
KCNH3 Loss-of-Function Variant Associated with Epilepsy and Neurodevelopmental Delay Enhances Kv12.2 Channel Inactivation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
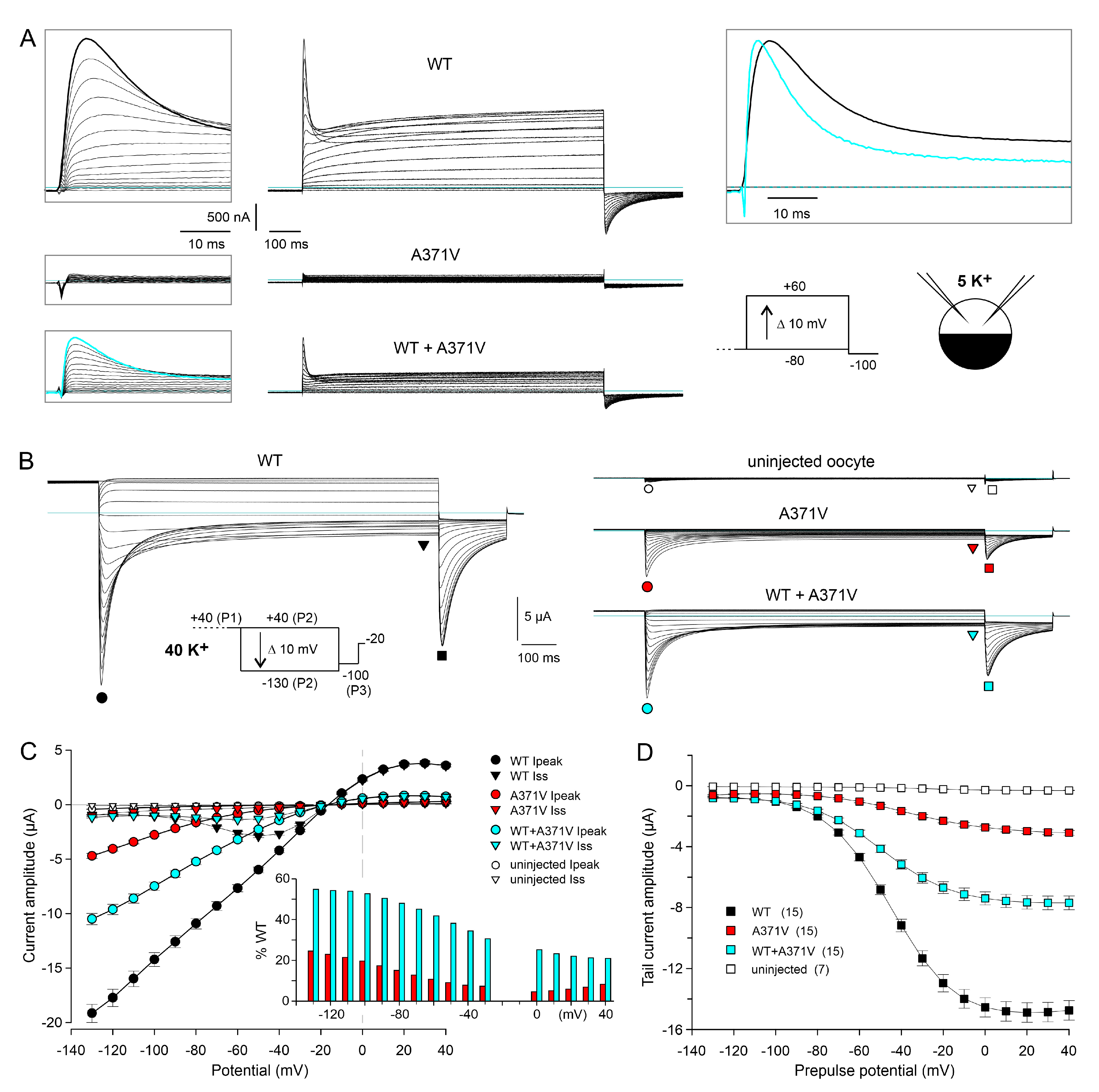
2.1. Functional Characterization of A371V Kv12.2 Channels in Xenopus Oocytes
2.1.1. Apparent Complete LoF Caused by the A371V Amino Acid Substitution
2.1.2. Elevated External K+ Allows the Detection of Tail Currents Mediated by Homomeric A371V Kv12.2 Channels
2.1.3. Voltage-Dependence of the A371V-Induced LoF Effect
2.2. Functional Characterization of A371V in a Mammalian Expression System
2.2.1. Enhancement in Kv12.2 Channel Expression in CHO Cells by Mild Hypothermia to Analyze the Effect of the A371V Amino Acid Substitution on Channel Availability
2.2.2. Moderate Effects of the A371V Amino Acid Substitution on Channel Activation
2.2.3. The A371V Amino Acid Substitution Drastically Enhances Channel Inactivation
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| WT | wild-type |
| EK | K+ equilibrium potential |
| LoF | loss of function |
| GHK | Goldman–Hodgkin–Katz |
| RMP | resting membrane potential |
| CHO | Chinese hamster ovary |
Appendix A

References
- Bauer, C.K.; Kortüm, F.; Mollring, A.; Grinstein, L.; Denecke, J.; Alawi, M.; Bähring, R.; Harms, F.L. Loss-of-function variant in KCNH3 is associated with global developmental delay, autistic behavior, insomnia, and nocturnal seizures. Seizure 2025, 129, 14–21. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Miyake, A.; Mochizuki, S.; Yokoi, H.; Kohda, M.; Furuichi, K. New ether-à-go-go K+ channel family members localized in human telencephalon. J. Biol. Chem. 1999, 274, 25018–25025. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bertaso, F.; Yoo, J.W.; Baumgartel, K.; Clancy, S.M.; Lee, V.; Cienfuegos, C.; Wilmot, C.; Avis, J.; Hunyh, T.; et al. Deletion of the potassium channel Kv12.2 causes hippocampal hyperexcitability and epilepsy. Nat. Neurosci. 2010, 13, 1056–1058. [Google Scholar] [CrossRef]
- Hermanstyne, T.O.; Yang, N.D.; Granados-Fuentes, D.; Li, X.; Mellor, R.L.; Jegla, T.; Herzog, E.D.; Nerbonne, J.M. Kv12-encoded K+ channels drive the day-night switch in the repetitive firing rates of SCN neurons. J. Gen. Physiol. 2023, 155, e202213310. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Kuzmicki, C.E.; Childs, R.R.; Hintz, C.J.; Delisle, B.P.; January, C.T. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 2014, 5, 5535. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Sanchez-Conde, F.G.; Jimenez-Vazquez, E.N.; Auerbach, D.S.; Jones, D.K. The ERG1 K+ Channel and Its Role in Neuronal Health and Disease. Front. Mol. Neurosci. 2022, 15, 890368. [Google Scholar] [CrossRef]
- Singh, V.; Auerbach, D.S. Neurocardiac pathologies associated with potassium channelopathies. Epilepsia 2024, 65, 2537–2552. [Google Scholar] [CrossRef]
- Yu, C.; Deng, X.J.; Xu, D. Gene mutations in comorbidity of epilepsy and arrhythmia. J. Neurol. 2023, 270, 1229–1248. [Google Scholar] [CrossRef]
- Li, G.; Shi, R.; Wu, J.; Han, W.; Zhang, A.; Cheng, G.; Xue, X.; Sun, C. Association of the hERG mutation with long-QT syndrome type 2, syncope and epilepsy. Mol. Med. Rep. 2016, 13, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Curran, M.E.; Spector, P.S.; Keating, M.T. Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc. Natl. Acad. Sci. USA 1996, 93, 2208–2212. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.; de la Pena, P.; Dominguez, P.; Sierra, L.M.; Pardo, L.A. The EAG voltage-dependent K+ channel subfamily: Similarities and differences in structural organization and gating. Front. Pharmacol. 2020, 11, 411. [Google Scholar] [CrossRef]
- Bauer, C.K.; Schwarz, J.R. Ether-à-go-go K+ channels: Effective modulators of neuronal excitability. J. Physiol. 2018, 596, 769–783. [Google Scholar] [CrossRef]
- Morais-Cabral, J.H.; Robertson, G.A. The enigmatic cytoplasmic regions of KCNH channels. J. Mol. Biol. 2015, 427, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Warmke, J.W.; Ganetzky, B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. USA 1994, 91, 3438–3442. [Google Scholar] [CrossRef]
- Noma, K.; Kimura, K.; Minatohara, K.; Nakashima, H.; Nagao, Y.; Mizoguchi, A.; Fujiyoshi, Y. Triple N-glycosylation in the long S5-P loop regulates the activation and trafficking of the Kv12.2 potassium channel. J. Biol. Chem. 2009, 284, 33139–33150. [Google Scholar] [CrossRef] [PubMed]
- Kurata, H.T.; Fedida, D. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 2006, 92, 185–208. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K+ channels: Structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef]
- Engeland, B.; Neu, A.; Ludwig, J.; Roeper, J.; Pongs, O. Cloning and functional expression of rat ether-à-go-go-like K+ channel genes. J. Physiol. 1998, 513 Pt 3, 647–654. [Google Scholar] [CrossRef]
- Becchetti, A.; De Fusco, M.; Crociani, O.; Cherubini, A.; Restano-Cassulini, R.; Lecchi, M.; Masi, A.; Arcangeli, A.; Casari, G.; Wanke, E. The functional properties of the human ether-à-go-go-like (HELK2) K+ channel. Eur. J. Neurosci. 2002, 16, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2001. [Google Scholar]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, M.C.; Titus, S.A.; Branchaw, J.L.; Ganetzky, B.; Robertson, G.A. Functional analysis of a mouse brain Elk-type K+ channel. J. Neurosci. 1999, 19, 2906–2918. [Google Scholar] [CrossRef] [PubMed]
- Barish, M.E. A transient calcium-dependent chloride current in the immature Xenopus oocyte. J. Physiol. 1983, 342, 309–325. [Google Scholar] [CrossRef]
- Tokimasa, T.; North, R.A. Effects of barium, lanthanum and gadolinium on endogenous chloride and potassium currents in Xenopus oocytes. J. Physiol. 1996, 496 Pt 3, 677–686. [Google Scholar] [CrossRef]
- Bauer, C.K.; Falk, T.; Schwarz, J.R. An endogenous inactivating inward-rectifying potassium current in oocytes of Xenopus laevis. Pflugers. Arch. 1996, 432, 812–820. [Google Scholar] [CrossRef]
- Clancy, S.M.; Chen, B.; Bertaso, F.; Mamet, J.; Jegla, T. KCNE1 and KCNE3 beta-subunits regulate membrane surface expression of Kv12.2 K+ channels in vitro and form a tripartite complex in vivo. PLoS ONE 2009, 4, e6330. [Google Scholar] [CrossRef]
- Ranjan, R.; Logette, E.; Marani, M.; Herzog, M.; Tache, V.; Scantamburlo, E.; Buchillier, V.; Markram, H. A kinetic map of the homomeric voltage-gated potassium channel (Kv) family. Front. Cell. Neurosci. 2019, 13, 358. [Google Scholar] [CrossRef]
- Li, X.; Anishkin, A.; Liu, H.; van Rossum, D.B.; Chintapalli, S.V.; Sassic, J.K.; Gallegos, D.; Pivaroff-Ward, K.; Jegla, T. Bimodal regulation of an Elk subfamily K+ channel by phosphatidylinositol 4,5-bisphosphate. J. Gen. Physiol. 2015, 146, 357–374. [Google Scholar] [CrossRef]
- Dai, G.; Zagotta, W.N. Molecular mechanism of voltage-dependent potentiation of KCNH potassium channels. elife 2017, 6, e26355. [Google Scholar] [CrossRef]
- Tan, P.S.; Perry, M.D.; Ng, C.A.; Vandenberg, J.I.; Hill, A.P. Voltage-sensing domain mode shift is coupled to the activation gate by the N-terminal tail of hERG channels. J. Gen. Physiol. 2012, 140, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Spector, P.S.; Curran, M.E.; Zou, A.; Keating, M.T.; Sanguinetti, M.C. Fast inactivation causes rectification of the IKr channel. J. Gen. Physiol. 1996, 107, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.; Lin, Z.; Humble, M.; Creech, C.D.; Wagoner, P.K.; Krafte, D.; Jegla, T.J.; Wickenden, A.D. Distribution and functional properties of human KCNH8 (Elk1) potassium channels. Am. J. Physiol.-Cell Physiol. 2003, 285, C1356–C1366. [Google Scholar] [CrossRef]
- Anderson, C.L.; Delisle, B.P.; Anson, B.D.; Kilby, J.A.; Will, M.L.; Tester, D.J.; Gong, Q.; Zhou, Z.; Ackerman, M.J.; January, C.T. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 2006, 113, 365–373. [Google Scholar] [CrossRef]
- Kagan, A.; Yu, Z.; Fishman, G.I.; McDonald, T.V. The dominant negative LQT2 mutation A561V reduces wild-type HERG expression. J. Biol. Chem. 2000, 275, 11241–11248. [Google Scholar] [CrossRef]
- Wulhfard, S.; Tissot, S.; Bouchet, S.; Cevey, J.; De Jesus, M.; Hacker, D.L.; Wurm, F.M. Mild hypothermia improves transient gene expression yields several fold in Chinese hamster ovary cells. Biotechnol. Prog. 2008, 24, 458–465. [Google Scholar] [CrossRef]
- Raimondo, J.V.; Burman, R.J.; Katz, A.A.; Akerman, C.J. Ion dynamics during seizures. Front. Cell. Neurosci. 2015, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Clay, J.R. Determining K+ channel activation curves from K+ channel currents often requires the Goldman-Hodgkin-Katz equation. Front. Cell. Neurosci. 2009, 3, 20. [Google Scholar] [CrossRef]
- Smith, P.L.; Baukrowitz, T.; Yellen, G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature 1996, 379, 833–836. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Varghese, A.; Lu, Y.; Bursill, J.A.; Mahaut-Smith, M.P.; Huang, C.L. Temperature dependence of human ether-à-go-go-related gene K+ currents. Am. J. Physiol.-Cell Physiol. 2006, 291, C165–C175. [Google Scholar] [CrossRef]
- Sturm, P.; Wimmers, S.; Schwarz, J.R.; Bauer, C.K. Extracellular potassium effects are conserved within the rat erg K+ channel family. J. Physiol. 2005, 564, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Sachse, F.B.; Sanguinetti, M.C. Tuning of EAG K+ channel inactivation: Molecular determinants of amplification by mutations and a small molecule. J. Gen. Physiol. 2012, 140, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Kortüm, F.; Caputo, V.; Bauer, C.K.; Stella, L.; Ciolfi, A.; Alawi, M.; Bocchinfuso, G.; Flex, E.; Paolacci, S.; Dentici, M.L.; et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat. Genet. 2015, 47, 661–667. [Google Scholar] [CrossRef]
- Schönherr, R.; Hehl, S.; Terlau, H.; Baumann, A.; Heinemann, S.H. Individual subunits contribute independently to slow gating of bovine EAG potassium channels. J. Biol. Chem. 1999, 274, 5362–5369. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S.; Morales, M.J.; Strauss, H.C.; Rasmusson, R.L. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J. Physiol. 1997, 502 Pt 1, 45–60. [Google Scholar] [CrossRef]
- Shibasaki, T. Conductance and kinetics of delayed rectifier potassium channels in nodal cells of the rabbit heart. J. Physiol. 1987, 387, 227–250. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Walker, B.D.; Campbell, T.J. HERG K+ channels: Friend and foe. Trends. Pharmacol. Sci. 2001, 22, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.E.; Hill, A.P.; Zhao, J.; Kondo, M.; Subbiah, R.N.; Campbell, T.J.; Vandenberg, J.I. Effect of S5P alpha-helix charge mutants on inactivation of hERG K+ channels. J. Physiol. 2006, 573, 291–304. [Google Scholar] [CrossRef]
- Wang, S.; Morales, M.J.; Liu, S.; Strauss, H.C.; Rasmusson, R.L. Time, voltage and ionic concentration dependence of rectification of h-erg expressed in Xenopus oocytes. FEBS Lett. 1996, 389, 167–173. [Google Scholar] [CrossRef]
- Butler, A.; Helliwell, M.V.; Zhang, Y.; Hancox, J.C.; Dempsey, C.E. An update on the structure of hERG. Front. Pharmacol. 2020, 10, 1572. [Google Scholar] [CrossRef]
- Butler, A.; Zhang, Y.; Stuart, A.G.; Dempsey, C.E.; Hancox, J.C. Functional and pharmacological characterization of an S5 domain hERG mutation associated with short QT syndrome. Heliyon 2019, 5, e01429. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; MacKinnon, R. Cryo-EM structure of the open human ether-à-go-go-related K+ channel hERG. Cell 2017, 169, 422–430.E10. [Google Scholar] [CrossRef]
- Perry, M.D.; Ng, C.A.; Vandenberg, J.I. Pore helices play a dynamic role as integrators of domain motion during Kv11.1 channel inactivation gating. J. Biol. Chem. 2013, 288, 11482–11491. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.; Sachse, F.B.; Abbruzzese, J.; Sanguinetti, M.C. PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc. Natl. Acad. Sci. USA 2009, 106, 20075–20080. [Google Scholar] [CrossRef] [PubMed]
- Qile, M.; Beekman, H.D.M.; Sprenkeler, D.J.; Houtman, M.J.C.; van Ham, W.B.; Stary-Weinzinger, A.; Beyl, S.; Hering, S.; van den Berg, D.J.; de Lange, E.C.M.; et al. LUF7244, an allosteric modulator/activator of Kv 11.1 channels, counteracts dofetilide-induced torsades de pointes arrhythmia in the chronic atrioventricular block dog model. Br. J. Pharmacol. 2019, 176, 3871–3885. [Google Scholar] [CrossRef]
- El Harchi, A.; Brincourt, O. Pharmacological activation of the hERG K+ channel for the management of the long QT syndrome: A review. J. Arrhythmia 2022, 38, 554–569. [Google Scholar] [CrossRef]
- Klein, P.; Kaminski, R.M.; Koepp, M.; Loscher, W. New epilepsy therapies in development. Nat. Rev. Drug Discov. 2024, 23, 682–708. [Google Scholar] [CrossRef]
- Gerlach, A.C.; Stoehr, S.J.; Castle, N.A. Pharmacological removal of human ether-a-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574). Mol. Pharmacol. 2010, 77, 58–68. [Google Scholar] [CrossRef]
- Garg, V.; Stary-Weinzinger, A.; Sanguinetti, M.C. ICA-105574 interacts with a common binding site to elicit opposite effects on inactivation gating of EAG and ERG potassium channels. Mol. Pharmacol. 2013, 83, 805–813. [Google Scholar] [CrossRef]
- Schuster, A.M.; Glassmeier, G.; Bauer, C.K. Strong activation of ether-à-go-go-related gene 1 K+ channel isoforms by NS1643 in human embryonic kidney 293 and Chinese hamster ovary cells. Mol. Pharmacol. 2011, 80, 930–942. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauer, C.K.; Bilet, A.; Harms, F.L.; Bähring, R. KCNH3 Loss-of-Function Variant Associated with Epilepsy and Neurodevelopmental Delay Enhances Kv12.2 Channel Inactivation. Int. J. Mol. Sci. 2025, 26, 4631. https://doi.org/10.3390/ijms26104631
Bauer CK, Bilet A, Harms FL, Bähring R. KCNH3 Loss-of-Function Variant Associated with Epilepsy and Neurodevelopmental Delay Enhances Kv12.2 Channel Inactivation. International Journal of Molecular Sciences. 2025; 26(10):4631. https://doi.org/10.3390/ijms26104631
Chicago/Turabian StyleBauer, Christiane K., Arne Bilet, Frederike L. Harms, and Robert Bähring. 2025. "KCNH3 Loss-of-Function Variant Associated with Epilepsy and Neurodevelopmental Delay Enhances Kv12.2 Channel Inactivation" International Journal of Molecular Sciences 26, no. 10: 4631. https://doi.org/10.3390/ijms26104631
APA StyleBauer, C. K., Bilet, A., Harms, F. L., & Bähring, R. (2025). KCNH3 Loss-of-Function Variant Associated with Epilepsy and Neurodevelopmental Delay Enhances Kv12.2 Channel Inactivation. International Journal of Molecular Sciences, 26(10), 4631. https://doi.org/10.3390/ijms26104631