A Brief Overview of the Epigenetic Regulatory Mechanisms in Plants



Abstract
1. Introduction
2. DNA Methylation
- Initial biogenesis of siRNA and ncRNA premature transcripts by RNA-polymerase II (POL II).
- Processing of pre-RNAs via a complicated and compartmentalized pathway involving DICER (DCL3) and ARGONAUTE (AGO4 and AGO6) systems.
- Recruitment of POL-IV and POL-V, two plant-specific RNA polymerases.
- Formation of the active ribonucleoprotein AGO-siRNA-ncRNA-POL V complex, which recruits DRM2 (domains rearranged methyltransferase 2), directed for specific methylation marked by the readers SUVH2 and SUVH9.
3. Histone Modifications
3.1. Histone Lysine Methylation
- Three homologs of E(z): CURLY LEAF (CLF), SWINGER (SWN), and MEDEA (MEA).
- Three homologs of Su(z)12: EMBRYONIC FLOWER 2 (EMF2), VERNALIZATION 2 (VRN2), and FERTILIZATION-INDEPENDENT SEED 2 (FIS2).
- One homolog of Esc: FERTILIZATION-INDEPENDENT ENDOSPERM (FIE).
- Five homologs of p55: MULTICOPY SUPPRESSOR OF IRA 1 (MSI1), MSI2, MSI3, MSI4/FVE, and MSI5.
3.2. Histone Arginine Methylation
3.3. Histone Acetylation
3.4. Histone Phosphorylation
3.5. MIR-Dependent Regulation of Gene Expression
4. Ubiquitination and Ubiquitin Ligase-Mediated Regulation
5. Histone Modifications During Seed Development and Germination
6. Stress-Related Epigenetic Aspects During Seed Development and Germination
6.1. Abiotic Stress Response Mechanisms
6.2. Biotic Stress Response Mechanisms
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Speybroeck, L.; De Waele, D.; Van De Vijver, G. Theories in early embryology. Ann. N. Y. Acad. Sci. 2002, 981, 7–49. [Google Scholar] [CrossRef]
- Ahmad, A.; Dong, Y.; Cao, X. Characterization of the PRMT gene family in rice reveals conservation of arginine methylation. PLoS ONE 2011, 8, e22664. [Google Scholar] [CrossRef] [PubMed]
- Ch, W. The epigenotype. Int. J. Epidemiol. 2011, 41, 10–13. [Google Scholar]
- Iwasaki, M.; Paszkowski, J. Identification of genes preventing transgenerational transmission of stress-induced epigenetic states. Proc. Natl. Acad. Sci. USA 2014, 111, 8547–8552. [Google Scholar] [CrossRef]
- Moore, D.S. The Developing Genome: An Introduction to Behavioral Epigenetics; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Singh, S.; Kumar, V.; Prasad, S.M. A review of plants strategies to resist biotic and abiotic environmental stressors. Environ. Res. 2023, 232, 116272. [Google Scholar]
- Du, Y.; Wang, Z.; Liu, Y.; Wang, Q.; Zhang, J. Strategies of plants to overcome abiotic and biotic stresses. Biol. Rev. 2024, 99, 13079. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E. Epigenetic modifications in plants: An evolutionary perspective. Curr. Opin. Plant Biol. 2011, 14, 179–186. [Google Scholar] [CrossRef]
- Henderson, I.; Jacobsen, S.E. Epigenetic inheritance in plants. Nature 2007, 447, 418–424. [Google Scholar] [CrossRef]
- Ryazansky, S.; Akulenko, N. Fraternity of old-timers: How ubiquitin regulates miRNA functions. Bioessays 2023, 45, e2200220. [Google Scholar] [CrossRef]
- Gupta, C.; Salgotra, R.K. Epigenetics and its role in effecting agronomical traits. Front Plant Sci. 2022, 13, 925688. [Google Scholar] [CrossRef]
- Kribelbauer, J.F.; Laptenko, O.; Chen, S.; Martini, G.D.; Freed-Pastor, W.A.; Prives, C.; Mann, R.S.; Bussemaker, H.J. Quantitative Analysis of the DNA Methylation Sensitivity of Transcription Factor Complexes. Cell Rep. 2017, 19, 2383–2395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Lang, Z.B.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T.; David, L.; Zhu, J. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA Glycosylase/Lyase. Cell 2002, 111, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Palanca, A.M.S.; Law, J.A. Locus-specific control of the de novo DNA methylation pathway in Arabidopsis by the CLASSY family. Nat. Genet. 2018, 50, 865–873. [Google Scholar] [CrossRef]
- Vergara, Z.; Gutierrez, C. Emerging roles of chromatin in the maintenance of genome organization and function in plants. Genome Biol. 2017, 18, 96. [Google Scholar] [CrossRef]
- Jeong, H.J.; Yang, J.; Yi, J.; An, G. Controlling flowering time by histone methylation and acet-ylation in arabidopsis and rice. J. Plant Biol. 2015, 58, 203–210. [Google Scholar] [CrossRef]
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone methylation in higher plants. Annu. Rev. Plant Biol. 2010, 61, 395–420. [Google Scholar] [CrossRef]
- Liu, J.; Shangguan, Y.; Tang, D.; Dai, Y. Histone succinylation and its function on the nucleosome. J. Cell. Mol. Med. 2021, 25, 7101–7109. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, Y.; Yang, K.; Zhao, Y.; Wen, C.; Yang, Y.; Zhang, W. Proteomic Analysis of Lysine Acetylation and Succinylation to Investigate the Pathogenicity of Virulent Pseudomonas syringae pv. tomato DC3000 and Avirulent Line Pseudomonas syringae pv. tomato DC3000 avrRpm1 on Arabidopsis thaliana. Genes 2024, 15, 499. [Google Scholar] [CrossRef]
- He, D.; Wang, Q.; Li, M.; Damaris, R.N.; Yi, X.; Cheng, Z.; Yang, P. Global Proteome Analyses of Lysine Acetylation and Succinylation Reveal the Widespread Involvement of both Modification in Metabolism in the Embryo of Germinating Rice Seed. J. Proteome Res. 2016, 15, 879–890. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, K.; Wang, Y.; Gu, Y. Comprehensive review of histone lactylation: Structure, function, and therapeutic targets. Biochem. Pharmacol. 2024, 225, 116331. [Google Scholar] [CrossRef] [PubMed]
- Beamer, Z.G.; Routray, P.; Choi, W.G.; Spangler, M.K.; Lokdarshi, A.; Roberts, D.M. Aquaporin family lactic acid channel NIP2;1 promotes plant survival under low oxygen stress in Arabidopsis. Plant Physiol. 2021, 187, 2262–2278. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin ligases: Structure, function, and regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Keren, I.; Lacroix, B.; Kohrman, A.; Citovsky, V. Histone Deubiquitinase OTU1 Epigenetically Regulates DA1 and DA2, Which Control Arabidopsis Seed and Organ Size. iScience 2020, 3, 100948. [Google Scholar] [CrossRef]
- Vaughan, N.; Scholz, N.; Lindon, C.; Licchesi, J.D. The E3 ubiquitin ligase HECTD1 contributes to cell proliferation through an effect on mitosis. Sci. Rep. 2022, 12, 13160. [Google Scholar] [CrossRef]
- Liu, R.; Wu, J.; Guo, H.; Yao, W.; Li, S.; Lu, Y.; Jia, Y.; Liang, X.; Tang, J.; Zhang, H. Post-translational modifications of histones: Mechanisms, biological functions, and therapeutic targets. MedComm 2023, 3, 292. [Google Scholar] [CrossRef]
- Schatlowski, N.; Creasey, K.; Goodrich, J.; Schubert, D. Keeping plants in shape: Polycomb-group genes and histone methylation. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2008; Volume 6, pp. 547–553. [Google Scholar]
- Molitor, A.; Shen, W.H. The Polycomb Complex PRC1: Composition and Function in Plants. J. Genet. Genom. 2013, 5, 231–238. [Google Scholar] [CrossRef]
- Sharma, S.; Ghoshal, C.; Arora, A.; Samar, W.; Nain, L.; Paul, D. Strain Improvement of Native Saccharomyces cerevisiae LN ITCC 8246 Strain Through Protoplast Fusion to Enhance Its Xylose Uptake. Appl. Biochem. Biotechnol. 2021, 193, 2455–2469. [Google Scholar] [CrossRef]
- Chen, L.; Hellmann, H. Plant E3 Ligases: Flexible Enzymes in a Sessile World. Mol. Plant 2013, 5, 1388–1404. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, T.-L.; Hu, C.-G.; Zhang, J.-Z. The Role of Drought and Temperature Stress in the Regulation of Flowering Time in Annuals and Perennials. Agronomy 2023, 13, 3034. [Google Scholar] [CrossRef]
- Qu, J.; Lin, Z. Autophagy Regulation by Crosstalk between miRNAs and Ubiquitination System. Int. J. Mol. Sci. 2021, 22, 11912. [Google Scholar] [CrossRef] [PubMed]
- Lucibelli, F.; Valoroso, M.C.; Aceto, S. Plant DNA Methylation: An Epigenetic Mark in Development, Environmental Interactions, and Evolution. Int. J. Mol. Sci. 2022, 23, 8299. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Bartolome, J. DNA methylation in plants: Mechanisms and tools for targeted manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef]
- Gallego-Bartolome, J.; Gardiner, J.; Liu, W.L.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef]
- Maeji, H.; Nishimura, T. Epigenetic Mechanisms in Plants. Adv. Bot. Res. 2018, 88, 21–47. [Google Scholar]
- Liu, W.; Duttke, S.H.; Hetzel, J.; Growth, M.; Feng, S.; Gallego-Bartolome, J.; Zhong, Z.; Kuo, H.Y.; Wang, Z.; Zhai, J.; et al. RNA-directed DNA methylation involves co-transcriptional small-RNA-guided slicing of polymerase V transcripts in Arabidopsis. Nat. Plants 2018, 4, 181–188. [Google Scholar] [CrossRef]
- Cuerda-Gil, D.; Slotkin, R.K. Non-canonical RNA-directed DNA methylation. Nat. Plants 2016, 2, 16163. [Google Scholar] [CrossRef]
- Martinez de Alba, A.E.; Elvira-Matelot, E.; Vaucheret, H. Gene silencing in plants: A diversity of pathways. Biochim. Biophys. Acta 2013, 1829, 1300–1308. [Google Scholar] [CrossRef]
- Li, X.; Qian, W.; Zhao, Y.; Wang, C.; Shen, J.; Zhu, J.; Gong, Z. Antisilencing role of the RNA-directed DNA methylation pathway and a histone acetyltransferase in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 11425–11430. [Google Scholar] [CrossRef]
- Deleris, A.; Halter, T.; Navarro, L. DNA methylation and demethylation in plant immunity. Annu. Rev. Phytopathol. 2016, 54, 579–603. [Google Scholar] [CrossRef]
- Gentry, M.; Hennig, L. Remodeling chromatin to shape development of plants. Exp. Cell Res. 2014, 321, 40–46. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Luna, E.; Bruce, T.J.; Roberts, M.R.; Flors, V.; Ton, J. Next-Generation systemic acquired resistance. Plant Physiol. 2011, 158, 844–853. [Google Scholar] [CrossRef]
- Choi, J.; Lyons, D.B.; Kim, M.Y.; Moore, J.D.; Zilberman, D. DNA Methylation and Histone H1 Jointly Repress Transposable Elements and Aberrant Intragenic Transcripts. Mol. Cell 2020, 77, 310–323. [Google Scholar] [CrossRef]
- Marunde, M.R.; Fuchs, H.A.; Burg, J.M.; Popova, I.K.; Vaidya, A.; Hall, N.W.; Weinzapfel, E.N.; Meiners, M.J.; Watson, R.; Gillespie, Z.B.; et al. Nucleosome conformation dictates the histone code. eLife 2024, 13, e78866. [Google Scholar] [CrossRef]
- Kumar, V.; Thakur, J.K.; Prasad, M. Histone acetylation dynamics regulating plant development and stress responses. Cell. Mol. Life Sci. 2021, 78, 4467–4486. [Google Scholar] [CrossRef]
- Zhao, T.; Zhan, Z.; Jiang, D. Histone modifications and their regulatory roles in plant development and environmental memory. J. Genet. Genom. 2019, 46, 467–476. [Google Scholar] [CrossRef]
- Julian, R.; Patrick, R.M.; Li, Y. Organ-specific characteristics govern the relationship between histone code dynamics and transcriptional reprogramming during nitrogen response in tomato. Commun. Biol. 2023, 6, 1225. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Tamburri, S.; Rustichelli, S.; Amato, S.; Pasini, D. Navigating the complexity of Polycomb repression: Enzymatic cores and regulatory modules. Mol. Cell. 2024, 84, 3381–3405. [Google Scholar] [CrossRef]
- Perianez-Rodriguez, J.; Manzano, C.; Moreno-Risueño, M.A. Post-embryonic organogenesis and plant regeneration from tissues: Two sides of the same coin? Front. Plant Sci. 2014, 5, 219. [Google Scholar] [CrossRef]
- Ornelas-Ayala, D.; Cortés-Quiñones, C.; Olvera-Herrera, J.; García-Ponce, B.; Garay-Arroyo, A.; Álvarez-Buylla, E.R.; Sanchez, M.d.l.P. A Green Light to Switch on Genes: Revisiting Trithorax on Plants. Plants 2023, 12, 75. [Google Scholar] [CrossRef]
- Mozgova, I.; Hennig, L. The Polycomb Group Protein Regulatory Network. Annu. Rev. Plant Biol. 2015, 66, 269–296. [Google Scholar] [CrossRef]
- Zhao, S.; Cheng, L.; Gao, Y.; Zhang, B.; Zheng, X.; Wang, L.; Li, P.; Sun, Q.; Li, H. Plant HP1 protein ADCP1 links multivalent H3K9 methylation readout to heterochromatin formation. Cell Res. 2018, 29, 54–66. [Google Scholar] [CrossRef]
- Antunez-Sanchez, J.; NaishJuan, M.; Ramirez-Prado, J.S.; Ohno, S.; Huang, Y.; Dawson, A.; Opassathian, K.; Manza-Mianza, D.; Ariel, F.; Raynaud, C.; et al. A new role for histone demethylases in the maintenance of plant genome integrity. eLife 2020, 9, e58533. [Google Scholar] [CrossRef]
- Li, Z.; Fu, X.; Wang, Y.; Liu, R.; He, Y. Polycomb-mediated gene silencing by the BAH-EMF1 complex in plants. Nat. Genet. 2018, 9, 1254–1261. [Google Scholar] [CrossRef]
- Li, Y.; Mukherjee, I.; Thum, K.E.; Tanurdzic, M.; Katari, M.S.; Obertello, M.; Edwards, M.B.; McCombie, W.R.; Martienssen, R.A.; Coruzzi, G.M. The histone methyltransferase SDG8 mediates the epigenetic modification of light and carbon responsive genes in plants. Genome Biol. 2015, 16, 79. [Google Scholar] [CrossRef]
- Berr, A.; Shafiq, S.; Pinon, V.; Dong, A.; Shen, W. The trxG family histone methyltransferase SET DOMAIN GROUP 26 promotes flowering via a distinctive genetic pathway. Plant J. 2014, 81, 316–328. [Google Scholar] [CrossRef]
- Walport, L.J.; Hopkinson, R.J.; Chowdhury, R.; Schiller, R.; Ge, W.; Kawamura, A.; Schofield, C.J. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat. Commun. 2016, 7, 11974. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Liu, X.; Yang, S.; Yu, C.-W.; Chen, C.-Y.; Wu, K. Chapter six—Histone Acetylation and Plant Development. Enzymes 2016, 40, 173–199. [Google Scholar]
- Pandey, R.; Müller, A.; Napoli, C.A.; Selinger, D.A.; Pikaard, C.S.; Richards, E.J.; Bender, J.; Mount, D.W.; Jorgensen, R.A. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 2002, 30, 5036–5055. [Google Scholar] [CrossRef]
- Aiese Cigliano, R.; Sanseverino, W.; Cremona, G.; Ercolano, M.R.; Conicella, C.; Consiglio, F.M. Genome-wide analysis of histone modifiers in tomato: Gaining an insight into their developmental roles. BMC Genom. 2013, 14, 57. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, J.; Armando Casas-Mollano, J.; Dou, Y.; Jia, S.; Yang, Q.; Zhang, C.; Cerutti, H. MLK4-mediated phosphorylation of histone H3T3 promotes flowering by transcriptional silencing of FLC/MAF in Arabidopsis thaliana. Plant J. 2021, 105, 1400–1412. [Google Scholar] [CrossRef]
- Huang, T.; Zhang, H.; Zhou, Y.; Su, Y.; Zheng, H.; Ding, Y. Phosphorylation of Histone H2A at Serine 95 Is Essential for Flowering Time and Development in Arabidopsis. Front. Plant Sci. 2021, 12, 761008. [Google Scholar] [CrossRef]
- Houben, A.; Demidov, D.; Caperta, A.D.; Karimi, R.; Agueci, F.; Vlasenko, L. Phosphorylation of histone H3 in plants—A dynamic affair. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 2007, 1769, 308–315. [Google Scholar] [CrossRef]
- Zhou, B.; Zeng, L. Conventional and unconventional ubiquitination in plant immunity. Mol. Plant Pathol. 2017, 18, 1313–1330. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target. Oncol. 2020, 3, 261–278. [Google Scholar] [CrossRef]
- Yang, H.; Cui, Y.; Feng, Y.; Hu, Y.; Liu, L.; Duan, L. Long Non-Coding RNAs of Plants in Response to Abiotic Stresses and Their Regulating Roles in Promoting Environmental Adaption. Cells 2023, 12, 729. [Google Scholar] [CrossRef]
- Jha, U.C.; Nayyar, H.; Jha, R.; Khurshid, M.; Zhou, M.; Mantri, N.; Siddique, K.H.M. Long non-coding RNAs: Emerging players regulating plant abiotic stress response and adaptation. BMC Plant Biol. 2020, 20, 466. [Google Scholar] [CrossRef]
- Urquiaga, M.C.O.; Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. From Trash to Luxury: The Potential Role of Plant LncRNA in DNA Methylation During Abiotic Stress. Front Plant Sci. 2020, 11, 603246. [Google Scholar] [CrossRef]
- Jin, X.; Wang, Z.; Li, X.; Ai, Q.; Wong, D.C.J.; Zhang, F.; Yang, J.; Zhang, N.; Si, H. Current perspectives of lncRNAs in abiotic and biotic stress tolerance in plants. Front Plant Sci. 2023, 14, 1334620. [Google Scholar] [CrossRef]
- Tremblay, B.J.M.; Questa, J.I. Mechanisms of epigenetic regulation of transcription by lncRNAs in plants. IUBMB Life 2023, 75, 427–439. [Google Scholar] [CrossRef]
- Ding, N.; Zhang, B. microRNA production in Arabidopsis. Front. Plant Sci. 2023, 14, 1096772. [Google Scholar] [CrossRef]
- Yadav, A.; Mathan, J.; Dubey, A.K.; Singh, A. The Emerging Role of Non-Coding RNAs (ncRNAs) in Plant Growth, Development, and Stress Response Signaling. Non-Coding RNA 2024, 10, 13. [Google Scholar] [CrossRef]
- Xiao, M.; Li, J.; Li, W.; Wang, Y.; Wu, F.; Xi, Y.; Zhang, L.; Ding, C.; Luo, H.; Li, Y.; et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017, 14, 1326–1334. [Google Scholar] [CrossRef]
- D’Ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big impact on plant development. Trends Plant Sci. 2017, 22, 1056–1068. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, Y.C.; Chen, X.M.; Chen, Y.Q. Plant noncoding RNAs: Hidden players in development and stress responses. Annu. Rev. Cell Dev. Biol. 2019, 35, 407–431. [Google Scholar] [CrossRef]
- Knop, K.; Stepien, A.; Barciszewska-Pacak, M.; Taube, M.; Bielewicz, D.; Michalak, M.; Borst, W.J.; Jarmolowski, A.; Szweykowska-Kulinska, Z. Active 5’ splice sites regulate the biogenesis efficiency of Arabidopsis microRNAs derived from intron-containing genes. Nucleic Acids Res. 2017, 45, 2757–2775. [Google Scholar] [CrossRef]
- Singh, A.; Gautam, V.; Singh, S.; Sarkar Das, S.; Verma, S.; Mishra, V.; Mukherjee, S.; Sarkar, A.K. Plant small RNAs: Advancement in the understanding of biogenesis and role in plant development. Planta 2018, 248, 545–558. [Google Scholar] [CrossRef]
- Kim, Y.J.; Zheng, B.L.; Yu, Y.; Won, S.Y.; Mo, B.X.; Chen, X.M. The role of mediator in small and long noncoding RNA production in Arabidopsis thaliana. EMBO J. 2011, 30, 814–822. [Google Scholar] [CrossRef]
- Xu, M.L.; Hu, T.Q.; Smith, M.R.; Poethig, R.S. Epigenetic regulation of vegetative phase change in Arabidopsis. Plant Cell 2016, 28, 28–41. [Google Scholar] [CrossRef]
- Li, S.J.; Liu, K.; Zhou, B.J.; Li, M.; Zhang, S.X.; Zeng, L.; Zhang, C.; Yu, B. MAC3A and MAC3B, two core subunits of the MOS4-associated complex, positively influence miRNA biogenesis. Plant Cell 2018, 30, 481–494. [Google Scholar] [CrossRef]
- Erdmann, R.M.; Picard, C.L. RNA-directed DNA methylation. PLoS Genet. 2020, 10, 1009034. [Google Scholar] [CrossRef]
- Zuin, A.; Isasa, M.; Crosas, B. Ubiquitin signaling: Extreme conservation as a source of diversity. Cells 2014, 3, 690–701. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Callis, J. The ubiquitination machinery of the ubiquitin system. Arab. Book 2014, 12, e0174. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, F.; Zhang, H.; He, H.; Ma, L. An insight into the roles of ubiquitin-specific proteases in plants: Development and growth, morphogenesis, and stress response. Front. Plant Sci. 2024, 15, 1396634. [Google Scholar]
- Xue, H.; Yao, T.; Cao, M.; Zhu, G.; Li, Y.; Yuan, G.; Chen, Y.; Lei, M.; Huang, J. Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature 2019, 573, 445–449. [Google Scholar] [CrossRef]
- Miricescu, A.; Goslin, K.; Graciet, E. Ubiquitylation in plants: Signaling hub for the integration of environmental signals. J. Exp. Bot. 2018, 69, 4511–4527. [Google Scholar] [CrossRef]
- Lee, J.; Kim, W.T. Regulation of abiotic stress signal transduction by E3 ubiquitin ligases in Arabidopsis. Mol. Cells 2011, 31, 201–208. [Google Scholar] [CrossRef]
- Ryu, M.Y.; Cho, S.K.; Hong, Y.; Kim, J.; Kim, J.H.; Kim, G.M.; Chen, Y.J.; Knoch, E.; Møller, B.L.; Kim, W.T.; et al. Classification of barley U-box E3 ligases and their expression patterns in response to drought and pathogen stresses. BMC Genom. 2019, 326, 4571. [Google Scholar] [CrossRef]
- Al-Saharin, R.; Hellmann, H.; Mooney, S. Plant E3 Ligases and Their Role in Abiotic Stress Response. Cells 2022, 11, 890. [Google Scholar] [CrossRef]
- Trenner, J.; Monaghan, J.; Saeed, B.; Quint, M.; Shabek, N.; Trujillo, M. Evolution and Functions of Plant U-Box Proteins: From Protein Quality Control to Signaling. Annu. Rev. Plant Biol. 2022, 73, 93–121. [Google Scholar] [CrossRef]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. The U box is a modified RING finger—A common domain in ubiquitination. CB 2000, 10, R132–R134. [Google Scholar] [CrossRef]
- Ban, Z.; Estelle, M. CUL3 E3 ligases in plant development and environmental response. Nat. Plants. 2021, 1, 6–16. [Google Scholar] [CrossRef]
- Shu, K.; Yang, W. E3 Ubiquitin ligases: Ubiquitous actors in plant development and abiotic stress responses. Plant Cell Physiol. 2017, 58, 1461–1476. [Google Scholar] [CrossRef]
- Isono, E.; Katsiarimpa, A.; Müller, I.K.; Anzenberger, F.; Stierhof, Y.; Geldner, N.; Chory, J.; Schwechheimer, C. The Deubiquitinating Enzyme AMSH3 Is Required for Intracellular Trafficking and Vacuole Biogenesis in Arabidopsis thaliana. Plant Cell 2010, 22, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Abdurakhmonov, I.Y. Genomics era for plants and crop species—Advances made and needed tasks ahead. In Plant Genomics; Abdurakhmonov, I.Y., Ed.; BoD—Books on Demand: Norderstedt, Germany, 2016. [Google Scholar] [CrossRef]
- Schatz, M.C.; Witkowski, J.; McCombie, W.R. Current challenges in de novo plant ge nome sequencing and assembly. Genome Biol. 2012, 13, 243. [Google Scholar] [CrossRef]
- Michael, T.P.; VanBuren, R. Progress, challenges and the future of crop genomes. Curr. Opin. Plant Biol. 2015, 24, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, A.; Michael, T.P.; Rivadavia, F.; Sousa, A.; Wang, W.; Temsch, E.M.; Greilhuber, J.; Müller, K.F.; Heubl, G. Evolution of genome size and chromosome number in the carnivorous plant genus Genlisea (Lentibulariaceae), with a new estimate of the minimum genome size in angiosperms. Ann. Bot. 2014, 114, 1651–1663. [Google Scholar] [CrossRef]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes 2018, 14, 88. [Google Scholar] [CrossRef]
- Ding, X.; Jia, X.; Xiang, Y.; Jiang, W. Histone Modification and Chromatin Remodeling During the Seed Life Cycle. Front. Plant Sci. 2022, 13, 865361. [Google Scholar] [CrossRef]
- Bouyer, D.; Roudier, F.; Heese, M.; Andersen, E.D.; Gey, D.; Nowack, M.K.; Goodrich, J.; Renou, J.P.; Grini, P.E.; Colot, V.; et al. Polycomb repressive complex 2 controls the embryo-to-seedling phase transition. PLoS Genet. 2011, 7, e1002014. [Google Scholar] [CrossRef]
- Wang, D.; Tyson, M.D.; Jackson, S.S.; Yadegari, R. Partially redundant functions of two SET-domain polycomb-group proteins in controlling initiation of seed development in Arabidopsis. Proc. Natl. Acad. Sci USA 2006, 103, 13244–13249. [Google Scholar] [CrossRef]
- Hehenberger, E.; Kradolfer, D.; Köhler, C. Endosperm cellularization defines an important developmental transition for embryo development. Development 2012, 139, 2031–2039. [Google Scholar] [CrossRef]
- Cheng, X.; Pan, M.E.Z.; Zhou, Y.; Niu, B.; Chen, C. The maternally expressed polycomb group gene OsEMF2a is essential for endosperm cellularization and imprinting in rice. Plant Commun. 2020, 2, 100092. [Google Scholar] [CrossRef]
- Tonosaki, K.; Ono, A.; Kunisada, M.; Nishino, M.; Nagata, H.; Sakamoto, S.; Kijima, S.T.; Furuumi, H.; Nonomura, K.-I.; Sato, Y.; et al. Mutation of the imprinted gene OsEMF2a induces autonomous endosperm development and delayed cellularization in rice. Plant Cell 2021, 33, 85–103. [Google Scholar] [PubMed]
- Footitt, S.; Müller, K.; Kermode, A.R.; Finch-Savage, W.E. Seed dormancy cycling in Arabidopsis: Chromatin remodelling and regulation of DOG1 in response to seasonal environmental signals. Plant J. 2015, 81, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, K.; Bartsch, M.; Xiang, Y.; Miatton, E.; Pellengahr, S.; Yano, R.; Soppe, W.J. The time required for dormancy release in Arabidopsis is determined by DELAY OF GERMINATION1 protein levels in freshly harvested seeds. Plant Cell 2012, 7, 2826–2838. [Google Scholar] [CrossRef]
- Müller, K.; Bouyer, D.; Schnittger, A.; Kermode, A.R. Evolutionarily conserved histone methylation dynamics during seed life-cycle transitions. PLoS ONE 2013, 7, e51532. [Google Scholar] [CrossRef]
- Chen, N.; Wang, H.; Abdelmageed, H.; Veerappan, V.; Tadege, M.; Allen, R.D. HSI2/VAL1 and HSL1/VAL2 function redundantly to repress DOG1 expression in Arabidopsis seeds and seedlings. New Phytol. 2020, 227, 840–856. [Google Scholar] [CrossRef]
- Zheng, J.; Chen, F.; Wang, Z.; Cao, H.; Li, X.; Deng, X.; Soppe, W.J.J.; Li, Y.; Liu, y. A novel role for histone methyltransferase KYP/SUVH4 in the control of Arabidopsis primary seed dormancy. New Phytol. 2012, 193, 605–616. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, P.; Zhang, F.; Luo, X.; Xie, J. Histone deacetylase HDA19 interacts with histone methyltransferase SUVH5 to regulate seed dormancy in Arabidopsis. Plant Biol. 2020, 22, 1062–1071. [Google Scholar] [CrossRef]
- Wu, K.; Tian, L.; Malik, K.; Brown, D.; Miki, B. Functional analysis of HD2 histone deacetylase homologues in Arabidopsis thaliana. Plant J. 2000, 22, 19–27. [Google Scholar] [CrossRef]
- Zhang, H.; Lu, Y.; Zhao, Y.; Zhou, D.X. OsSRT1 is involved in rice seed development through regulation of starch metabolism gene expression. Plant Sci. 2016, 248, 28–36. [Google Scholar] [CrossRef]
- Forestan, C.; Farinati, S.; Rouster, J.; Lassagne, H.; Lauria, M.; Dal Ferro, N.; Varotto, S. Control of maize vegetative and reproductive development, fertility, and rRNAs silencing by HISTONE DEACETYLASE. Genetics 2018, 108, 1443–1466. [Google Scholar] [CrossRef]
- Miller, T.; Krogan, N.J.; Dover, J.; Erdjument-Bromage, H.; Tempst, P.; Johnston, M.; Greenblatt, J.F.; Shilatifard, A. COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci USA 2001, 98, 12902–12907. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.W.; Allis, C.D. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 2002, 418, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Zografidis, A.; Kapolas, G.; Podia, V.; Beri, D.; Papadopoulou, K.K.; Milioni, D.; Haralampidis, K. Transcriptional regulation and functional involvement of the Arabidopsis pescadillo ortholog AtPES in root development. Plant Sci. 2014, 229, 53–65. [Google Scholar] [CrossRef]
- Fiorucci, A.S.; Bourbousse, C.; Concia, L.; Rougée, M.; Deton-Cabanillas, A.F.; Zabulon, G.; Layat, E.; Latrasse, D.; Kim, S.K.; Chaumont, N.; et al. Arabidopsis S2Lb links AtCOMPASS-like and SDG2 activity in H3K4me3 independently from histone H2B monoubiquitination. Genome Biol. 2019, 20, 1–21. [Google Scholar] [CrossRef]
- Narro-Diego, L.; López-González, L.; Jarillo, J.A.; Piñeiro, M. The PHD-containing protein EARLY BOLTING IN SHORT DAYS regulates seed dormancy in Arabidopsis. Plant Cell Environ. 2017, 40, 2393–2405. [Google Scholar] [CrossRef]
- Colville, A.; Alhattab, R.; Hu, M.; Labbé, H.; Xing, T.; Miki, B. Role of HD2genes in seed germination and early seedling growth in Arabidopsis. Plant Cell Rep. 2011, 30, 1969–1979. [Google Scholar] [CrossRef]
- Luo, M.; Wang, Y.Y.; Liu, X.; Yang, S.; Lu, Q.; Cui, Y.; Wu, K. HD2C interacts with HDA6 and is involved in ABA and salt stress response in Arabidopsis. J. Exp. Bot. 2012, 63, 3297–3306. [Google Scholar] [CrossRef]
- Chen, L.T.; Wu, K. Role of histone deacetylases HDA6 and HDA19 in ABA and abiotic stress response. Plant Signal. Behav. 2010, 5, 1318–1320. [Google Scholar] [CrossRef]
- Moon, Y.H.; Chen, L.; Pan, R.L.; Chang, H.S.; Zhu, T.; Maffeo, D.M.; Sung, Z.R. EMFgenes maintain vegetative development by repressing the flower program in Arabidopsis. Plant Cell 2003, 15, 681–693. [Google Scholar] [CrossRef]
- Tang, X.; Lim, M.H.; Pelletier, J.; Tang, M.; Nguyen, V.; Keller, W.A.; Tsang, E.W.T.; Wang, A.; Rothstein, S.J.; Harada, J.J.; et al. Synergistic repression of the embryonic programme by SET DOMAIN GROUP8 and EMBRYONIC FLOWER2 in Arabidopsis seedlings. J. Exp. Bot. 2012, 63, 1391–1404. [Google Scholar] [CrossRef]
- Graeber, K.; Voegele, A.; Büttner-Mainik, A.; Sperber, K.; Mummenhoff, K.; Leubner-Metzger, G. Spatiotemporal seed development analysis provides insight into primary dormancy induction and evolution of the Lepidium delay of germination1 genes. Plant Physiol. 2013, 161, 1903–1917. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Shafiq, S.; Xu, W.; Sun, Q. EARLYFLOWERINGINSHORT DAYS (EFS) regulates the seed size in Arabidopsis. Sci. China Life Sci. 2018, 61, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wei, X.; Sheng, Z.; Jiao, G.; Tang, S.; Luo, J.; Hu, P. Polycomb protein OsFIE2 affects plant height and grain yield in rice. PLoS ONE 2016, 11, e0164748. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Ji, R.; He, C.; Peng, T.; Zhang, M.; Duan, J.; Xiong, C.; Liu, X. Arabidopsis histone methyltransferase SUVH5 is a positive regulator of light-mediated seed germination. Front. Plant. Sci. 2019, 10, 841. [Google Scholar] [CrossRef]
- Cho, J.N.; Ryu, J.Y.; Jeong, Y.M.; Park, J.; Song, J.J.; Amasino, R.M.; Noh, B.; Noh, Y.-S. Control of seed germination by light-induced histone arginine demethylation activity. Dev. Cell 2012, 22, 736–748. [Google Scholar] [CrossRef]
- Chen, H.; Tong, J.; Fu, W.; Liang, Z.; Ruan, J.; Yu, Y.; Song, X.; Yuan, L.; Xiao, L.; Liu, J.; et al. The H3K27me3 demethylase RELATIVE OFE EARLY FLOWERING6 suppressesse eddormancy by inducing abscisic acid catabolism. Plant Physiol. 2020, 184, 1969–1978. [Google Scholar] [CrossRef]
- Singh, D.; Laxmi, A. Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 2015, 6, 895. [Google Scholar] [CrossRef]
- He, M.; He, C.Q.; Ding, N.-Z. Abiotic stress: General defenses of land plants and chances for engineering multistress tolerance. Front. Plant Sci. 2018, 9, 1771. [Google Scholar] [CrossRef]
- Nunez-Vazquez, R.; Desvoyes, B.; Gutierrez, C. Histone variants and modifications during abiotic stress response. Front. Plant 2022, 13, 984702. [Google Scholar] [CrossRef]
- Earley, K.W.; Shook, M.S.; Brower-Toland, B.; Hicks, L.; Pikaard, C.S. In vitro specificities of Arabidopsis co-activator histone acetyltransferases: Implications for histone hyperacetylation in gene activation. Plant J. Cell Mol. Biol. 2007, 52, 615–626. [Google Scholar] [CrossRef]
- Pecinka, A.; Dinh, H.Q.; Baubec, T.; Rosa, M.; Lettner, N.; Scheid, O.M. Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis. Plant Cell 2010, 22, 3118–3129. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Mittelsten Scheid, O. Stress-induced structural changes in plant chromatin. Curr. Opin. Plant Biol. 2015, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lim, C.J.; Shen, M.; Park, H.J.; Cha, J.-Y.; Iniesto, E.; Rubio, V.; Mengiste, T.; Zhu, J.-K.; Bressan, R.A.; et al. Epigenetic switch from repressive to permissive chromatin in response to cold stress. Proc. Natl. Acad. Sci. USA 2018, 115, E5400–E5409. [Google Scholar] [CrossRef] [PubMed]
- Bäurle, I.; Trindade, I. Chromatin regulation of somatic abiotic stress memory. J. Exp. Bot. 2020, 71, 5269–5279. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Liu, B.; Xu, Z.Y. Dynamic regulation of DNA methylation and histone modifications in response to abiotic stresses in plants. J. Integr. Plant Biol. 2022, 64, 2252–2274. [Google Scholar] [CrossRef]
- Jia, H.; Suzuki, M.; McCarty, D.R. Regulation of the seed to seedling developmental phase transition by the LAFL and VAL transcription factor networks. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 135–145. [Google Scholar] [CrossRef]
- Shu, K.; Liu, X.D.; Xie, Q.; He, Z.H. Two faces of one seed: Hormonal regulation of dormancy and germination. Mol. Plant 2016, 9, 34–45. [Google Scholar] [CrossRef]
- Jacobsen, J.V.; Pearce, D.W.; Poole, A.T.; Pharis, R.P.; Mander, L.N. Abscisic acid, phaseic acid and gibberellin contents associated with dormancy and germination in barley. Physiol. Plant 2002, 115, 428–441. [Google Scholar] [CrossRef]
- Chang, G.; Wang, C.; Kong, X.; Chen, Q.; Yang, Y.; Hu, X. AFP2 as the novel regulator breaks high-temperature-induced seeds secondary dormancy through ABI5 and SOM in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2018, 501, 232–238. [Google Scholar] [CrossRef]
- Vaistij, F.E.; Barros-Galvão, T.; Cole, A.F.; Gilday, A.D.; He, Z.; Li, Y.; Harvey, D.; Larson, T.R.; Graham, I.A. MOTHER-OF-FT-AND-TFL1 represses seed germination under far-red light by modulating phytohormone responses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2018, 115, 8442–8447. [Google Scholar] [CrossRef]
- Luo, Q.; Lian, H.L.; He, S.B.; Li, L.; Jia, K.P.; Yang, H.Q. COP1 and phyB physically interact with PIL1 to regulate its stability and photomorphogenic development in Arabidopsis. Plant Cell 2014, 26, 2441–2456. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Chen, C.Y.; Zhao, M.; Zhao, L.; Duan, X.; Duan, J.; Wu, K.; Liu, X. Identification of HDA15-PIF1 as a key repression module directing the transcriptional network of seed germination in the dark. Nucleic. Acids Res. 2017, 45, 7137–7150. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, Q.; He, D.; Zhou, Y.; Ni, H.; Tian, D.; Chang, G.; Jing, Y.; Lin, R.; Huang, J.; et al. AGAMOUSLIKE67 cooperates with the histone mark reader EBS to modulate seed germination under high temperature. Plant Physiol. 2020, 184, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, Z.; Huang, Y.; Chang, G.; Li, P.; Wei, J.; Yuan, X.; Huang, J.; Hu, X. Powerdress as the novel regulator enhances Arabidopsis seeds germination tolerance to high temperature stress by histone modification of SOM locus. Plant Sci. 2019, 284, 91–98. [Google Scholar] [CrossRef]
- Perella, G.; Lopez-Vernaza, M.A.; Carr, C.; Sani, E.; Gosselé, V.; Verduyn, C.; Kellermeier, F.; Hannah, M.A.; Amtmann, A. Histone deacetylase complex1 expression level titrates plant growth and abscisic acid sensitivity in Arabidopsis. Plant Cell 2013, 25, 3491–3505. [Google Scholar] [CrossRef]
- Chalot, M.; Puschenreiter, M. Exploring Plant Rhizosphere, Phyllosphere and Endosphere Microbial Communities to Improve the Management of Polluted Sites. Front. Microbiol. 2021, 12, 763566. [Google Scholar] [CrossRef]
- Jacoby, R.P.; Chen, L.; Schwier, M.; Koprivova, A.; Kopriva, S. Recent advances in the role of plant metabolites in shaping the root microbiome. F1000Research 2020, 9, F1000 Faculty Rev-151. [Google Scholar] [CrossRef]
- Rizaludin, M.S.; Stopnisek, N.; Raaijmakers, J.M.; Garbeva, P. The chemistry of stress: Understanding the ‘cry for Help’ of plant roots. Metabolites 2021, 11, 357. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Monohon, S.J.; Manter, D.K.; Vivanco, J.M. Conditioned soils reveal plant-selected microbial communities that impact plant drought response. Sci. Rep. 2021, 11, 21153. [Google Scholar] [CrossRef]
- Mönchgesang, S.; Strehmel, N.; Schmidt, S.; Westphal, L.; Taruttis, F.; Müller, E.; Herklotz, S.; Neumann, S.; Scheel, D. Natural variation of root exudates in Arabidopsis thaliana-linking metabolomic and genomic data. Sci. Rep. 2016, 6, 29033. [Google Scholar] [CrossRef] [PubMed]
- Tracanna, V.; Ossowicki, A.; Petrus, M.L.C.; Overdium, S.; Telouw, B.R.; Lund, G.; Robinson, S.L.; Warris, S.; Schijlien, E.G.; Van Wezel, G.P.; et al. Dissecting disease-suppressive rhizosphere microbiomes by functional amplicon sequencing and 10× metagenomics. mSystems 2021, 6, e01116-20. [Google Scholar] [CrossRef] [PubMed]
- Kaleh, A.M.; Singh, P.; Ooi Chua, K.; Harikrishna, J.A. Modulation of plant transcription factors and priming of stress tolerance by plant growth-promoting bacteria: A systematic review. Ann. Bot. 2025, 135, 387–402. [Google Scholar] [CrossRef]
- Glick, B.R. Plant growth-promoting bacteria: Mechanisms and applications. Scientifica 2012, 2012, 963401. [Google Scholar] [CrossRef]
- Chen, C.; Wang, M.; Zhu, J.; Tang, Y.; Zhang, H.; Zhao, Q.; Jing, M.; Chen, Y.; Xu, X.; Jiang, J.; et al. Long-term effect of epigenetic modification in plant–microbe interactions: Modification of DNA methylation induced by plant growth-promoting bacteria mediates promotion process. Microbiome 2022, 10, 36. [Google Scholar] [CrossRef]
- Martínez-Medina, A.; Flors, V.; Heil, M.; Mauch-Mani, B.; Pieterse, C.M.J.; Pozo, M.J.; Ton, J.; van Dam, N.M.; Conrath, U. Recognizing plant defense priming. Trends Plant Sci. 2016, 21, 818–822. [Google Scholar] [CrossRef]
- Pieterse, C.M.; Zamioudis, C.; Berendsen, L.R.; Weller, D.M.; van Wees, S.C.; Bakker, P.A. Induced systemic resistance by beneficial microbes. Annu. Rev. Phytopathol. 2014, 52, 347–375. [Google Scholar] [CrossRef]
- Alwutayd, K.M.; Rawat, A.A.; Sheikh, A.H.; Bhat, J.A.; Elango, D.; Alsubaie, O.; Alzubaidy, H.; Jalal, R.; Saad, M.M.; Hirt, H. Microbe-induced drought tolerance by ABA-mediated root architecture and epigenetic reprogramming. EMBO Rep. 2023, 24, e56754. [Google Scholar] [CrossRef]
- Siddika, A.; Rashid, A.A.; Khan, S.N.; Khatun, A.; Karim, M.M.; Prasad, P.V.V.; Hasanuzzaman, M. Harnessing plant growth-promoting rhizobacteria, Bacillus subtilis and Bacillus aryabhattai to combat salt stress in rice: Regulation of antioxidant defense, ion homeostasis, and photosynthetic parameters. Front. Plant Sci. 2024, 15, 1419764. [Google Scholar] [CrossRef]
- Fukami, J.; Cerezini, P.; Hungria, M. Azospirillum: Benefits that go far beyond biological nitrogen fixation. AMB Expr. 2018, 8, 73. [Google Scholar] [CrossRef]
- Satgé, C.; Moreau, S.; Sallet, E.; Lefort, G.; Auriac, M.C.; Remblière, C.; Cottret, L.; Gallardo, K.; Noirot, C.; Jardinaud, M.F.; et al. Reprogramming of DNA methylation is critical for nodule development in Medicago truncatula. Nat. Plants 2016, 2, 16166. [Google Scholar] [CrossRef]
- Vílchez, J.I.; Varotto, S.; Jung, H.W. Editorial: Epigenetic regulation behind plant-microbe interactions. Front. Plant Sci. 2024, 15, 1385356. [Google Scholar] [CrossRef]
- Li, R.; Ma, X.Y.; Zhang, Y.J.; Zhu, H.; Shao, S.N.; Zhang, D.D.; Klosterman, S.J.; Dai, X.F.; Subbarao, K.V.; Chen, J.Y. Genome-wide identification and analysis of a cotton secretome reveals its role in resistance against Verticillium dahliae. BMC Biol. 2023, 21, 166. [Google Scholar] [CrossRef]
- Romero-Rodríguez, B.; Petek, M.; Jiao, C.; Križnik, M.; Zagorščak, M.; Fei, Z.; Bejarano, E.R.; Gruden, K.; Castillo, A.G. Transcriptional and epigenetic changes during tomato yellow leaf curl virus infection in tomato. BMC Plant Biol. 2023, 23, 651. [Google Scholar] [CrossRef]
- Kuźnicki, D.; Meller, B.; Arasimowicz-Jelonek, M.; Braszewska-Zalewska, A.; Drozda, A.; Floryszak-Wieczorek, J. BABA-Induced DNA Methylome Adjustment to Intergenerational Defense Priming in Potato to Phytophthora infestans. Front. Plant Sci. 2019, 10, 650. [Google Scholar] [CrossRef]
- Hewezi, T. Epigenetic mechanisms in nematode-plant interactions. Annu. Rev. Phytopathol. 2017, 55, 199–220. [Google Scholar] [CrossRef]
- Lai, Y.; Eulgem, T. Transcript-level expression control of plant NLR genes. Mol. Plant Pathol. 2018, 19, 1267–1281. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Jin, H. Coordinated Epigenetic Regulation in Plants: A Potent Managerial Tool to Conquer Biotic Stress. Front. Plant Sci. 2022, 12, 795274. [Google Scholar] [CrossRef]
- De La Peña, C.; Rangel-Cano, A.; Alvarez-Venegas, R. Regulation of disease-responsive genes mediated by epigenetic factors: Interaction of Arabidopsis–Pseudomonas. Mol. Plant Pathol. 2012, 13, 388–398. [Google Scholar] [CrossRef]
- Lopez, S.A.; Stassen, J.H.Μ.; Furci, L.; Smith, L.M.; Ton, J. The role of DNA (de)methylation in immune responsiveness of Arabidopsis. Plant J. 2016, 88, 361–374. [Google Scholar] [CrossRef]
- Xia, S.; Cheng, Y.T.; Huang, S.; Wi, J.; Soards, A.; Jinn, T.L.; Jones, J.D.; Kamoun, S.; Chen, S.; Zhang, Y.; et al. Regulation of transcription of nucleotide-binding leucine-rich repeat-encoding genes SNC1 and RPP4 via H3K4 trimethylation. Plant Physiol. 2013, 162, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Cambiagno, D.A.; Torres, J.R.; Alvarez, M.E. Convergent epigenetic mechanisms avoid constitutive expression of immune receptor gene subsets. Front. Plant Sci. 2021, 12, 703667. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Zimmerli, L. The histone demethylase IBM1 positively regulates Arabidopsis immunity by control of defense gene expression. Front. Plant Sci. 2019, 10, 1587. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Fu, F.; Xu, S.; Lee, S.Y.; Yun, D.-J.; Mengiste, T. Global Regulation of Plant Immunity by Histone Lysine Methyl Transferases. Plant Cell 2016, 28, 1640–1661. [Google Scholar] [CrossRef]
- Moran-Diez, M.E.; Martinez de Alba, A.E.; Rubio, M.B.; Hermosa, R.; Monte, E. Trichoderma and the plant heritable priming responses. J. Fungi 2021, 7, 318. [Google Scholar] [CrossRef]

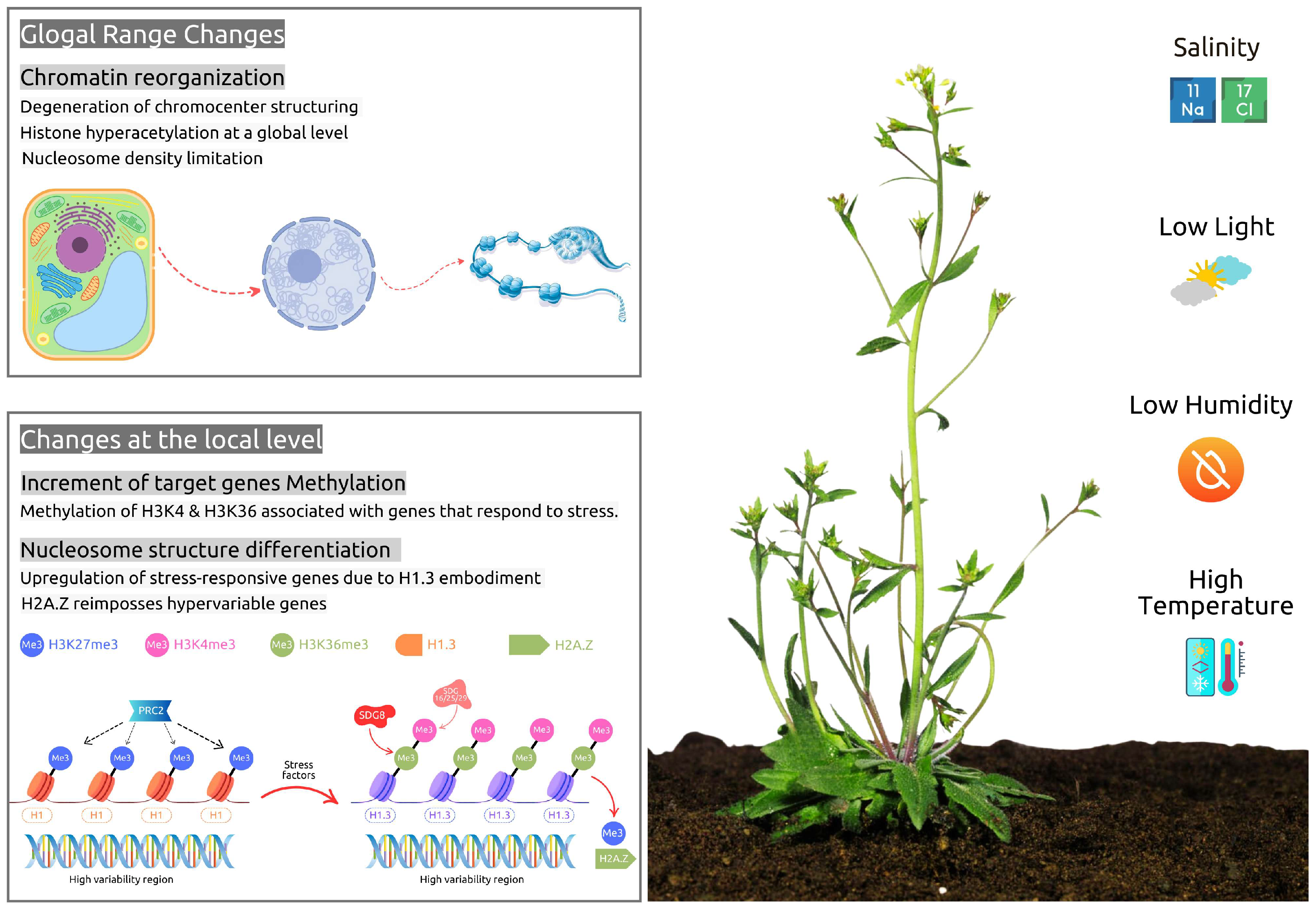


{kind=link}
{kind=link}
{kind=link}
| Targeted Genomic Region | Specific Function | References |
|---|---|---|
| Gene regulatory regions (promoters and enhancers) | Transcriptional gene silencing (TGS) | [35] |
| Intronic TEs and DNA repeats | Alternative mRNA splicing and alternative mRNA polyadenylation | [13] |
| Euchromatin in regions read by SUVH1 and SUVH3 methyl-readers | Proximal gene expression activation of ROS1 and DNAJ1 and 2 | [13,15,39] |
| In GC-rich areas of gene exonic regions | Suppresses intragenic antisense transcripts | [48] |
| Methylation | Readers | Writers | Erasers | Biological Role |
|---|---|---|---|---|
| H3K9me1 and 2 | ADCP1/AGDP1 | KRYPTONITE (KYP)/SUVH4, SUVH5, and SUVH6 | JmiC | Repressive state of heterochromatin, transposon silencing |
| H3K27me1 | ARABIDOPSIS TRITHORAX-RELATED PROTEIN 5 (ATXR5) and ATXR6 | JmiC | Heterochromatin active state | |
| H3K27me3 | PRC1, EBS, and SHL | PRC2 | JmiC group (REF6, ELF6, JMJ13) | Repressive state of euchromatin |
| H3K4me1/2/3 | COMPASS-1 | LSD1, JmiC group | Euchromatin activation | |
| H3K36me3 | SDG8 and SDG26 | Gene activation |
| Histone | Residue | Type of Modification | References |
|---|---|---|---|
| H3 | LYS9 (K9) | Me1 and me2 | [51] |
| H3 | LYS27 | Me1 and me3 | [56,59,62] |
| H3 | LYS4 | Me1,2,3 | [51,62] |
| H3 | LYS36 | Me3 | [62] |
| H3 | LYS9,14,23,27 | Acetylation | [50,65] |
| H3 | SER10 and SER28, THR3 and THR11 | Phosphorylation | [70] |
| H4 | LYS5,8,12,16 | Acetylation | [65] |
| H2A | SER95 | Phosphorylation | [68] |
| H2A | LYS48,63 and MET1 | Monoubiquitination | [24] |
| H2A | LYS48,63 | Polyubiquitination | [24] |
| H2B | LYS6,11,27,29,33 | Monoubiquitination | [71] |
| Gene | Activity | Functional Outcome | References |
|---|---|---|---|
| HD2A | Deacetylase | Regulates seed development and germination | [129] |
| SD2C | Deacetylase | Regulates seed germination | [130] |
| HDA6 | Deacetylase | Regulates seed germination and establishment | [130,131] |
| EMF 1/2 | Embryonic flowering reduces H3K27me3 methylation | Seed enlargement | [109,132] |
| Repression of maturation following germination | [13,133] | ||
| DOG1 | Delay of dormancy gene 1 | Temperature detector for dormancy state | [115,134] |
| EFS | Early flowering in short days, H3K36 writer | Regulator of seed germination and seedling establishment | [135] |
| CLF | Curly leaf gene, PRC2 component, H3K27me3 writer | Involved in regulation of seed dormancy, development, and seedling establishment | [136] |
| SUVH 4,5 | HeK9me2 writers | Regulates seed dormancy and seed germination | [137] |
| ATX1 | Arabidopsis trithorax related H3K4 reader | Counteracts CLF repression during seedling establishment | [109] |
| JMJ family | Histone demethylases | Enhances ABA response, positive regulation of seed germination | [109,138] |
| REF6 | Relative of early flowering 6, H3K27me3 demethylases | Decreases rate of seed dormancy | [139] |
| HUB1,2 | Histone monoubiquitination | Activates DOG1 and seed dormancy | [109] |
| FIS2 | Fertilization-independent seed complexes with PRC2 | Enhance seed development | [133] |
| EBS | Early bolting in short days, H3K4me2/3 writer, interacts with HDAC | Increases seed dormancy and decreases seed generation | [109,128] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tresas, T.; Isaioglou, I.; Roussis, A.; Haralampidis, K. A Brief Overview of the Epigenetic Regulatory Mechanisms in Plants. Int. J. Mol. Sci. 2025, 26, 4700. https://doi.org/10.3390/ijms26104700
Tresas T, Isaioglou I, Roussis A, Haralampidis K. A Brief Overview of the Epigenetic Regulatory Mechanisms in Plants. International Journal of Molecular Sciences. 2025; 26(10):4700. https://doi.org/10.3390/ijms26104700
Chicago/Turabian StyleTresas, Theodoros, Ioannis Isaioglou, Andreas Roussis, and Kosmas Haralampidis. 2025. "A Brief Overview of the Epigenetic Regulatory Mechanisms in Plants" International Journal of Molecular Sciences 26, no. 10: 4700. https://doi.org/10.3390/ijms26104700
APA StyleTresas, T., Isaioglou, I., Roussis, A., & Haralampidis, K. (2025). A Brief Overview of the Epigenetic Regulatory Mechanisms in Plants. International Journal of Molecular Sciences, 26(10), 4700. https://doi.org/10.3390/ijms26104700