
Clinical and Biochemical Characterization of Hereditary ATTR Amyloidosis Caused by a Novel Transthyretin Variant V121A (p.V141A)

 , , , , and
, , , , and
Abstract
1. Introduction
2. Patients
3. Results
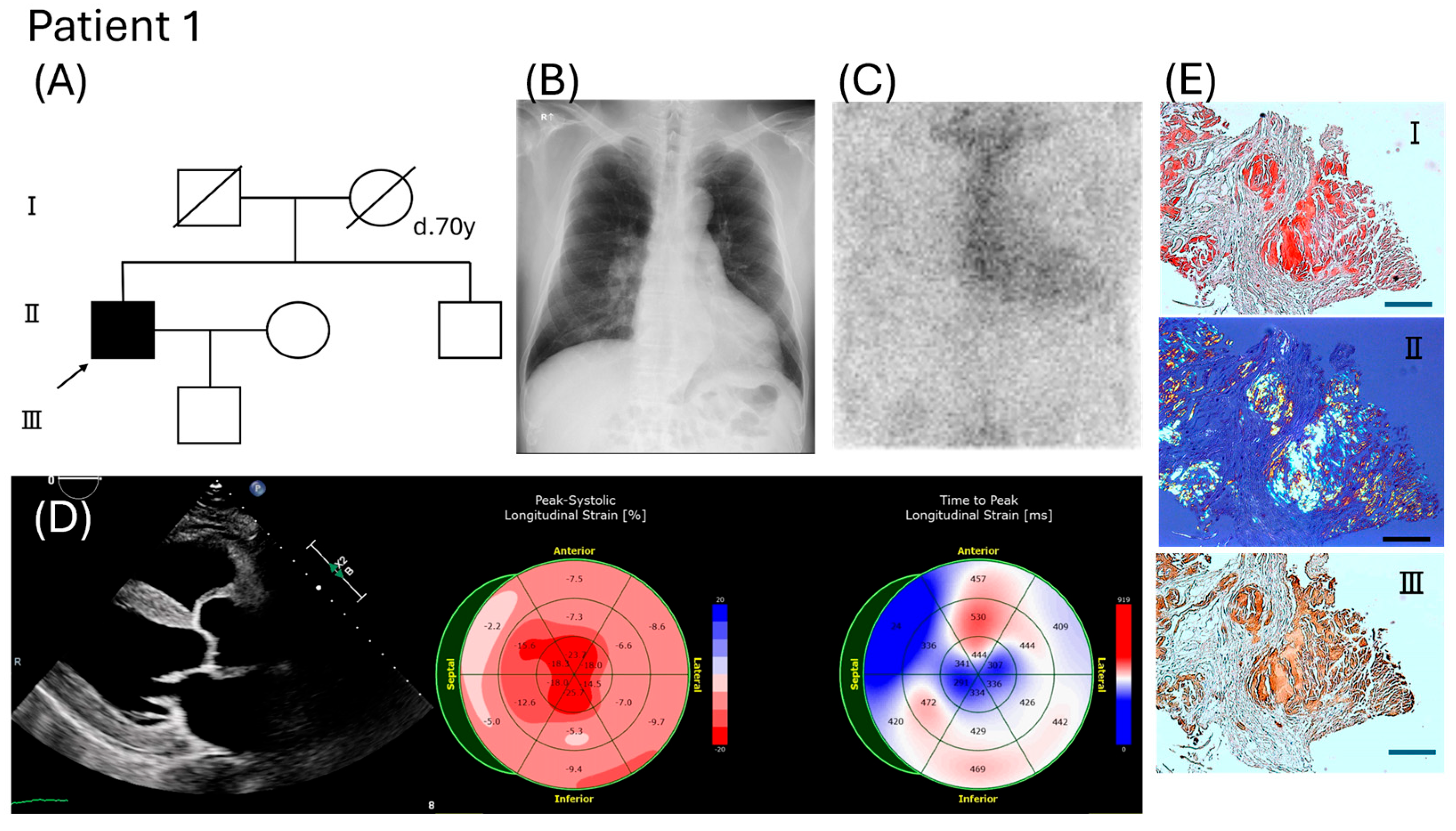
3.1. Clinical Features of Two Patients
3.2. Evaluation of Pathogenicity Associated with This Novel TTR Variant
3.2.1. In Silico Analysis of V121A
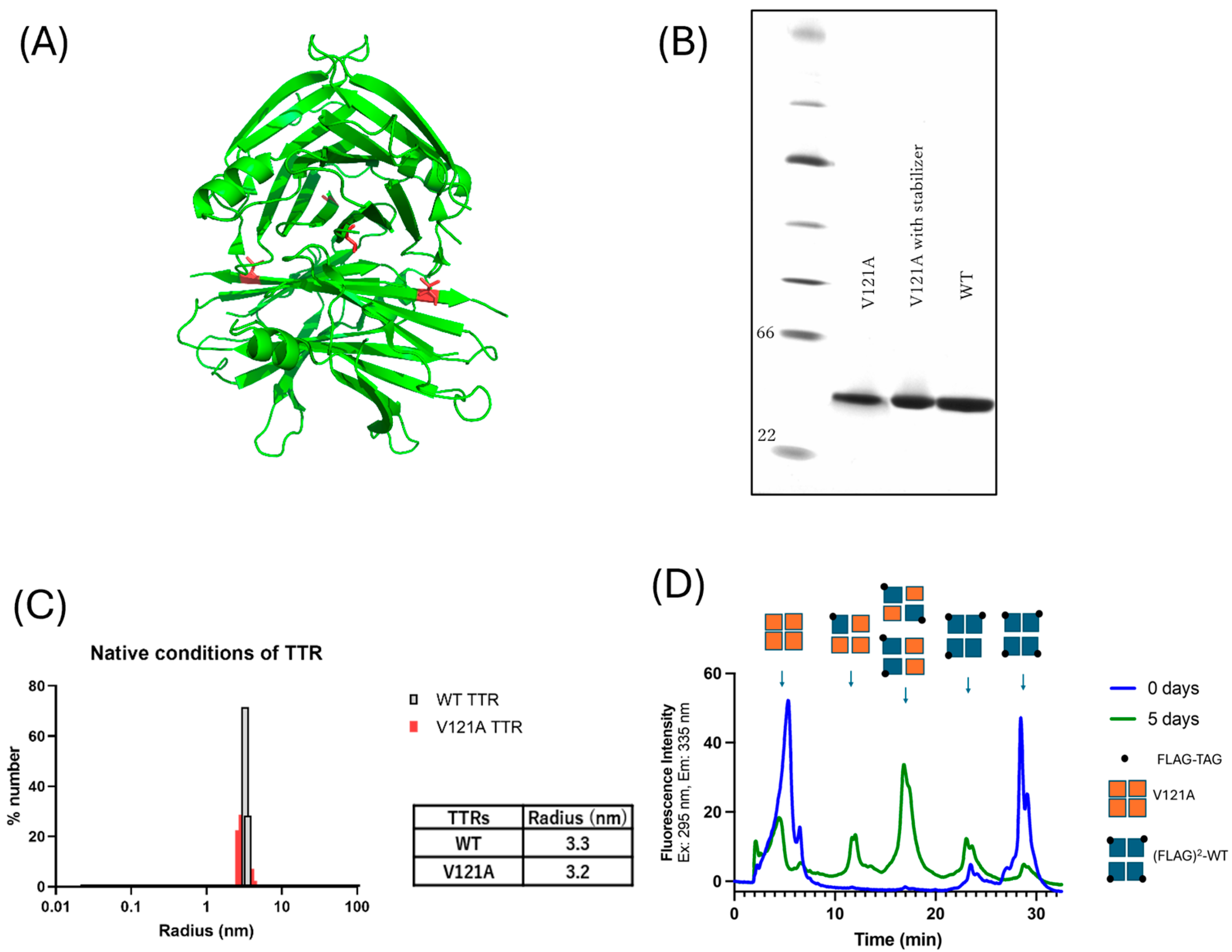
3.2.2. Analyses of Tetramer Formation Ability in the V121A(p.V141A) Mutation
3.2.3. V121A (p.V141A) Aggregates Under Acidic Conditions Despite the Presence of Kinetic Stabilizers
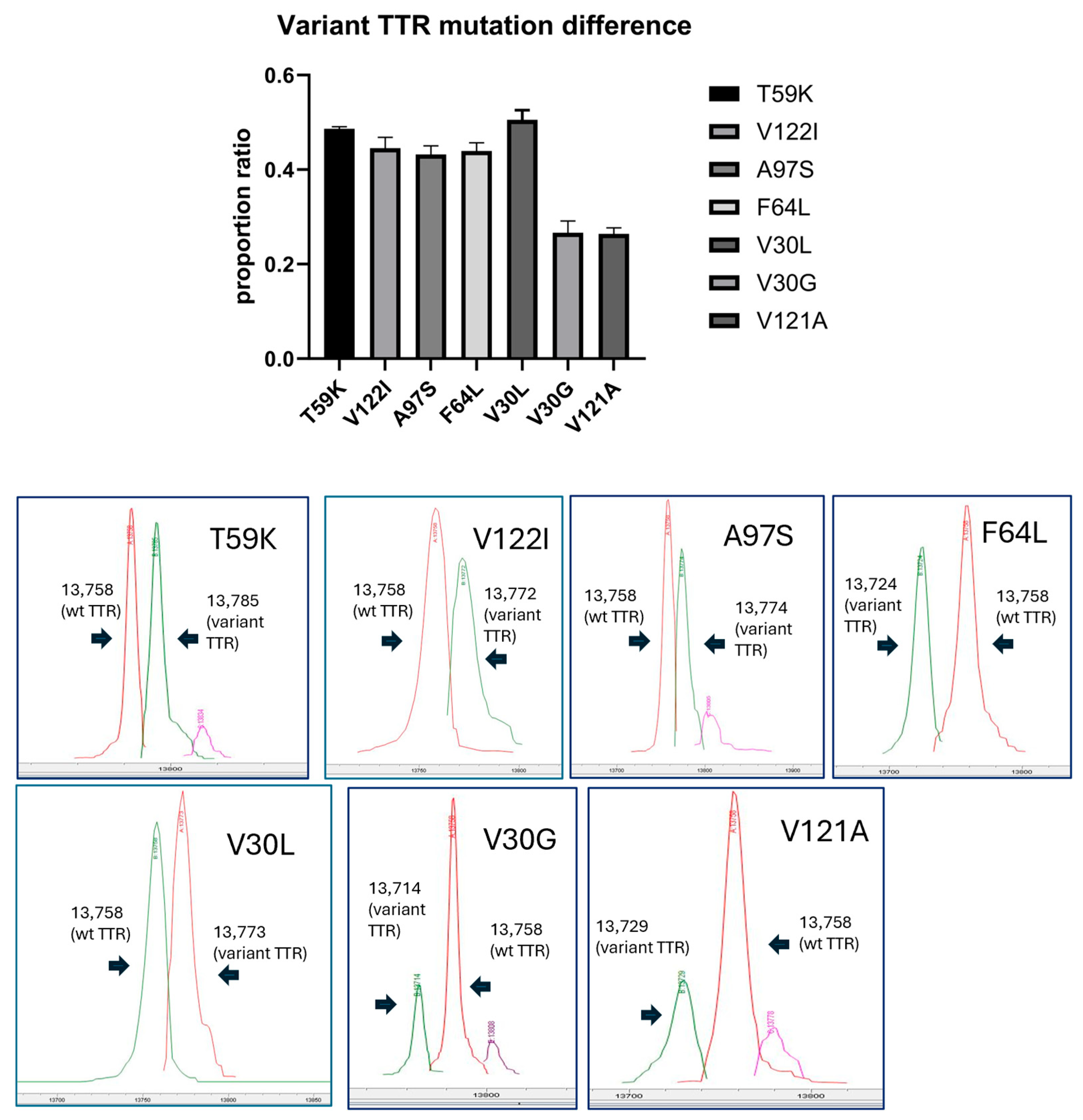
3.2.4. Proportion Ratio of TTRs in Serum Variant Mutants Including V121A (p.V141A)
4. Discussion
5. Methods
5.1. Evaluation of Pathogenicity Associated with This Novel TTR Variant
5.1.1. Survey in Public Databases and Genetic Analysis In Silico
5.1.2. 3D Structural Prediction
5.1.3. Expression and Purification of Recombinant TTR Protein
5.1.4. Non-Denatured Electrophoresis (Native PAGE)
5.1.5. Measurement of Particle Size (Dynamic Light Scattering)
5.1.6. Endpoint of Subunit Exchange Assay
5.1.7. TTR Aggregation Measured by Turbidity
5.1.8. Evaluation of TTR Aggregation with Known TTR Stabilizers
5.1.9. Isothermal Titration Calorimetry
5.1.10. Proportion of Variant TTR to Wild-Type TTR in Serum
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TTR | Transthyretin |
| ATTRv amyloidosis | Hereditary transthyretin amyloidosis |
| ATTRwt amyloidosis | Wild-type transthyretin amyloidosis |
| WT | Wild-type |
| 99mTc-PYP | 99mTechnetium-pyrophosphate |
References
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Eisenberg, D.S.; Fändrich, M.; McPhail, E.D.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Westermark, P. Amyloid nomenclature 2024: Update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2024, 31, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.; Geisow, M.J.; Oatley, S.J.; Rérat, B.; Rérat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J. Mol. Biol. 1978, 121, 339–356. [Google Scholar] [CrossRef] [PubMed]
- White, J.T.; Kelly, J.W. Support for the multigenic hypothesis of amyloidosis: The binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 13019–13024. [Google Scholar] [CrossRef]
- Arvanitis, M.; Koch, C.M.; Chan, G.G.; Torres-Arancivia, C.; LaValley, M.P.; Jacobson, D.R.; Berk, J.L.; Connors, L.H.; Ruberg, F.L. Identification of Transthyretin Cardiac Amyloidosis Using Serum Retinol-Binding Protein 4 and a Clinical Prediction Model. JAMA Cardiol. 2017, 2, 305–313. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Monteiro, C.; Fearns, C.; Encalada, S.E.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 759–780. [Google Scholar] [CrossRef]
- Rappley, I.; Monteiro, C.; Novais, M.; Baranczak, A.; Solis, G.; Wiseman, R.L.; Helmke, S.; Maurer, M.S.; Coelho, T.; Powers, E.T.; et al. Quantification of transthyretin kinetic stability in human plasma using subunit exchange. Biochemistry 2014, 53, 1993–2006. [Google Scholar] [CrossRef]
- Nelson, L.T.; Paxman, R.J.; Xu, J.; Webb, B.; Powers, E.T.; Kelly, J.W. Blinded potency comparison of transthyretin kinetic stabilisers by subunit exchange in human plasma. Amyloid 2021, 28, 24–29. [Google Scholar] [CrossRef]
- Ueda, M.; Horibata, Y.; Shono, M.; Misumi, Y.; Oshima, T.; Su, Y.; Tasaki, M.; Shinriki, S.; Kawahara, S.; Jono, H.; et al. Clinicopathological features of senile systemic amyloidosis: An ante- and post-mortem study. Mod. Pathol. 2011, 24, 1533–1544. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA J. Am. Med. Assoc. 2013, 310, 2658–2667. [Google Scholar] [CrossRef]
- Coelho, T.; Maia, L.F.; Martins da Silva, A.; Waddington Cruz, M.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceicao, I.M.; Schmidt, H.H.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Sant’Anna, R.; Gallego, P.; Robinson, L.Z.; Pereira-Henriques, A.; Ferreira, N.; Pinheiro, F.; Esperante, S.; Pallares, I.; Huertas, O.; Almeida, M.R.; et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun. 2016, 7, 10787. [Google Scholar] [CrossRef] [PubMed]
- Penchala, S.C.; Connelly, S.; Wang, Y.; Park, M.S.; Zhao, L.; Baranczak, A.; Rappley, I.; Vogel, H.; Liedtke, M.; Witteles, R.M.; et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. USA 2013, 110, 9992–9997. [Google Scholar] [CrossRef]
- Hammarström, P.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 2003, 299, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. New Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Planté-Bordeneuve, V.; Berk, J.L.; González-Duarte, A.; Gillmore, J.D.; Low, S.C.; Sekijima, Y.; et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: A randomized clinical trial. Amyloid 2023, 30, 18–26. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. New Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Coelho, T.; Marques, W.; Dasgupta, N.R., Jr.; Chao, C.C.; Parman, Y.; França, M.C.; Guo, Y.C.; Wixner, J.; Ro, L.S.; Calandra, C.R.; et al. Eplontersen for Hereditary Transthyretin Amyloidosis with Polyneuropathy. JAMA J. Am. Med. Assoc. 2023, 330, 1448–1458. [Google Scholar] [CrossRef]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef]
- Nomura, T.; Ueda, M.; Tasaki, M.; Misumi, Y.; Masuda, T.; Inoue, Y.; Tsuda, Y.; Okada, M.; Okazaki, T.; Kanenawa, K.; et al. New simple and quick method to analyze serum variant transthyretins: Direct MALDI method for the screening of hereditary transthyretin amyloidosis. Orphanet J. Rare Dis. 2019, 14, 116. [Google Scholar] [CrossRef]
- Hörnberg, A.; Eneqvist, T.; Olofsson, A.; Lundgren, E.; Sauer-Eriksson, A.E. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J. Mol. Biol. 2000, 302, 649–669. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.R.; Tan, S.Y.; Hawkins, P.N.; Pepys, M.B.; Frustaci, A. A novel variant of transthyretin, 59Thr→Lys, associated with autosomal dominant cardiac amyloidosis in an Italian family. Circulation 1995, 91, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Maurer, M.S.; Judge, D.P.; Zeldenrust, S.; Skinner, M.; Kim, A.Y.; Falk, R.H.; Cheung, K.N.; Patel, A.R.; Pano, A.; et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS). Am. Heart J. 2012, 164, 222–228.e1. [Google Scholar] [CrossRef]
- Tachibana, N.; Tokuda, T.; Yoshida, K.; Taketomi, T.; Nakazato, M.; Li, Y.F.; Masuda, Y.; Ikeda, S.I. Usefulness of MALDI/TOF mass spectrometry of immunoprecipitated serum variant transthyretin in the diagnosis of familial amyloid polyneuropathy. Amyloid 1999, 6, 282–288. [Google Scholar] [CrossRef]
- Ii, S.; Minnerath, S.; Ii, K.; Dyck, P.J.; Sommer, S.S. Two-tiered DNA-based diagnosis of transthyretin amyloidosis reveals two novel point mutations. Neurology 1991, 41, 893–898. [Google Scholar] [CrossRef]
- Murakami, T.; Atsumi, T.; Maeda, S.; Tanase, S.; Ishikawa, K.; Mita, S.; Kumamoto, T.; Araki, S.; Ando, M. A novel transthyretin mutation at position 30 (Leu for Val) associated with familial amyloidotic polyneuropathy. Biochem. Biophys. Res. Commun. 1992, 187, 397–403. [Google Scholar] [CrossRef]
- Petersen, R.B.; Goren, H.; Cohen, M.; Richardson, S.L.; Tresser, N.; Lynn, A.; Gali, M.; Estes, M.; Gambetti, P. Transthyretin amyloidosis: A new mutation associated with dementia. Ann. Neurol. 1997, 41, 307–313. [Google Scholar] [CrossRef]
- Jacobson, D.R.; Pastore, R.D.; Yaghoubian, R.; Kane, I.; Gallo, G.; Buck, F.S.; Buxbaum, J.N. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. New Engl. J. Med. 1997, 336, 466–473. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef]
- Falk, R.H. Diagnosis and management of the cardiac amyloidoses. Circulation 2005, 112, 2047–2060. [Google Scholar] [CrossRef]
- Sekijima, Y.; Hammarström, P.; Matsumura, M.; Shimizu, Y.; Iwata, M.; Tokuda, T.; Ikeda, S.I.; Kelly, J.W. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab. Investig. 2003, 83, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Klimtchuk, E.S.; Prokaeva, T.; Frame, N.M.; Abdullahi, H.A.; Spencer, B.; Dasari, S.; Cui, H.; Berk, J.L.; Kurtin, P.J.; Connors, L.H.; et al. Unusual duplication mutation in a surface loop of human transthyretin leads to an aggressive drug-resistant amyloid disease. Proc. Natl. Acad. Sci. USA 2018, 115, E6428–E6436. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Misumi, Y.; Mizuguchi, M.; Nakamura, M.; Yamashita, T.; Sekijima, Y.; Ota, K.; Shinriki, S.; Jono, H.; Ikeda, S.I.; et al. SELDI-TOF mass spectrometry evaluation of variant transthyretins for diagnosis and pathogenesis of familial amyloidotic polyneuropathy. Clin. Chem. 2009, 55, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Aono, Y.; Hamatani, Y.; Katoh, N.; Nakagawa, M.; Nakamura, K.; Yazaki, M.; Kametani, F.; Iguchi, M.; Murakami, I.; Ogawa, H.; et al. Late-onset Hereditary ATTR Amyloidosis with a Novel p.P63S (P43S) Transthyretin Variant. Intern. Med. 2021, 60, 557–561. [Google Scholar] [CrossRef]
- Choi, S.; Ong, D.S.; Kelly, J.W. A stilbene that binds selectively to transthyretin in cells and remains dark until it undergoes a chemoselective reaction to create a bright blue fluorescent conjugate. J. Am. Chem. Soc. 2010, 132, 16043–16051. [Google Scholar] [CrossRef]
- Heegaard, N.H.; Hansen, M.Z.; Sen, J.W.; Christiansen, M.; Westermark, P. Immunoaffinity chromatographic and immunoprecipitation methods combined with mass spectrometry for characterization of circulating transthyretin. J. Sep. Sci. 2006, 29, 371–377. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | ||
|---|---|---|
| 1 | 2 | |
| Age of diagnosis (sex) | 67 (M) | 71 (M) |
| Age of onset | 62 | 70 |
| Ethnicity | Japanese | Japanese |
| Positive family history | none | none |
| Cardiac dysfunction | (+) | (+) |
| NYHA class | II | III |
| BNP (pg/mL) (<18.4 pg/mL) | 92.8 | NA |
| NT-proBNP (pg/mL) (<125 pg/mL) | 1,183 | 10,988 |
| Hs-cTnT (ng/mL) (<0.014 ng/ml) | 0.033 | 0.207 |
| ECG | ||
| Low QRS voltage | (+) | (+) |
| Pseudoinfarct pattern | (+) | (+) |
| Echocardiography | ||
| LVEF (%) (>55%) | 39 | 53 |
| LAD (mm) (19–40 mm) | 44 | 59 |
| LVDd (mm) (39–55 mm) | 57 | NA |
| LVDs (mm) (22–42 mm) | 46 | NA |
| LVPWTd (mm) (8–12 mm) | 12.5 | 16 |
| IVSd (mm) (<12 mm) | 13 | 20 |
| Increased RV wall thickness | (+) | (−) |
| Restrictive LV filling pattern | (+) | (+) |
| Valvular abnormalities | (−) | (−) |
| Pericardial effusion | (−) | (−) |
| Autonomic dysfunction | (−) | (−) |
| Orthostatic hypotension | (−) | (−) |
| Gastrointestinal manifestations | (−) | (−) |
| Loss of weight | (−) | (−) |
| Polyneuropathy | (−) | (−) |
| CNS manifestations | (−) | (−) |
| Small fiber neuropathy | (+) * | (−) |
| Carpal tunnel syndrome | (−) | (−) |
| Biceps tendon rapture | (−) | (−) |
| Spinal canal stenosis | (+) * | (−) |
| Ocular manifestations | (−) | (−) |
| Chronic kidney disease | (−) | (+) |
| TTR concentration (22−40 mg/dL) | 4 mg/dL | 10 mg/dL |
| Proportion ratio variant TTR: wild TTRof amyloid in cardiac specimens | 50% | variant TTR peptide detected ** |
| Outcome | Survival | Death |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshinaga, T.; Yoshioka, Y.; Tsai, F.J.; Nelson, L.; Cheng, M.; Ito, R.; Fujita, S.; Ishikawa, E.; Kametani, F.; Aoyagi, R.; et al. Clinical and Biochemical Characterization of Hereditary ATTR Amyloidosis Caused by a Novel Transthyretin Variant V121A (p.V141A). Int. J. Mol. Sci. 2025, 26, 4659. https://doi.org/10.3390/ijms26104659
Yoshinaga T, Yoshioka Y, Tsai FJ, Nelson L, Cheng M, Ito R, Fujita S, Ishikawa E, Kametani F, Aoyagi R, et al. Clinical and Biochemical Characterization of Hereditary ATTR Amyloidosis Caused by a Novel Transthyretin Variant V121A (p.V141A). International Journal of Molecular Sciences. 2025; 26(10):4659. https://doi.org/10.3390/ijms26104659
Chicago/Turabian StyleYoshinaga, Tsuneaki, Yuuki Yoshioka, Felix J. Tsai, Luke Nelson, Ming Cheng, Ryota Ito, Satoshi Fujita, Eri Ishikawa, Fuyuki Kametani, Ryuzi Aoyagi, and et al. 2025. "Clinical and Biochemical Characterization of Hereditary ATTR Amyloidosis Caused by a Novel Transthyretin Variant V121A (p.V141A)" International Journal of Molecular Sciences 26, no. 10: 4659. https://doi.org/10.3390/ijms26104659
APA StyleYoshinaga, T., Yoshioka, Y., Tsai, F. J., Nelson, L., Cheng, M., Ito, R., Fujita, S., Ishikawa, E., Kametani, F., Aoyagi, R., Okumura, T., Murohara, T., Yazaki, M., & Sekijima, Y. (2025). Clinical and Biochemical Characterization of Hereditary ATTR Amyloidosis Caused by a Novel Transthyretin Variant V121A (p.V141A). International Journal of Molecular Sciences, 26(10), 4659. https://doi.org/10.3390/ijms26104659