


Melatonin Improves Lipid Homeostasis, Mitochondrial Biogenesis, and Antioxidant Defenses in the Liver of Prediabetic Rats
 , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Results
2.1. Sucrose Solution Consumption Alters Daily Feed Intake in Prediabetic Wistar Rats
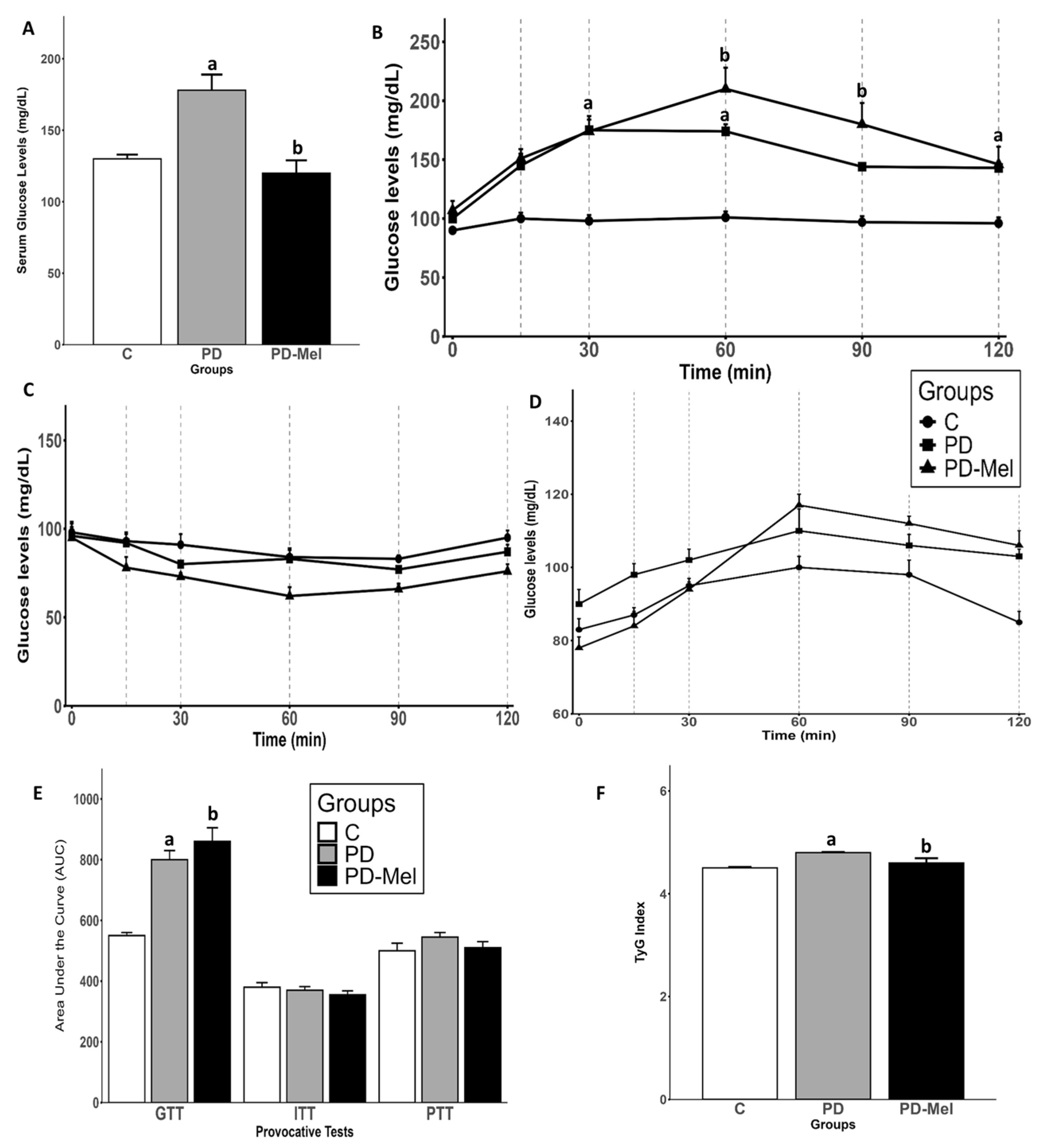
2.2. Melatonin Reduces Serum Glucose but Does Not Improve Glucose Intolerance
2.3. Melatonin Reduces Fat Deposits and Influences Serum Lipid Profiles
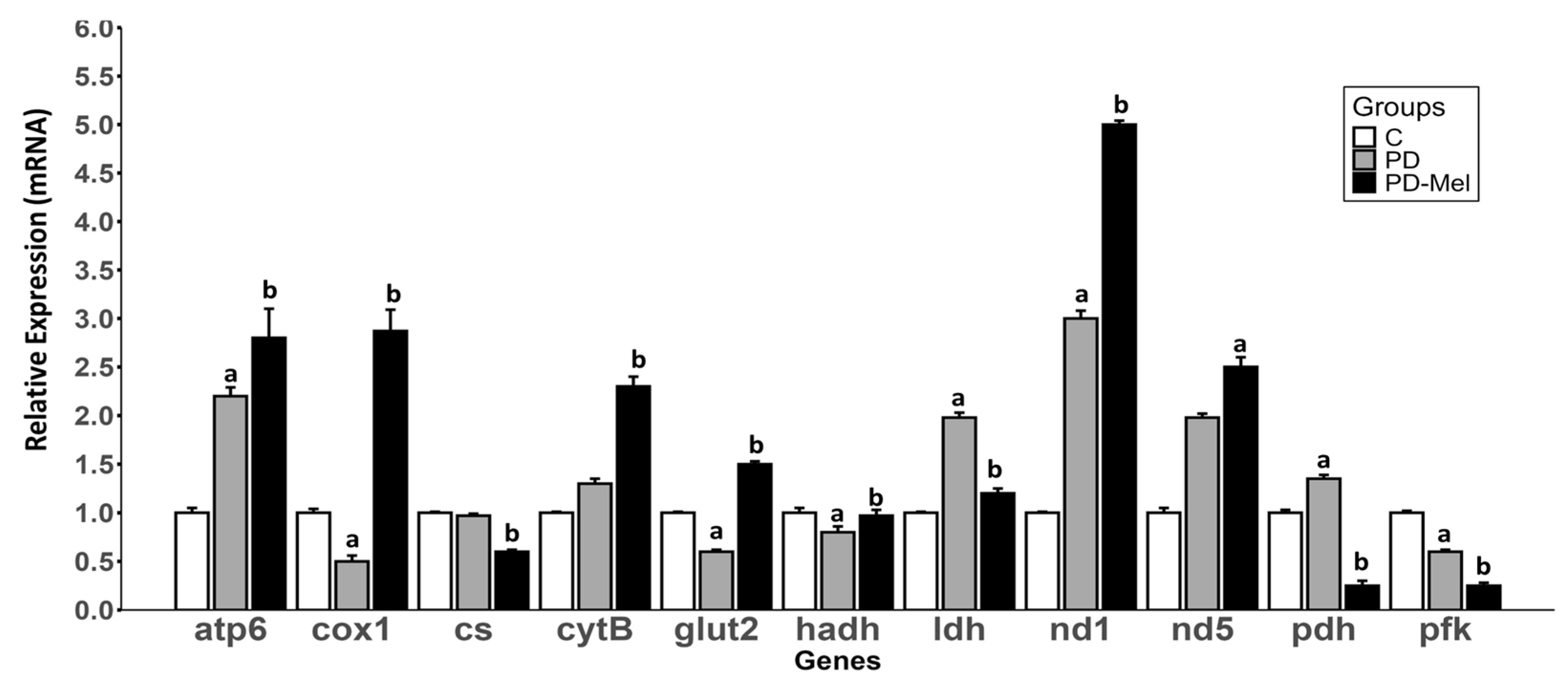
2.4. Melatonin Modulates the Expression of Genes Related to Energy Metabolism
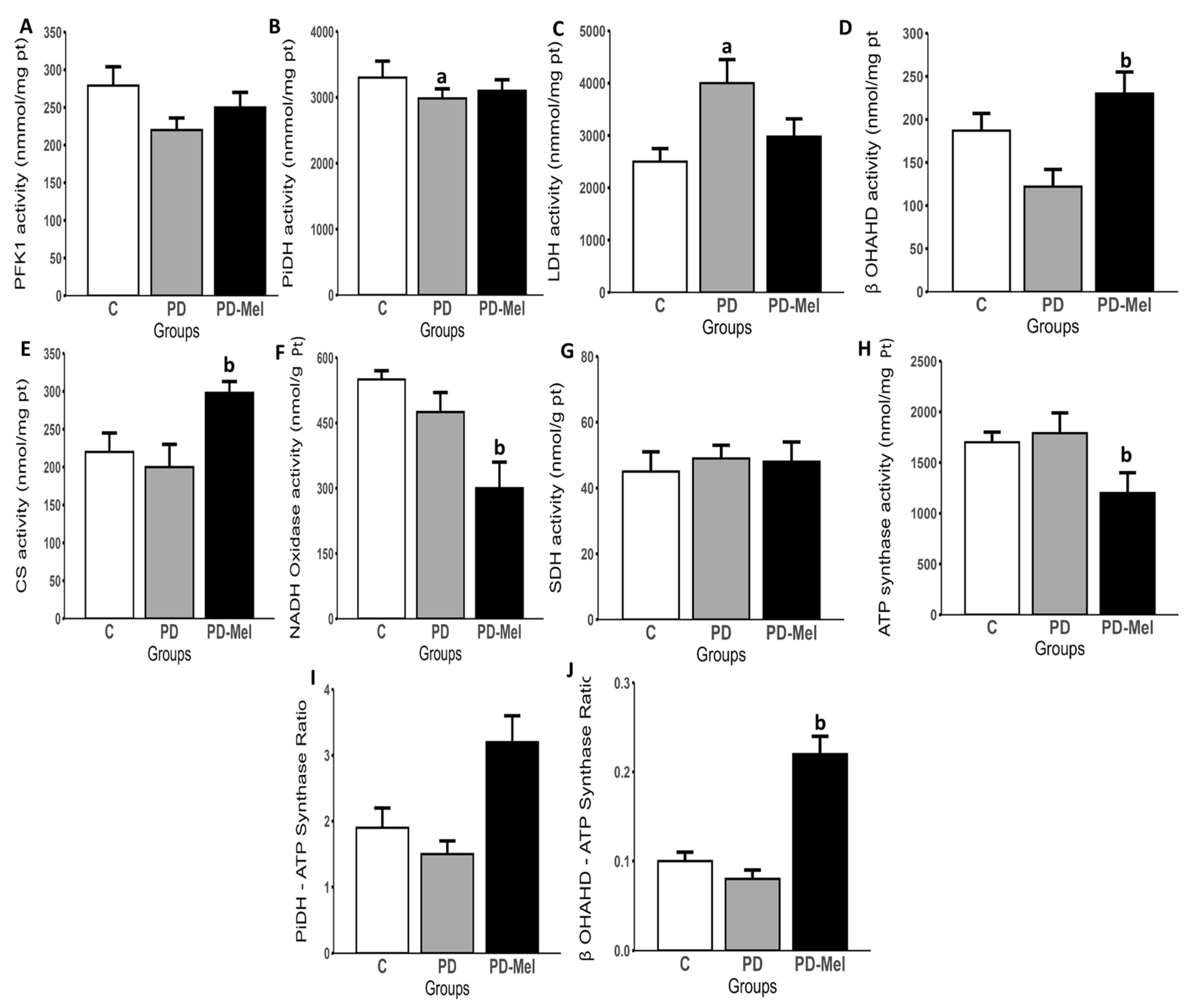
2.5. Melatonin Modulates the Activity of Enzymes Related to Energy Metabolism
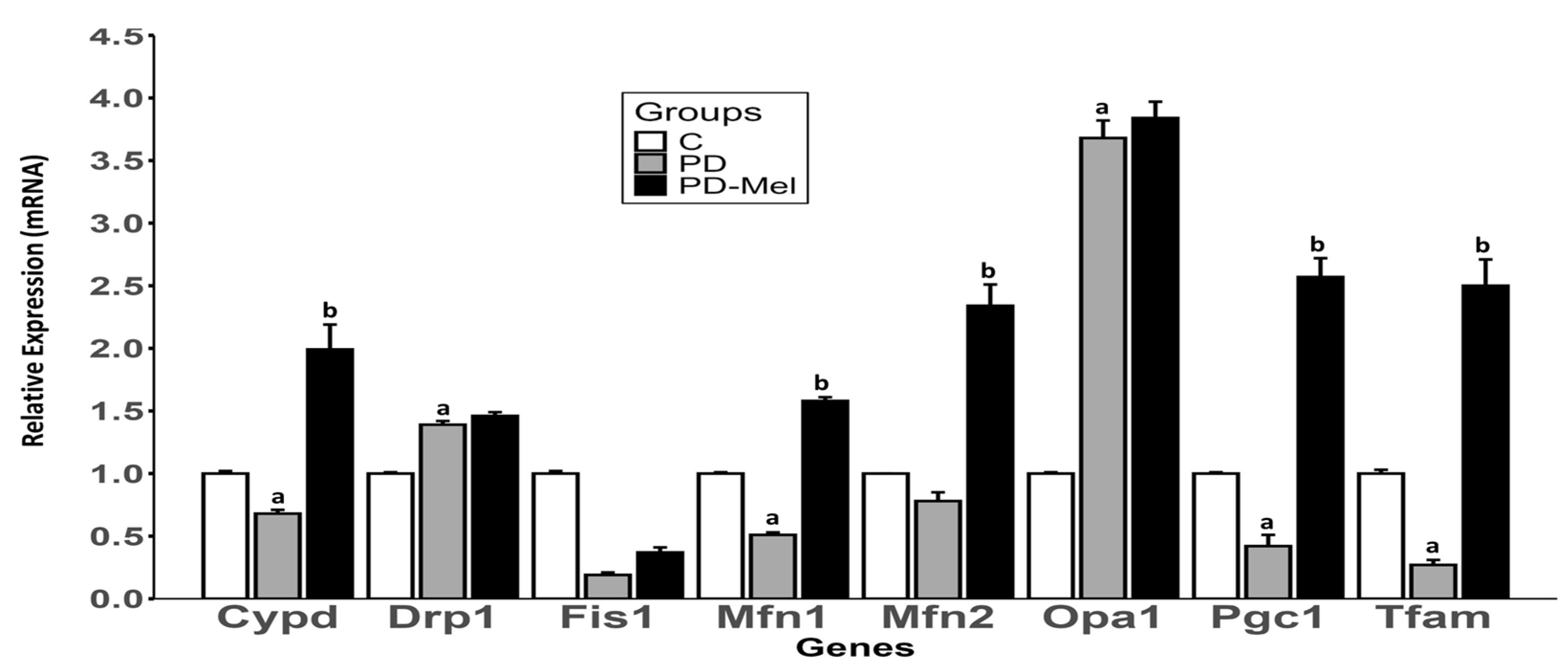
2.6. Melatonin Alters the Expression of Mitochondrial Biogenesis and Dynamics Markers
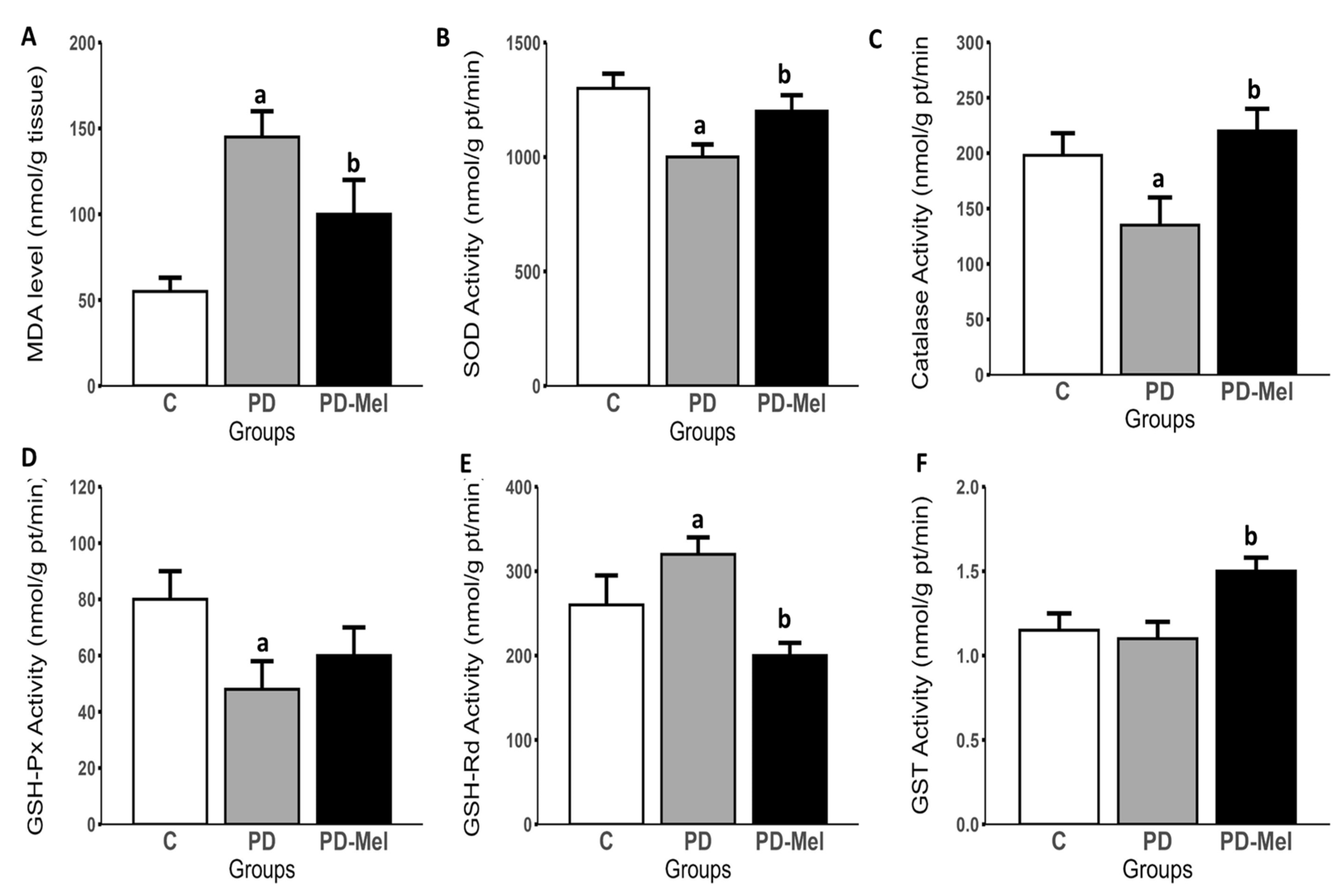
2.7. Melatonin Reduces Hepatic Oxidative Stress
2.8. Prediabetic Animals Exhibit Increased Collagen Deposition and Enlarged Hepatocyte Diameter
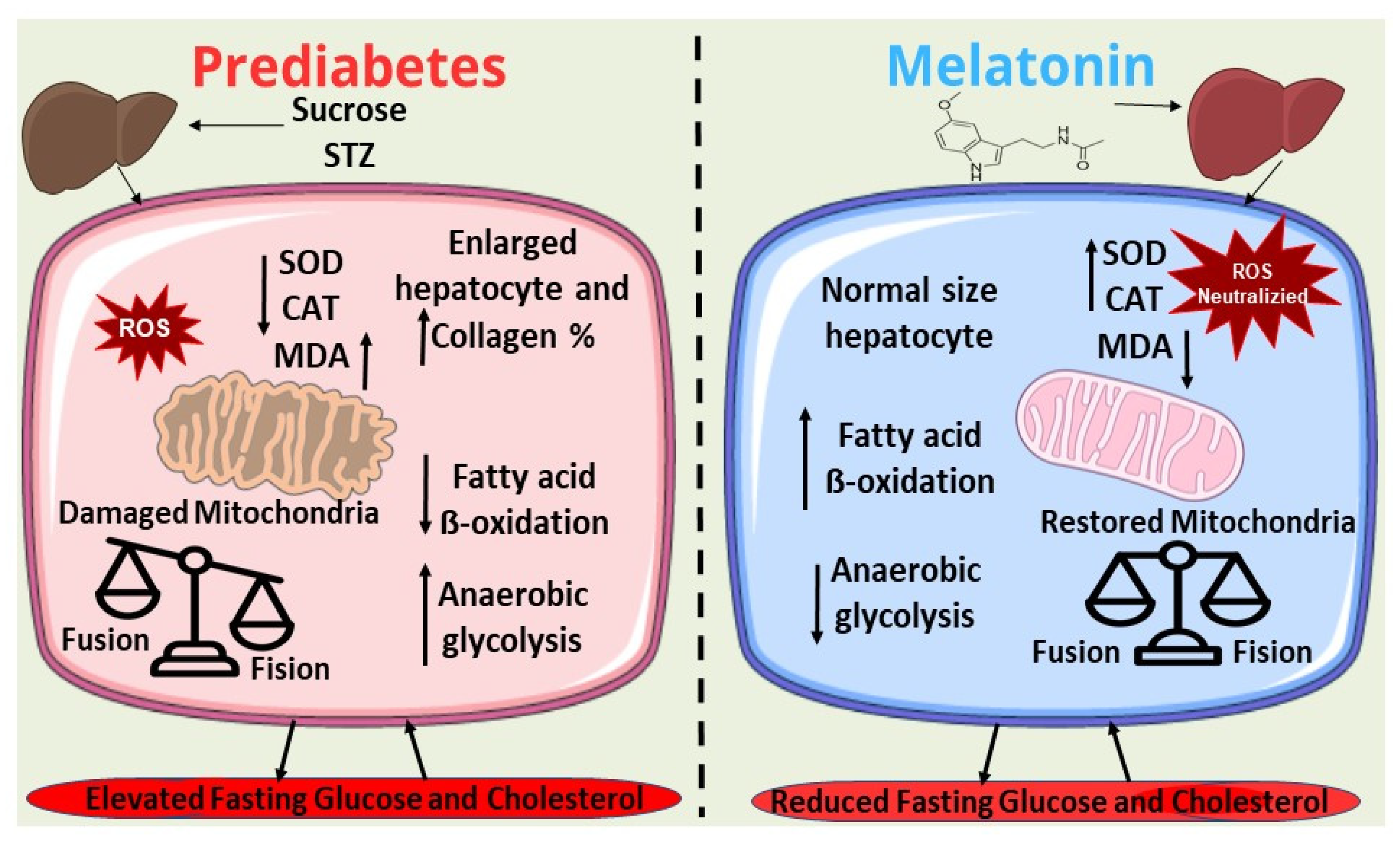
3. Discussion
Study Limitations
4. Materials and Methods
4.1. Animals and Experimental Protocol
4.2. Nutritional and Morphometric Parameters
4.3. Intraperitoneal Glucose, Insulin, and Pyruvate Tolerance Tests
4.4. Collection of Biological Material
4.5. Total RNA Extraction and cDNA Synthesis
4.6. Gene Expression Analysis
4.7. Hepatic Energy Metabolism
- −
- PFK: 50 mmol/L Tris-HCl buffer (pH 8.0), 10 mmol/L MgCl2, 1U glyceraldehyde-3-phosphate dehydrogenase, 1 U aldolase, 2.5 U triose phosphate isomerase, 0.12 mmol/L NADH, 0.75 mmol/L ATP, and 6 mmol/L fructose-6-phosphate.
- −
- CS: 50 mmol/L Tris-HCl buffer (pH 8.0), 0.3 mmol/L acetyl-CoA, 0.1 mmol/L 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB), and 0.5 mmol/L oxaloacetate.
- −
- LDH: 50 mmol/L Tris-HCl buffer (pH 7.4), 0.14 mmol/L NADH, and 1 mmol/L sodium pyruvate.
- −
- β-OHADH: 0.1 mol/L Tris-HCl buffer (pH 7.0), 5 mmol/L EDTA, 0.05 mmol/L acetoacetyl-coenzyme A, and 0.1 mmol/L NADH.
- −
- PiDH: 2.5 mmol/L NAD, 0.1 mmol/L coenzyme A, 0.2 mmol/L thiamine pyrophosphate, 0.3 mmol/L dithiothreitol (DTT), 1 mmol/L MgCl2, 5 mmol/L pyruvate, 0.08 mmol/L nitrotetrazolium blue (NBT), and 1 mg bovine serum albumin (BSA), with 3 mmol/L sodium pyruvate and 0.05 mmol/L phenazine metasulfate in 50 mmol/L potassium phosphate buffer (pH 7.4).
- −
- SDH: 50 mmol/L potassium phosphate buffer (pH 7.4), 10 mmol/L sodium succinate, 0.36 mmol/L phenazine metasulfate, and 0.12 mmol/L dichlorophenolindophenol (DPIP).
- −
- NADH-oxidase: 80 mmol/L sodium phosphate buffer (pH 7.4), 50 mmol/L EDTA, and 0.2 mmol/L NADH.
- −
- ATP synthase: 0.11 mmol/L MgCl2, 0.02 mmol/L NADH, 0.25 mmol/L phosphoenolpyruvate (PEP), 0.2 mmol/L ATP, 1686 U LDH, and 4001.18 U pyruvate kinase.
4.8. Hepatic Oxidative Stress
4.9. Histopathological Analysis
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alam, S.; Hasan, M.K.; Neaz, S.; Hussain, N.; Hossain, M.F.; Rahman, T. Diabetes Mellitus: Insights from Epidemiology, Biochemistry, Risk Factors, Diagnosis, Complications and Comprehensive Management. Diabetology 2021, 2, 36–50. [Google Scholar] [CrossRef]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 Diabetes Mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- World Health Organization. Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycemia. 2006. Available online: https://www.who.int/publications/i/item/definition-and-diagnosis-of-diabetes-mellitus-and-intermediate-hyperglycaemia (accessed on 1 March 2024).
- American Diabetes Association Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2014, 37, 81–90. [CrossRef] [PubMed]
- Rosinger, A.; Herrick, K.; Gahche, J.; Park, S. Sugar-Sweetened Beverage Consumption Among U.S. Adults, 2011–2014; NCHS Data Brief No. 270. 2017; pp. 1–8. Available online: https://www.cdc.gov/nchs/products/databriefs/db270.htm (accessed on 1 March 2024).
- Blüher, M. Obesity: Global Epidemiology and Pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Echouffo-Tcheugui, J.B.; Perreault, L.; Ji, L.; Dagogo-Jack, S. Diagnosis and Management of Prediabetes: A Review. JAMA 2023, 329, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Young, J.L.; Wang, K.; Qian, Y.; Cai, L. Diabetic-Induced Alterations in Hepatic Glucose and Lipid Metabolism: The Role of Type 1 and Type 2 Diabetes Mellitus (Review). Mol. Med. Rep. 2020, 22, 603–611. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Proteins: From Biogenesis to Functional Networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial Biogenesis: An Update. J. Cell Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Chen, G.; Chen, Q. Crosstalk between Mitochondrial Biogenesis and Mitophagy to Maintain Mitochondrial Homeostasis. J. Biomed. Sci. 2023, 30, 86. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial Dynamics in Health and Disease: Mechanisms and Potential Targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M.; Region, V. Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Veluthakal, R.; Esparza, D.; Hoolachan, J.M.; Balakrishnan, R.; Ahn, M.; Oh, E.; Jayasena, C.S.; Thurmond, D.C. Mitochondrial Dysfunction, Oxidative Stress, and Inter-Organ Miscommunications in T2D Progression. Int. J. Mol. Sci. 2024, 25, 1504. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Chronic Pharmacological Modulation of Mitochondrial Dynamics Alleviates Prediabetes-Induced Myocardial Ischemia–Reperfusion Injury by Preventing Mitochondrial Dysfunction and Programmed Apoptosis. Cardiovasc. Drugs Ther. 2023, 37, 89–105. [Google Scholar] [CrossRef]
- Park, I.R.; Chung, Y.G.; Won, K.C. Overcoming β-Cell Dysfunction in Type 2 Diabetes Mellitus: CD36 Inhibition and Antioxidant System. Diabetes Metab. J. 2025, 49, 1–12. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R.J. Melatonin as a Potent and Inducible Endogenous Antioxidant: Synthesis and Metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [CrossRef] [PubMed]
- Souza Cruz, E.M.; Bitencourt de Morais, J.M.; Dalto da Rosa, C.V.; da Silva Simões, M.; Comar, J.F.; de Almeida Chuffa, L.G.; Seiva, F.R.F. Long-Term Sucrose Solution Consumption Causes Metabolic Alterations and Affects Hepatic Oxidative Stress in Wistar Rats. Biol. Open 2020, 9, bio047282. [Google Scholar] [CrossRef]
- de Morais, J.M.B.; Cruz, E.M.S.; da Rosa, C.V.D.; Cesário, R.C.; Comar, J.F.; Moreira, C.C.L.; de Almeida Chuffa, L.G.; Seiva, F.R.F. Pterostilbene Influences Glycemia and Lipidemia and Enhances Antioxidant Status in the Liver of Rats That Consumed Sucrose Solution. Life Sci. 2021, 269, 119048. [Google Scholar] [CrossRef]
- Patel, R.; Parmar, N.; Pramanik Palit, S.; Rathwa, N.; Ramachandran, A.V.; Begum, R. Diabetes Mellitus and Melatonin: Where Are We? Biochimie 2022, 202, 2–14. [Google Scholar] [CrossRef]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an Antioxidant: Under Promises but over Delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Shieh, J.M.; Wu, H.T.; Cheng, K.C.; Cheng, J.T. Melatonin Ameliorates High Fat Diet-Induced Diabetes and Stimulates Glycogen Synthesis via a PKCzeta-Akt-GSK3beta Pathway in Hepatic Cells. J. Pineal Res. 2009, 47, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Fernández Vázquez, G.; Reiter, R.J.; Agil, A. Melatonin Increases Brown Adipose Tissue Mass and Function in Zücker Diabetic Fatty Rats: Implications for Obesity Control. J. Pineal Res. 2018, 64, e12472. [Google Scholar] [CrossRef] [PubMed]
- Hidayat, M.; Mahar, Y.; Wasim, B. Effect of Melatonin on Serum Glucose and Body Weights in Strptozotocin Induced Diabetes in Albino Rats. J. Ayub Med. Coll. Abbottabad 2015, 27, 274–276. [Google Scholar]
- Hadjzadeh, M.A.R.; Alikhani, V.; Hosseinian, S.; Zarei, B.; Keshavarzi, Z. The Effect of Melatonin against Gastric Oxidative Stress and Dyslipidemia in Streptozotocin-Induced Diabetic Rats. Acta Endocrinol. 2018, 14, 453. [Google Scholar] [CrossRef]
- Uluışık, D.; Keskin, E.; Hatipoğlu, D. A Study on Effect of Melatonin in Dyslipidemia Caused by Experimentally Diabetes. Turk. J. Sport Exerc. 2019, 21, 117–121. [Google Scholar] [CrossRef]
- Mirunalini, S.; Jayakumar, P.; Gomathy, T. Modulation of Antioxidant Status, Carbohydrate and Lipid Metabolism by Melatonin on Streptozotocin Induced Diabetic Rats. J. Biochem. Technol. 2012, 4, 524–530. [Google Scholar]
- Malik, V.S.; Hu, F.B. The Role of Sugar-Sweetened Beverages in the Global Epidemics of Obesity and Chronic Diseases. Nat. Rev. Endocrinol. 2022, 18, 205–218. [Google Scholar] [CrossRef]
- Munsters, M.J.M.; Saris, W.H.M. The Effect of Sugar-Sweetened Beverage Intake on Energy Intake in an Ad Libitum 6-Month Low-Fat High-Carbohydrate Diet. Ann. Nutr. Metab. 2010, 57, 116–123. [Google Scholar] [CrossRef]
- Dimeglio, D.P.; Mattes, R.D. Liquid versus Solid Carbohydrate: Effects on Food Intake and Body Weight. Int. J. Obes. 2000, 24, 794–800. [Google Scholar] [CrossRef]
- De Magalhães, D.A.; Kume, W.T.; Correia, F.S.; Queiroz, T.S.; Allebrandt Neto, E.W.; Dos Santos, M.P.; Kawashita, N.H.; De França, S.A. High-Fat Diet and Streptozotocin in the Induction of Type 2 Diabetes Mellitus: A New Proposal. An. Acad. Bras. Cienc. 2019, 91, e20180314. [Google Scholar] [CrossRef]
- von Krüchten, R.; Lorbeer, R.; Müller-Peltzer, K.; Rospleszcz, S.; Storz, C.; Askani, E.; Kulka, C.; Schuppert, C.; Rathmann, W.; Peters, A.; et al. Association between Adipose Tissue Depots and Dyslipidemia: The KORA-MRI Population-Based Study. Nutrients 2022, 14, 797. [Google Scholar] [CrossRef] [PubMed]
- Abdulwahab, D.A.; El-Missiry, M.A.; Shabana, S.; Othman, A.I.; Amer, M.E. Melatonin Protects the Heart and Pancreas by Improving Glucose Homeostasis, Oxidative Stress, Inflammation and Apoptosis in T2DM-Induced Rats. Heliyon 2021, 7, e06474. [Google Scholar] [CrossRef]
- Wan, X.D.; Li, S.Q.; Xi, S.M.; Wang, J.F.; Guo, Y.C.; Wang, X.M. Long-Term Melatonin Administration Improves Glucose Homeostasis and Insulin Resistance State in High-Fat-Diet Fed Rats. Cent. Eur. J. Biol. 2013, 8, 958–967. [Google Scholar] [CrossRef]
- Ausina, P.; Da Silva, D.; Majerowicz, D.; Zancan, P.; Sola-Penna, M. Insulin Specifically Regulates Expression of Liver and Muscle Phosphofructokinase Isoforms. Biomed. Pharmacother. 2018, 103, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Manz, A.; Volk, T.; Kleber, A. Study of Melatonin-Mediated Effects on Various Hepatic Inflammatory Responses Stimulated by IL-6 in a New HepG2-on-a-Chip Platform. Biomed. Microdevices 2018, 20, 54. [Google Scholar] [CrossRef]
- Hassani-Nezhad-Gashti, F.; Rysä, J.; Kummu, O.; Näpänkangas, J.; Buler, M.; Karpale, M.; Hukkanen, J.; Hakkola, J. Activation of Nuclear Receptor PXR Impairs Glucose Tolerance and Dysregulates GLUT2 Expression and Subcellular Localization in Liver. Biochem. Pharmacol. 2018, 148, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin Action and Resistance in Obesity and Type 2 Diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Cruz, E.M.S.; Concato, V.M.; de Morais, J.M.B.; Silva, T.F.; Inoue, F.S.R.; de Souza Cremer, M.; Bidóia, D.L.; Machado, R.R.B.; de Almeida Chuffa, L.G.; Mantovani, M.S.; et al. Melatonin Modulates the Warburg Effect and Alters the Morphology of Hepatocellular Carcinoma Cell Line Resulting in Reduced Viability and Migratory Potential. Life Sci. 2023, 319, 121530. [Google Scholar] [CrossRef]
- Wang, G.S.; Hoyte, C. Review of Biguanide (Metformin) Toxicity. J. Intensive Care Med. 2019, 34, 863–876. [Google Scholar] [CrossRef]
- Defronzo, R.; Fleming, G.A.; Chen, K.; Bicsak, T.A. Metformin-Associated Lactic Acidosis: Current Perspectives on Causes and Risk. Metabolism 2016, 65, 20–29. [Google Scholar] [CrossRef]
- Akhter, M.S.; Uppal, P. Toxicity of Metformin and Hypoglycemic Therapies. Adv. Chronic Kidney Dis. 2020, 27, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. Staying Current PGC-1: A Key Regulator of Energy Metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Wilson-Fritch, L.; Nicoloro, S.; Chouinard, M.; Lazar, M.A.; Chui, P.C.; Leszyk, J.; Straubhaar, J.; Czech, M.P.; Corvera, S. Mitochondrial Remodeling in Adipose Tissue Associated with Obesity and Treatment with Rosiglitazone. J. Clin. Investig. 2004, 114, 1281–1289. [Google Scholar] [CrossRef]
- Hu, B.; Chen, Z.; Liang, L.; Zheng, M.; Chen, X.; Zeng, Q. Melatonin Promotes Mitochondrial Biogenesis and Mitochondrial Degradation in Hepatocytes During Sepsis. Altern. Ther. Health Med. 2023, 29, 284–289. [Google Scholar]
- Cooper, I.D.; Brookler, K.H.; Kyriakidou, Y.; Elliott, B.T.; Crofts, C.A.P. Metabolic Phenotypes and Step by Step Evolution of Type 2 Diabetes: A New Paradigm. Biomedicines 2021, 9, 800. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Manjarrés-Raza, I.; Vicente-Gutiérrez, C.; Corrado, M.; Bolaños, J.P.; Scorrano, L. Opa1 Relies on Cristae Preservation and ATP Synthase to Curtail Reactive Oxygen Species Accumulation in Mitochondria. Redox Biol. 2021, 41, 101944. [Google Scholar] [CrossRef] [PubMed]
- Maechler, P.; Jornot, L.; Wollheim, C.B. Hydrogen Peroxide Alters Mitochondrial Activation and Insulin Secretion in Pancreatic Beta Cells. J. Biol. Chem. 1999, 274, 27905–27913. [Google Scholar] [CrossRef] [PubMed]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, M.F.; Harakeh, S.; et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells 2022, 11, 552. [Google Scholar] [CrossRef]
- Brand, M.D. The Sites and Topology of Mitochondrial Superoxide Production. Exp. Gerontol. 2010, 45, 466. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and the Electron Transport Chain. Cell. Mol. Life Sci. 2017, 74, 3883–3896. [Google Scholar] [CrossRef]
- Grigorov, I.; Bogojević, D.; Jovanović, S.; Petrović, A.; Ivanović-Matić, S.; Zolotarevski, L.; Poznanović, G.; Martinović, V. Hepatoprotective Effects of Melatonin against Pronecrotic Cellular Events in Streptozotocin-Induced Diabetic Rats. J. Physiol. Biochem. 2014, 70, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Kahya, M.C.; Naziroǧlu, M. Melatonin Reduces Lens Oxidative Stress Level in STZ-Induced Diabetic Rats through Supporting Glutathione Peroxidase and Reduced Glutathione Values. J. Cell. Neurosci. Oxidative Stress 2016, 8, 588–594. [Google Scholar] [CrossRef]
- Agarkov, A.A.; Popova, T.N.; Matasova, L.V. Effects of Melatonin on the Activity of the Glutathione Antioxidant System and Various NADPH-Generating Enzymes in the Liver and Blood of Rats with Type 2 Diabetes Mellitus. Pharm. Chem. J. 2011, 45, 385–388. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, Q.; Hua, C.; Ci, X. Melatonin Alleviates Particulate Matter-Induced Liver Fibrosis by Inhibiting ROS-Mediated Mitophagy and Inflammation via Nrf2 Activation. Ecotoxicol. Environ. Saf. 2023, 268, 115717. [Google Scholar] [CrossRef]
- Salido, E.M.; Bordone, M.; De Laurentiis, A.; Chianelli, M.; Keller Sarmiento, M.I.; Dorfman, D.; Rosenstein, R.E. Therapeutic Efficacy of Melatonin in Reducing Retinal Damage in an Experimental Model of Early Type 2 Diabetes in Rats. J. Pineal Res. 2013, 54, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol. 2015, 70, 5.47.1–5.47.20. [Google Scholar] [CrossRef]
- Barrière, D.A.; Noll, C.; Roussy, G.; Lizotte, F.; Kessai, A.; Kirby, K.; Belleville, K.; Beaudet, N.; Longpré, J.M.; Carpentier, A.C.; et al. Combination of High-Fat/High-Fructose Diet and Low-Dose Streptozotocin to Model Long-Term Type-2 Diabetes Complications. Sci. Rep. 2018, 8, 424. [Google Scholar] [CrossRef]
- Macêdo, A.P.A.; Cordeiro, G.S.; Santos, L.S.; Santo, D.A.E.; Perez, G.S.; Couto, R.D.; Machado, M.E.P.C.; Medeiros, J.M.B. Murinometric Measurements and Retroperitoneal Adipose Tissue in Young Rats Exposed to the High-Fat Diet: Is There Correlation? Braz. J. Biol. 2021, 81, 246–250. [Google Scholar] [CrossRef]
- Seiva, F.R.F.; Berbert, C.M.S.; Souza, G.A.; Rocha, K.K.H.; Ebaid, G.M.X.; Burneiko, R.C.M.; Novelli, E.L.B. Energy Expenditure, Lipid Profile, Oxidative Stress, and Cardiac Energy Metabolism after Growth Hormone Treatment in Obese Young Rats. Horm. Metab. Res. 2010, 42, 496–501. [Google Scholar] [CrossRef]
- Zheng, K.; Wu, L.; He, Z.; Yang, B.; Yang, Y. Measurement of the Total Protein in Serum by Biuret Method with Uncertainty Evaluation. Measurement 2017, 112, 16–21. [Google Scholar] [CrossRef]
- Alisik, M.; Neselioglu, S.; Erel, O. A Colorimetric Method to Measure Oxidized, Reduced and Total Glutathione Levels in Erythrocytes. J. Lab. Med. 2019, 43, 269–277. [Google Scholar] [CrossRef]
- Hopkins, J.; Tudhope, G.R. Glutathione Peroxidase in Human Redcells in Health and Disease. J. Haematol. 1973, 25, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Keen, J.; Habig, W.H.; Jakoby, W.B. Mechanism for Several Activities of the Glutathione-S-Transferase. J. Biol. Chem. 1976, 20, 6183–6188. [Google Scholar] [CrossRef]
- Crouch, R.K.; Gandy, S.E.; Kimsey, G.; Galbraith, R.A.; Galbraith, G.M.; Buse, M.G. The Inhibition of Islet Superoxide Dismutase by Diabetogenic Drugs. Diabetes 1981, 30, 235–241. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in Vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Groups | ||
|---|---|---|---|
| C | PD | PD-Mel | |
| Body weight gain (g) | 212.3 ± 18.27 | 244.4 ± 20.39 | 197.7 ± 76.36 |
| BMI (g/cm2) | 0.63 ± 0.02 | 0.63 ± 0.01 | 0.55 ± 0.04 |
| Lee index (g/cm) | 11.71 ± 0.22 | 11.85 ± 0.37 | 11.08 ± 0.76 |
| Abdominal circumference (cm) | 16.1 ± 1.37 | 16.00 ± 0.81 | 15.22 ± 1.3 |
| Body length (cm) | 24.5 ± 0.53 | 24.38 ± 0.74 | 25.13 ± 1.8 |
| Food consumption (g/day) | 27.87 ± 0.47 | 15.51 ± 0.5 a | 16.85 ± 1.18 |
| Liquid consumption (mL/day) | 52.7 ± 1.47 | 50.14 ± 2.4 | 65 ± 7.57 |
| Energy intake from food (kcal/day) | 83.98 ± 1.45 | 47.06 ± 2.27 a | 50.89 ± 2.55 |
| Energy intake from liquid (kcal/day) | 0 ± 0.00 | 87.53 ± 4.23 a | 95.6 ± 3.77 |
| Total energy intake (kcal/day) | 83.98 ± 1.45 | 145.5 ± 8.73 a | 149.1 ± 6.64 |
| Parameters | Groups | ||
|---|---|---|---|
| C | PD | PD-Mel | |
| Epididymal fat deposit (mg) | 3.28 ± 0.22 | 5.75 ± 0.38 a | 2.08 ± 0.49 b |
| Visceral fat deposit (mg) | 2.33 ± 0.18 | 4.14 ± 0.34 | 2.31 ± 0.57 |
| Retroperitoneal fat deposit (mg) | 2.24 ± 0.31 | 6.54 ± 0.57 a | 2.48 ± 0.81 b |
| Sum of fat depots (mg) | 7.84 ± 1.85 | 16.45 ± 3 a | 6.4 ± 4.89 b |
| Serum triglycerides levels (mg/dL) | 101 ± 9.50 | 143 ± 29.31 a | 118 ± 34.11 |
| Serum cholesterol levels (mg/dL) | 105 ± 2.8 | 128 ± 4.51 a | 112 ± 2.41 b |
| Serum HDL levels (mg/dL) | 55 ± 2.81 | 61 ± 3.49 | 50 ± 5.51 |
| Serum LDL levels (mg/dL) | 28 ± 4.17 | 36 ± 3.35 | 27 ± 3.00 |
| Serum ALT levels (U/L) | 29 ± 2.07 | 20 ± 2.44 | 24 ± 4.35 |
| Serum AST levels (U/L | 43 ± 2.42 | 44 ± 2.74 | 30 ± 4.81 b |
| Liver triglycerides levels (mg/dL) | 87 ± 2.28 | 103 ± 4.02 a | 100 ± 3.10 |
| Liver cholesterol levels (mg/dL) | 94 ± 0.84 | 97 ± 1.17 | 95 ± 0.76 |
| Liver HDL levels (mg/dL) | 21 ± 0.17 | 20 ± 0.12 a | 20 ± 0.11 |
| Liver LDL levels (mg/dL) | 57 ± 7.43 | 55 ± 5.34 | 53 ± 2.22 |
| Parameters | Groups | ||
|---|---|---|---|
| C | PD | PD-Mel | |
| Total Glutathione (nmol/mg tissue) | 2.97 ± 0.10 | 2.21 ± 0.15 | 1.93 ± 0.29 |
| GSH (nmol/mg tissue) | 2.11 ± 0.08 | 1.31 ± 0.07 a | 1.33 ± 0.15 |
| GSSG (nmol/mg tissue) | 0.52 ± 0.04 | 0.45 ± 0.02 | 0.3 ± 0.04 b |
| OSI (%) | 10.55 ± 0.72 | 14.47 ± 1.75 a | 10.34 ± 1.56 b |
| Parameters | Groups | |||
|---|---|---|---|---|
| C | PD | PD-Mel | ||
| Hepatocyte number | 237.3 ± 6.86 | 221.6 ± 8.99 | 232.6 ± 10.85 | |
| Hepatocyte morphometry (µm) | 2.96 ± 0.07 | 3.64 ± 0.09 a | 2.82 ± 0.10 b | |
| Collagen (%) | 0.31 ± 0.02 | 1.40 ± 0.19 a | 1.08 ± 0.14 | |
| Gene (ID) | Primer | 5’–3’ Sequence | Cycle Condition | Product Size (bp) |
|---|---|---|---|---|
| ACTB (81822) | Forward | CCTCTATGCCAACACAGTGC | 95 °C—8 s; 61.5 °C—8 s; 72 °C—8 s | 206 |
| Reverse | CCTGCTTGCTGATCCACATC | |||
| Slc2a2 (25351) | Forward | TGTGAAAGTCATGCATGTGGC | 95 °C—8 s; 61.5 °C—8 s; 72 °C—8 s | 104 |
| Reverse | AGGCCAGGGATTGGTGTTAAA | |||
| Hadh (113965) | Forward | AGTCTGGACTTGACCTTCTTGG | 95 °C—8 s; 61.5 °C—8 s; 72 °C—8 s | 138 |
| Reverse | TCTGAGGGCCCATTTTGATGT | |||
| Pdh1 (29554) | Forward | TCCCCAGCTTGGCTTATTGT | 95 °C—8 s; 61.5 °C—8 s; 72 °C—8 s | 132 |
| Reverse | GGGTGGCTTCAAGTTTGCTTT | |||
| Pfkl (25741) | Forward | AACAATGTCCCTGGCACTGA | 95 °C—8 s; 61.5 °C—8 s; 72 °C—8 s | 189 |
| Reverse | ACTCTCCATTGCAGCGTTGA | |||
| Cs (170587) | Forward | ACCATGACGGTGGCAATGTA | 95 °C—8 s; 61.5 °C—8 s; 72 °C—8 s | 210 |
| Reverse | TGGTTTGCTAGTCCATGCAGA | |||
| Ldha (24533) | Forward | TCCTCAGCGTCCCATGTATC | 95 °C—8 s; 61.5 °C—8 s; 72 °C—8 s | 193 |
| Reverse | TCCATAGAAACCCTGCTGCA | |||
| Tfam (21780) | Forward | TCCACAGAACAGCTACCCAA | 95 °C—8 s; 66 °C—8 s; 72 °C—8 s | 84 |
| Reverse | CCACAGGGCTGCAATTTTCC | |||
| Cypd (105675) | Forward | CCCTCCTGGTCACTGTGAAT | 95 °C—8 s; 66 °C—8 s; 72 °C—8 s | 186 |
| Reverse | GCCAGAAGACACTTCCCTCT | |||
| Nd1 (17716) | Forward | ATTACTTCTGCCAGCCTGACC | 95 °C—8 s; 66 °C—8 s; 72 °C—8 s | 70 |
| Reverse | GGCCCGGTTTGTTTCTGCTA | |||
| Nd5 (4540) | Forward | CCTGGCACTGAGTCACCATA | 95 °C—8 s; 66 °C—8 s; 72 °C—8 s | 214 |
| Reverse | TTGTTGGCTGAGGTGAGGAT | |||
| Atp6 (100503946) | Forward | TCCCAATCGTTGTAGCCATCA | 95 °C—8 s; 66 °C—8 s; 72 °C—8 s | 76 |
| Reverse | AGACGGTTGTTGATTAGGCGT | |||
| Cox1 (17708) | Forward | ATCACTACCAGTGCTAGCCG | 95 °C—8 s; 66 °C—8 s; 72 °C—8 s | 84 |
| Reverse | CCTCCAGCGGGATCAAAGAA | |||
| Drp1 (74006) | Forward | GTCGAGTCCCCATTCATTGC | 95 °C—8 s; 66 °C—8 s; 72 °C—8 s | 151 |
| Reverse | ACTACGACGATCTGAGGCAG | |||
| Mfn1 (67414) | Forward | GCAGACAGCACATGGAGAGA | 95 °C—8 s; 61 °C—8 s; 72 °C—8 s | 83 |
| Reverse | GATCCGATTCCGAGCTTCCG | |||
| Mfn2 (170731) | Forward | TGCACCGCCATATAGAGGAAG | 95 °C—8 s; 61 °C—8 s; 72 °C—8 s | 78 |
| Reverse | TCTGCAGTGAACTGGCAATG | |||
| Opa1 (74143) | Forward | ACCTTGCCAGTTTAGCTCCC | 95 °C—8 s; 61 °C—8 s; 72 °C—8 s | 82 |
| Reverse | TTGGGACCTGCAGTGAAGAA | |||
| Fis1 (66437) | Forward | CAAAGAGGAACAGCGGGACT | 95 °C—8 s; 61 °C—8 s; 72 °C—8 s | 95 |
| Reverse | ACAGCCCTCGCACATACTTT | |||
| Cytb (17711) | Forward | GGCTACGTCCTTCCATGAGG | 95 °C—8 s; 61 °C—8 s; 72 °C—8 s | 75 |
| Reverse | TGGGATGGCTGATAGGAGGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, M.C.; Agneis, M.L.G.; das Neves, K.A.; de Almeida, M.R.; Feltran, G.d.S.; Souza Cruz, E.M.; Schoffen, J.P.F.; Chuffa, L.G.d.A.; Seiva, F.R.F. Melatonin Improves Lipid Homeostasis, Mitochondrial Biogenesis, and Antioxidant Defenses in the Liver of Prediabetic Rats. Int. J. Mol. Sci. 2025, 26, 4652. https://doi.org/10.3390/ijms26104652
de Souza MC, Agneis MLG, das Neves KA, de Almeida MR, Feltran GdS, Souza Cruz EM, Schoffen JPF, Chuffa LGdA, Seiva FRF. Melatonin Improves Lipid Homeostasis, Mitochondrial Biogenesis, and Antioxidant Defenses in the Liver of Prediabetic Rats. International Journal of Molecular Sciences. 2025; 26(10):4652. https://doi.org/10.3390/ijms26104652
Chicago/Turabian Stylede Souza, Milena Cremer, Maria Luisa Gonçalves Agneis, Karoliny Alves das Neves, Matheus Ribas de Almeida, Geórgia da Silva Feltran, Ellen Mayara Souza Cruz, João Paulo Ferreira Schoffen, Luiz Gustavo de Almeida Chuffa, and Fábio Rodrigues Ferreira Seiva. 2025. "Melatonin Improves Lipid Homeostasis, Mitochondrial Biogenesis, and Antioxidant Defenses in the Liver of Prediabetic Rats" International Journal of Molecular Sciences 26, no. 10: 4652. https://doi.org/10.3390/ijms26104652
APA Stylede Souza, M. C., Agneis, M. L. G., das Neves, K. A., de Almeida, M. R., Feltran, G. d. S., Souza Cruz, E. M., Schoffen, J. P. F., Chuffa, L. G. d. A., & Seiva, F. R. F. (2025). Melatonin Improves Lipid Homeostasis, Mitochondrial Biogenesis, and Antioxidant Defenses in the Liver of Prediabetic Rats. International Journal of Molecular Sciences, 26(10), 4652. https://doi.org/10.3390/ijms26104652