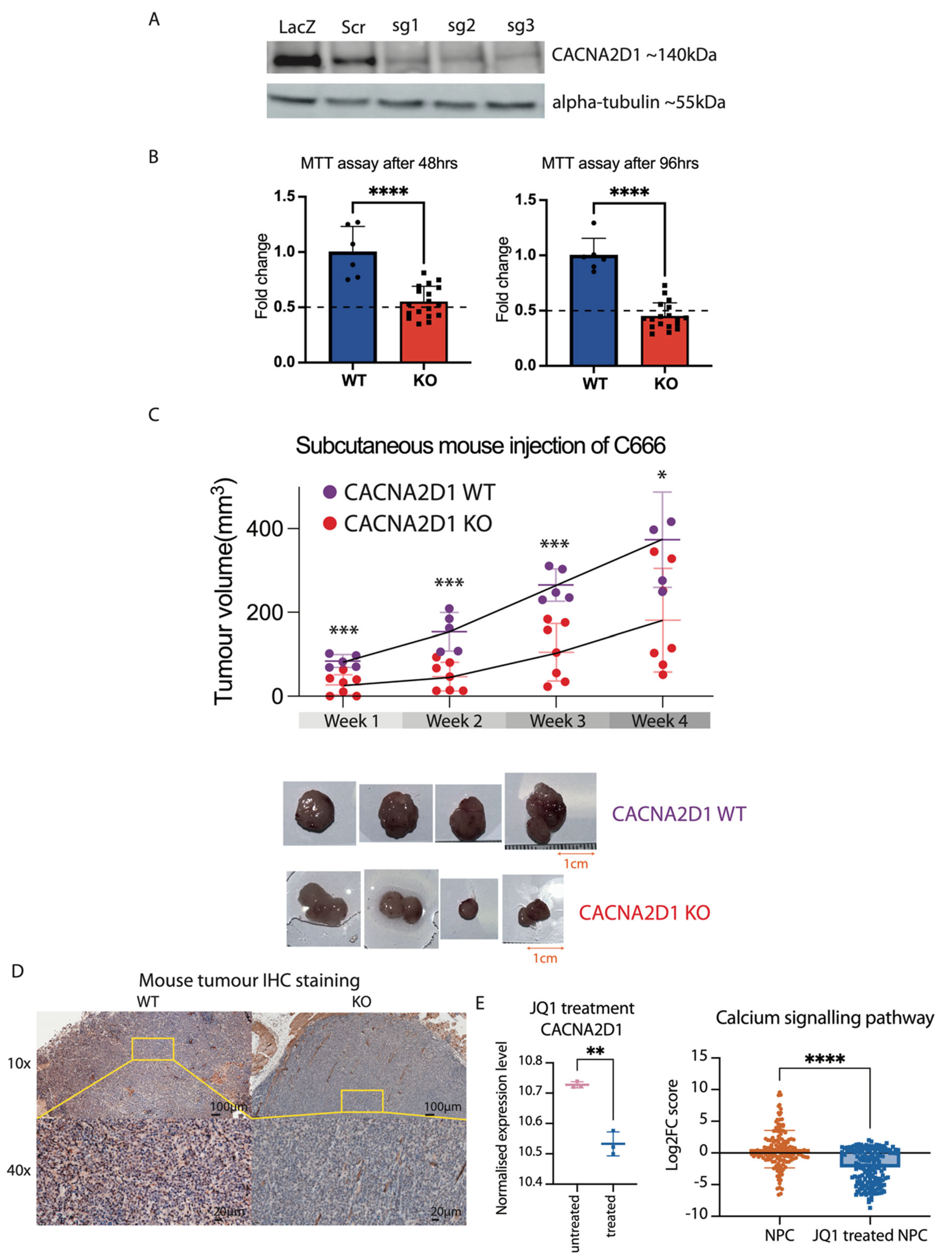
2.4. CACNA2D1 Knockout Suppressed Cell Viability and Tumour Growth
3. Discussion
4. Materials and Methods
4.1. Bulk Transcriptomic Dataset Selection and Preprocessing
4.2. Differential Expression Analysis and Meta-Analysis
4.3. Target Prioritisation by PandaOmics
4.4. Single-Cell RNA-seq (scRNA-seq) Data Analysis
4.5. Hallmark Gene Set Enrichment Analysis
4.6. Target Selection (CACNA2D1, NTRK2, PTGS2, ROBO1, DUSP10)
4.7. Cell Lines
4.8. Modified Hi-C to Identify EBV-Interacting Regions (EBVIRs)
4.9. Cleavage Under Targets and Release Using Nuclease with Sequencing (CUT&RUN-seq)
4.10. The Assay for Transposase-Accessible Chromatin with Sequencing (ATAC-seq)
4.11. Validation of CACNA2D1 in NPC Cell Lines by Western Blotting
4.12. Bulk RNA Sequencing (RNAseq) for Clinical Samples
4.13. Digital Spatial Profiling (DSP) for Spatial Transcriptome Analysis
4.14. NanoString CosMxTM Single-Cell Molecular Imaging (SMI) for Single-Cell Spatial Analysis
4.15. Immunohistochemistry (IHC) Staining for Clinical Samples and Mouse Tumours
4.16. Establishment of Functional Knockout CACNA2D1 by Genome Editing CRISPR-Cas9 System
4.17. MTT Assay
4.18. Subcutaneous Injection of Mouse
4.19. Ethical Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chua, M.L.; Wee, J.T.; Hui, E.P.; Chan, A.T. Nasopharyngeal carcinoma. Lancet 2016, 387, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.I.; Sham, J.S. Nasopharyngeal carcinoma. Lancet 2005, 365, 2041–2054. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Chan, A.T.; Le, Q.T.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.; Zell, J.; Ziogas, A.; Anton-Culver, H. Epidemiology of nasopharyngeal carcinoma in the United States: Improved survival of Chinese patients within the keratinizing squamous cell carcinoma histology. Ann. Oncol. 2006, 18, 29–35. [Google Scholar] [CrossRef]
- Wan, S.K.; Chan, J.K.; Lau, W.H.; Yip, T.T. Basaloid-squamous carcinoma of the nasopharynx. An Epstein-Barr virus-associated neoplasm compared with morphologically identical tumors occurring in other sites. Cancer 1995, 76, 1689–1693. [Google Scholar] [CrossRef]
- Chai, A.W.Y.; Yee, S.M.; Lee, H.M.; Aziz, N.A.; Yee, P.S.; Marzuki, M.; Wong, K.W.; Chiang, A.K.; Chow, L.K.-Y.; Dai, W.; et al. Establishment and Characterization of an Epstein-Barr Virus–positive Cell Line from a Non-keratinizing Differentiated Primary Nasopharyngeal Carcinoma. Cancer Res. Commun. 2024, 4, 645–659. [Google Scholar] [CrossRef]
- Wong, K.C.W.; Hui, E.P.; Lo, K.-W.; Lam, W.K.J.; Johnson, D.; Li, L.; Tao, Q.; Chan, K.C.A.; To, K.-F.; King, A.D.; et al. Nasopharyngeal carcinoma: An evolving paradigm. Nat. Rev. Clin. Oncol. 2021, 18, 679–695. [Google Scholar] [CrossRef]
- Campion, N.J.; Ally, M.; Jank, B.J.; Ahmed, J.; Alusi, G. The molecular march of primary and recurrent nasopharyngeal carcinoma. Oncogene 2021, 40, 1757–1774. [Google Scholar] [CrossRef]
- Cao, Y. EBV based cancer prevention and therapy in nasopharyngeal carcinoma. NPJ Precis. Oncol. 2017, 1, 10. [Google Scholar] [CrossRef]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Alami, I.E.; Khaali, W.; Jalbout, M.; Gihbid, A.; Ayoub, W.B.; Benider, A.; Brahim, S.M.; Cherif, M.H.; Benchakroun, N.; El Mzibri, M.; et al. Genetic variations in CYP2A6, CYP2E1, GSTM1, GSTT1 genes and the risk of Nasopharyngeal carcinoma in North African population. Ann. Hum. Genet. 2024. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Winkler, C.A.; Li, J.; Guan, L.; Tang, M.; Liao, J.; Deng, H.; de Thé, G.; Zeng, Y.; O’brien, S.J. Evaluation and Integration of Genetic Signature for Prediction Risk of Nasopharyngeal Carcinoma in Southern China. BioMed Res. Int. 2014, 2014, 434072. [Google Scholar] [CrossRef] [PubMed]
- Lao, T.D.; Nguyen, T.N.; Le, T.A.H. Promoter Hypermethylation of Tumor Suppressor Genes Located on Short Arm of the Chromosome 3 as Potential Biomarker for the Diagnosis of Nasopharyngeal Carcinoma. Diagnostics 2021, 11, 1404. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.-S.; Hwang, C.-I.; Somerville, T.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef]
- Akhtar-Zaidi, B.; Cowper-Sal·lari, R.; Corradin, O.; Saiakhova, A.; Bartels, C.F.; Balasubramanian, D.; Myeroff, L.; Lutterbaugh, J.; Jarrar, A.; Kalady, M.F.; et al. Epigenomic Enhancer Profiling Defines a Signature of Colon Cancer. Science 2012, 336, 736–739. [Google Scholar] [CrossRef]
- Mizokami, H.; Okabe, A.; Choudhary, R.; Mima, M.; Saeda, K.; Fukuyo, M.; Rahmutulla, B.; Seki, M.; Goh, B.-C.; Kondo, S.; et al. Enhancer infestation drives tumorigenic activation of inactive B compartment in Epstein-Barr virus-positive nasopharyngeal carcinoma. EBioMedicine 2024, 102, 105057. [Google Scholar] [CrossRef]
- Okabe, A.; Kaneda, A. Transcriptional dysregulation by aberrant enhancer activation and rewiring in cancer. Cancer Sci. 2021, 112, 2081–2088. [Google Scholar] [CrossRef]
- Baloche, V.; Ferrand, F.-R.; Makowska, A.; Even, C.; Kontny, U.; Busson, P. Emerging therapeutic targets for nasopharyngeal carcinoma: Opportunities and challenges. Expert Opin. Ther. Targets 2020, 24, 545–558. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Pun, F.W.; Ozerov, I.V.; Zhavoronkov, A. AI-powered therapeutic target discovery. Trends Pharmacol. Sci. 2023, 44, 561–572. [Google Scholar] [CrossRef]
- Kamya, P.; Ozerov, I.V.; Pun, F.W.; Tretina, K.; Fokina, T.; Chen, S.; Naumov, V.; Long, X.; Lin, S.; Korzinkin, M.; et al. PandaOmics: An AI-Driven Platform for Therapeutic Target and Biomarker Discovery. J. Chem. Inf. Model. 2024, 64, 3961–3969. [Google Scholar] [CrossRef] [PubMed]
- Pun, F.W.; Leung, G.H.D.; Leung, H.W.; Rice, J.; Schmauck-Medina, T.; Lautrup, S.; Long, X.; Liu, B.H.M.; Wong, C.W.; Ozerov, I.V.; et al. A comprehensive AI-driven analysis of large-scale omic datasets reveals novel dual-purpose targets for the treatment of cancer and aging. Aging Cell 2023, 22, e14017. [Google Scholar] [CrossRef] [PubMed]
- Pun, F.W.; Liu, B.H.M.; Long, X.; Leung, H.W.; Leung, G.H.D.; Mewborne, Q.T.; Gao, J.; Shneyderman, A.; Ozerov, I.V.; Wang, J.; et al. Identification of Therapeutic Targets for Amyotrophic Lateral Sclerosis Using PandaOmics—An AI-Enabled Biological Target Discovery Platform. Front. Aging Neurosci. 2022, 14, 914017. [Google Scholar] [CrossRef]
- Ren, F.; Aliper, A.; Chen, J.; Zhao, H.; Rao, S.; Kuppe, C.; Ozerov, I.V.; Zhang, M.; Witte, K.; Kruse, C.; et al. A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nat. Biotechnol. 2024, 43, 63–75. [Google Scholar] [CrossRef]
- Pun, F.W.; Leung, G.H.D.; Leung, H.W.; Liu, B.H.M.; Long, X.; Ozerov, I.V.; Wang, J.; Ren, F.; Aliper, A.; Izumchenko, E.; et al. Hallmarks of aging-based dual-purpose disease and age-associated targets predicted using PandaOmics AI-powered discovery engine. Aging 2022, 14, 2475–2506. [Google Scholar] [CrossRef]
- Yang, L.; Liu, G.; Li, Y.; Pan, Y. The emergence of tumor-infiltrating lymphocytes in nasopharyngeal carcinoma: Predictive value and immunotherapy implications. Genes Dis. 2021, 9, 1208–1219. [Google Scholar] [CrossRef]
- Huang, S.C.M.; Tsao, S.W.; Tsang, C.M. Interplay of Viral Infection, Host Cell Factors and Tumor Microenvironment in the Pathogenesis of Nasopharyngeal Carcinoma. Cancers 2018, 10, 106. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Hu, C.; Wei, W.; Chen, X.; Woodman, C.B.; Yao, Y.; Nicholls, J.M.; Joab, I.; Sihota, S.K.; Shao, J.Y.; Derkaoui, K.D.; et al. A global view of the oncogenic landscape in nasopharyngeal carcinoma: An integrated analysis at the genetic and expression levels. PLoS ONE 2012, 7, e41055. [Google Scholar] [CrossRef]
- Tay, J.K.; Zhu, C.; Shin, J.H.; Zhu, S.X.; Varma, S.; Foley, J.W.; Vennam, S.; Yip, Y.L.; Goh, C.K.; Wang, D.Y.; et al. The microdissected gene expression landscape of nasopharyngeal cancer reveals vulnerabilities in FGF and noncanonical NF-κB signaling. Sci. Adv. 2022, 8, eabh2445. [Google Scholar] [CrossRef]
- Lin, C.; Zong, J.; Lin, W.; Wang, M.; Xu, Y.; Zhou, R.; Lin, S.; Guo, Q.; Chen, H.; Ye, Y.; et al. EBV-miR-BART8-3p induces epithelial-mesenchymal transition and promotes metastasis of nasopharyngeal carcinoma cells through activating NF-κB and Erk1/2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Den Boon, J.A.; Chen, I.H.; Newton, M.A.; Dahl, D.B.; Chen, M.; Cheng, Y.J.; Westra, W.H.; Chen, C.J.; Hildesheim, A.; et al. Genome-wide expression profiling reveals EBV-associated inhibition of MHC class I expression in nasopharyngeal carcinoma. Cancer Res. 2006, 66, 7999–8006. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Chung, G.T.-Y.; Lou, W.P.-K.; Chow, C.; To, K.-F.; Choy, K.-W.; Leung, A.W.-C.; Tong, C.Y.-K.; Yuen, J.W.-F.; Ko, C.-W.; Yip, T.T.-C.; et al. Constitutive activation of distinct NF-κB signals in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 311–322. [Google Scholar] [CrossRef]
- Bruce, J.P.; To, K.-F.; Lui, V.W.Y.; Chung, G.T.Y.; Chan, Y.-Y.; Tsang, C.M.; Yip, K.Y.; Ma, B.B.Y.; Woo, J.K.S.; Hui, E.P.; et al. Author Correction: Whole-genome profiling of nasopharyngeal carcinoma reveals viral-host co-operation in inflammatory NF-κB activation and immune escape. Nat. Commun. 2022, 13, 4353. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, Z.; Wu, X.; Zhou, Q.; Liao, C.; Liu, Y.; Li, D.; Shen, L.; Feng, D.; Yang, L. LMP1 promotes nasopharyngeal carcinoma metastasis through NTRK2-mediated anoikis resistance. Am. J. Cancer Res. 2020, 10, 2083. [Google Scholar]
- Sui, X.; Geng, J.-H.; Li, Y.-H.; Zhu, G.-Y.; Wang, W.-H. Calcium channel α2δ1 subunit (CACNA2D1) enhances radioresistance in cancer stem-like cells in non-small cell lung cancer cell lines. Cancer Manag. Res. 2018, 10, 5009–5018. [Google Scholar] [CrossRef]
- Liu, X.; Wang, S.; Xu, J.; Kou, B.; Chen, D.; Wang, Y.; Zhu, X. Extract of Stellerachamaejasme L(ESC) inhibits growth and metastasis of human hepatocellular carcinoma via regulating microRNA expression. BMC Complement. Altern. Med. 2018, 18, 99. [Google Scholar] [CrossRef]
- Song, Q.; An, Q.; Niu, B.; Lu, X.; Zhang, N.; Cao, X. Role of miR-221/222 in Tumor Development and the Underlying Mechanism. J. Oncol. 2019. 2019, 7252013. [Google Scholar] [CrossRef]
- Chen, H.; Yao, X.; Di, X.; Zhang, Y.; Zhu, H.; Liu, S.; Chen, T.; Yu, D.; Sun, X. MiR-450a-5p inhibits autophagy and enhances radiosensitivity by targeting dual-specificity phosphatase 10 in esophageal squamous cell carcinoma. Cancer Lett. 2020, 483, 114–126. [Google Scholar] [CrossRef]
- Jiménez-Martínez, M.; Ostalé, C.M.; van der Burg, L.R.; Galán-Martínez, J.; Hardwick, J.C.H.; López-Pérez, R.; Hawinkels, L.J.A.C.; Stamatakis, K.; Fresno, M. DUSP10 Is a Regulator of YAP1 Activity Promoting Cell Proliferation and Colorectal Cancer Progression. Cancers 2019, 11, 1767. [Google Scholar] [CrossRef]
- Mkrtchyan, G.V.; Veviorskiy, A.; Izumchenko, E.; Shneyderman, A.; Pun, F.W.; Ozerov, I.V.; Aliper, A.; Zhavoronkov, A.; Scheibye-Knudsen, M. High-confidence cancer patient stratification through multiomics investigation of DNA repair disorders. Cell Death Dis. 2022, 13, 999. [Google Scholar] [CrossRef]
- Liu, Y.; He, S.; Wang, X.-L.; Peng, W.; Chen, Q.-Y.; Chi, D.-M.; Chen, J.-R.; Han, B.-W.; Lin, G.-W.; Li, Y.-Q.; et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat. Commun. 2021, 12, 741. [Google Scholar] [CrossRef]
- Gong, L.; Kwong, D.L.-W.; Dai, W.; Wu, P.; Li, S.; Yan, Q.; Zhang, Y.; Zhang, B.; Fang, X.; Liu, L.; et al. Comprehensive single-cell sequencing reveals the stromal dynamics and tumor-specific characteristics in the microenvironment of nasopharyngeal carcinoma. Nat. Commun. 2021, 12, 1540. [Google Scholar] [CrossRef]
- Chen, Y.-P.; Yin, J.-H.; Li, W.-F.; Li, H.-J.; Chen, D.-P.; Zhang, C.-J.; Lv, J.-W.; Wang, Y.-Q.; Li, X.-M.; Li, J.-Y.; et al. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell Res. 2020, 30, 1024–1042. [Google Scholar] [CrossRef]
- Dai, W.; Zheng, H.; Cheung, A.K.; Lung, M.L. Genetic and epigenetic landscape of nasopharyngeal carcinoma. Chin. Clin. Oncol. 2016, 5, 16. [Google Scholar] [CrossRef]
- Dai, W.; Chung, D.L.-S.; Chow, L.K.-Y.; Yu, V.Z.; Lei, L.C.; Leong, M.M.-L.; Chan, C.K.-C.; Ko, J.M.-Y.; Lung, M.L. Clinical Outcome–Related Mutational Signatures Identified by Integrative Genomic Analysis in Nasopharyngeal Carcinoma. Clin. Cancer Res. 2020, 26, 6494–6504. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Zhou, X.; Zhu, S.; Kaya, N.A.; Chan, Y.S.; Ma, L.; Xu, M.; Zhai, W. An Integrative Analysis of Nasopharyngeal Carcinoma Genomes Unraveled Unique Processes Driving a Viral-Positive Cancer. Cancers 2023, 15, 1243. [Google Scholar] [CrossRef]
- Ma, B.B.Y.; Goh, B.C.; Lim, W.T.; Hui, E.P.; Tan, E.H.; Lopes, G.d.L.; Lo, K.W.; Li, L.; Loong, H.; Foster, N.R.; et al. Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Investig. New Drugs 2015, 33, 985–991. [Google Scholar] [CrossRef]
- Zhao, S.-J.; Shen, Y.-F.; Li, Q.; He, Y.-J.; Zhang, Y.-K.; Hu, L.-P.; Jiang, Y.-Q.; Xu, N.-W.; Wang, Y.-J.; Li, J.; et al. SLIT2/ROBO1 axis contributes to the Warburg effect in osteosarcoma through activation of SRC/ERK/c-MYC/PFKFB2 pathway. Cell Death Dis. 2018, 9, 390. [Google Scholar] [CrossRef]
- Inoue, H.; Shiozaki, A.; Kosuga, T.; Shimizu, H.; Kudou, M.; Ohashi, T.; Arita, T.; Konishi, H.; Komatsu, S.; Kubota, T.; et al. Functions and Clinical Significance of CACNA2D1 in Gastric Cancer. Ann. Surg. Oncol. 2022, 29, 4522–4535. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Adams, R.C.; Rashidieh, B. Can computers conceive the complexity of cancer to cure it? Using artificial intelligence technology in cancer modelling and drug discovery. Math. Biosci. Eng. 2020, 17, 6515–6530. [Google Scholar] [CrossRef]
- Guan, S.; Wei, J.; Huang, L.; Wu, L. Chemotherapy and chemo-resistance in nasopharyngeal carcinoma. Eur. J. Med. Chem. 2020, 207, 112758. [Google Scholar] [CrossRef]
- Waldron, J.; Tin, M.M.; Keller, A.; Lum, C.; Japp, B.; Sellmann, S.; van Prooijen, M.; Gitterman, L.; Blend, R.; Payne, D.; et al. Limitation of conventional two dimensional radiation therapy planning in nasopharyngeal carcinoma. Radiother. Oncol. 2003, 68, 153–161. [Google Scholar] [CrossRef]
- Kang, Y.; He, W.; Ren, C.; Qiao, J.; Guo, Q.; Hu, J.; Xu, H.; Jiang, X.; Wang, L. Advances in targeted therapy mainly based on signal pathways for nasopharyngeal carcinoma. Signal Transduct. Target. Ther. 2020, 5, 245. [Google Scholar] [CrossRef]
- Li, J.-B.; Guo, S.-S.; Tang, L.-Q.; Guo, L.; Mo, H.-Y.; Chen, Q.-Y.; Mai, H.-Q. Longitudinal Trend of Health-Related Quality of Life During Concurrent Chemoradiotherapy and Survival in Patients with Stage II–IVb Nasopharyngeal Carcinoma. Front. Oncol. 2020, 10, 579292. [Google Scholar] [CrossRef]
- Lewis, M.W.; Wisniewska, K.; King, C.M.; Li, S.; Coffey, A.; Kelly, M.R.; Regner, M.J.; Franco, H.L. Enhancer RNA Transcription Is Essential for a Novel CSF1 Enhancer in Triple-Negative Breast Cancer. Cancers 2022, 14, 1852. [Google Scholar] [CrossRef]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- Herz, H.-M.; Hu, D.; Shilatifard, A. Enhancer Malfunction in Cancer. Mol. Cell 2014, 53, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Okabe, A.; Huang, K.K.; Matsusaka, K.; Fukuyo, M.; Xing, M.; Ong, X.; Hoshii, T.; Usui, G.; Seki, M.; Mano, Y.; et al. Cross-species chromatin interactions drive transcriptional rewiring in Epstein–Barr virus–positive gastric adenocarcinoma. Nat. Genet. 2020, 52, 919–930. [Google Scholar] [CrossRef]
- Dolphin, A.C. Calcium channel auxiliary α2δ and β subunits: Trafficking and one step beyond. Nat. Rev. Neurosci. 2012, 13, 542–555. [Google Scholar] [CrossRef]
- Ablinger, C.; Geisler, S.M.; Stanika, R.I.; Klein, C.T.; Obermair, G.J. Neuronal α2δ proteins and brain disorders. Pflugers Arch 2020, 472, 845–863. [Google Scholar] [CrossRef]
- Chintalapudi, S.R.; Maria, D.; Di Wang, X.; Bailey, J.N.C.; Hysi, P.G.; Wiggs, J.L.; Williams, R.W.; Jablonski, M.M. Systems genetics identifies a role for Cacna2d1 regulation in elevated intraocular pressure and glaucoma susceptibility. Nat. Commun. 2017, 8, 1755. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Z.; Zhang, K.; Dong, Y.; Zhang, A.; Lu, C.; Liu, L. MicroRNA-107 inhibits proliferation and invasion of laryngeal squamous cell carcinoma cells by targeting CACNA2D1 in vitro. Anti-Cancer Drugs 2020, 31, 260–271. [Google Scholar] [CrossRef]
- Liang, J.; Deng, Y.; Zhang, Y.; Wu, B.; Zhou, J. PRLR and CACNA2D1 Impact the Prognosis of Breast Cancer by Regulating Tumor Immunity. J. Pers. Med. 2022, 12, 2086. [Google Scholar] [CrossRef]
- Yu, D.; Holm, R.; Goscinski, M.A.; Trope, C.G.; Nesland, J.M.; Suo, Z. Prognostic and clinicopathological significance of Cacna2d1 expression in epithelial ovarian cancers: A retrospective study. 2016, 6, 2088–2097.
- Huang, T.; Feng, X.; Wang, J.; Zhou, J.; Wang, J. Calcium-Related Genes Predicting Outcomes and Serving as Therapeutic Targets in Endometrial Cancer. Cells 2022, 11, 3156. [Google Scholar] [CrossRef]
- Inoue, H.; Shiozaki, A.; Kosuga, T.; Shimizu, H.; Kudou, M.; Arita, T.; Konishi, H.; Komatsu, S.; Kuriu, Y.; Kubota, T.; et al. CACNA2D1 regulates the progression and influences the microenvironment of colon cancer. J. Gastroenterol. 2024, 59, 556–571. [Google Scholar] [CrossRef]
- Yarilina, A.; Xu, K.; Chen, J.; Ivashkiv, L.B. TNF activates calcium–nuclear factor of activated T cells (NFAT)c1 signaling pathways in human macrophages. Proc. Natl. Acad. Sci. USA 2011, 108, 1573–1578. [Google Scholar] [CrossRef]
- Grewal, S.S.; Fass, D.M.; Yao, H.; Ellig, C.L.; Goodman, R.H.; Stork, P.J.S. Calcium and cAMP Signals Differentially Regulate cAMP-responsive Element-binding Protein Function via a Rap1-Extracellular Signal-regulated Kinase Pathway. J. Biol. Chem. 2000, 275, 34433–34441. [Google Scholar] [CrossRef]
- Maki, A.; Berezesky, I.K.; Fargnoli, J.; Holbrook, N.J.; Trump, B.F. Role of [Ca2+]i in induction of c-fos, c-jun, and c-myc mRNA in rat PTE after oxidative stress. FASEB J. 1992, 6, 919–924. [Google Scholar] [CrossRef]
- Li, D.W.-C.; Liu, J.-P.; Mao, Y.W.; Xiang, H.; Wang, J.; Ma, W.-Y.; Dong, Z.; Pike, H.M.; Brown, R.E.; Reed, J.C. Calcium-activated RAF/MEK/ERK Signaling Pathway Mediates p53-dependent Apoptosis and Is Abrogated by αB-Crystallin through Inhibition of RAS Activation. Mol. Biol. Cell 2005, 16, 4437–4453. [Google Scholar] [CrossRef]
- Berry, C.T.; May, M.J.; Freedman, B.D. STIM- and Orai-mediated calcium entry controls NF-κB activity and function in lymphocytes. Cell Calcium 2018, 74, 131–143. [Google Scholar] [CrossRef]
- Dolmetsch, R.E.; Xu, K.; Lewis, R.S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature 1998, 392, 933–936. [Google Scholar] [CrossRef]
- Lee, A.W.; Fee, W.E.; Ng, W.; Chan, L. Nasopharyngeal carcinoma: Salvage of local recurrence. Oral Oncol. 2012, 48, 768–774. [Google Scholar] [CrossRef]
- Lin, S.; Tham, I.W.M.; Pan, J.; Han, L.; Chen, Q.; Lu, J.J.M. Combined High-dose Radiation Therapy and Systemic Chemotherapy Improves Survival in Patients with Newly Diagnosed Metastatic Nasopharyngeal Cancer. Am. J. Clin. Oncol. 2012, 35, 474–479. [Google Scholar] [CrossRef]
- Chen, W.-M.; Chen, S.-X.; Jiang, L.; Shu, G.-G.; Yin, Y.-X.; Quan, Z.-P.; Zhou, Z.-Y.; Shen, M.-J.; Qin, Y.-T.; Yang, C.-L.; et al. Establishment of a visualized mouse orthotopic xenograft model of nasopharyngeal carcinoma. Cancer Biol. Ther. 2024, 25, 2382531. [Google Scholar] [CrossRef]
- Bruford, E.A.; Braschi, B.; Denny, P.; Jones, T.E.M.; Seal, R.L.; Tweedie, S. Guidelines for human gene nomenclature. Nat. Genet. 2020, 52, 754–758. [Google Scholar] [CrossRef]
- Stouffer, S.A.; Suchman, E.A.; Devinney, L.C.; Star, S.A.; Williams, R.M., Jr. The American Soldier: Adjustment During Army Life. (Studies in Social Psychology in World War II); Princeton University Press: Princeton, NJ, USA, 1949. [Google Scholar]
- Liu, B.H.M.; Lin, Y.; Long, X.; Hung, S.W.; Gaponova, A.; Ren, F.; Zhavoronkov, A.; Pun, F.W.; Wang, C.C. Utilizing AI for the Identification and Validation of Novel Therapeutic Targets and Repurposed Drugs for Endometriosis. Adv. Sci. 2024, 12, e2406565. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Dolgalev, I. msigdbr, version 7.5.1; msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2019. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, D.L.-S.; Leung, G.H.D.; Liu, S.; Lok, S.W.Y.; Xin, Y.; Xia, Y.; Zhavoronkov, A.; Pun, F.W.; Ng, W.-T.; Dai, W. AI-Driven Drug Target Screening Platform Identified Oncogene CACNA2D1 Activated by Enhancer Infestation in Epstein-Barr Virus-Associated Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2025, 26, 4697. https://doi.org/10.3390/ijms26104697
Chung DL-S, Leung GHD, Liu S, Lok SWY, Xin Y, Xia Y, Zhavoronkov A, Pun FW, Ng W-T, Dai W. AI-Driven Drug Target Screening Platform Identified Oncogene CACNA2D1 Activated by Enhancer Infestation in Epstein-Barr Virus-Associated Nasopharyngeal Carcinoma. International Journal of Molecular Sciences. 2025; 26(10):4697. https://doi.org/10.3390/ijms26104697
Chicago/Turabian StyleChung, Dittman Lai-Shun, Geoffrey Ho Duen Leung, Songran Liu, Sarah Wing Yan Lok, Ying Xin, Yunfei Xia, Alex Zhavoronkov, Frank W. Pun, Wai-Tong Ng, and Wei Dai. 2025. "AI-Driven Drug Target Screening Platform Identified Oncogene CACNA2D1 Activated by Enhancer Infestation in Epstein-Barr Virus-Associated Nasopharyngeal Carcinoma" International Journal of Molecular Sciences 26, no. 10: 4697. https://doi.org/10.3390/ijms26104697
APA StyleChung, D. L.-S., Leung, G. H. D., Liu, S., Lok, S. W. Y., Xin, Y., Xia, Y., Zhavoronkov, A., Pun, F. W., Ng, W.-T., & Dai, W. (2025). AI-Driven Drug Target Screening Platform Identified Oncogene CACNA2D1 Activated by Enhancer Infestation in Epstein-Barr Virus-Associated Nasopharyngeal Carcinoma. International Journal of Molecular Sciences, 26(10), 4697. https://doi.org/10.3390/ijms26104697