
In Silico Approach to Design of New Multi-Targeted Inhibitors Based on Quinoline Ring with Potential Anticancer Properties

 ,
,  , and
, and 
Abstract

1. Introduction
2. Results and Discussion
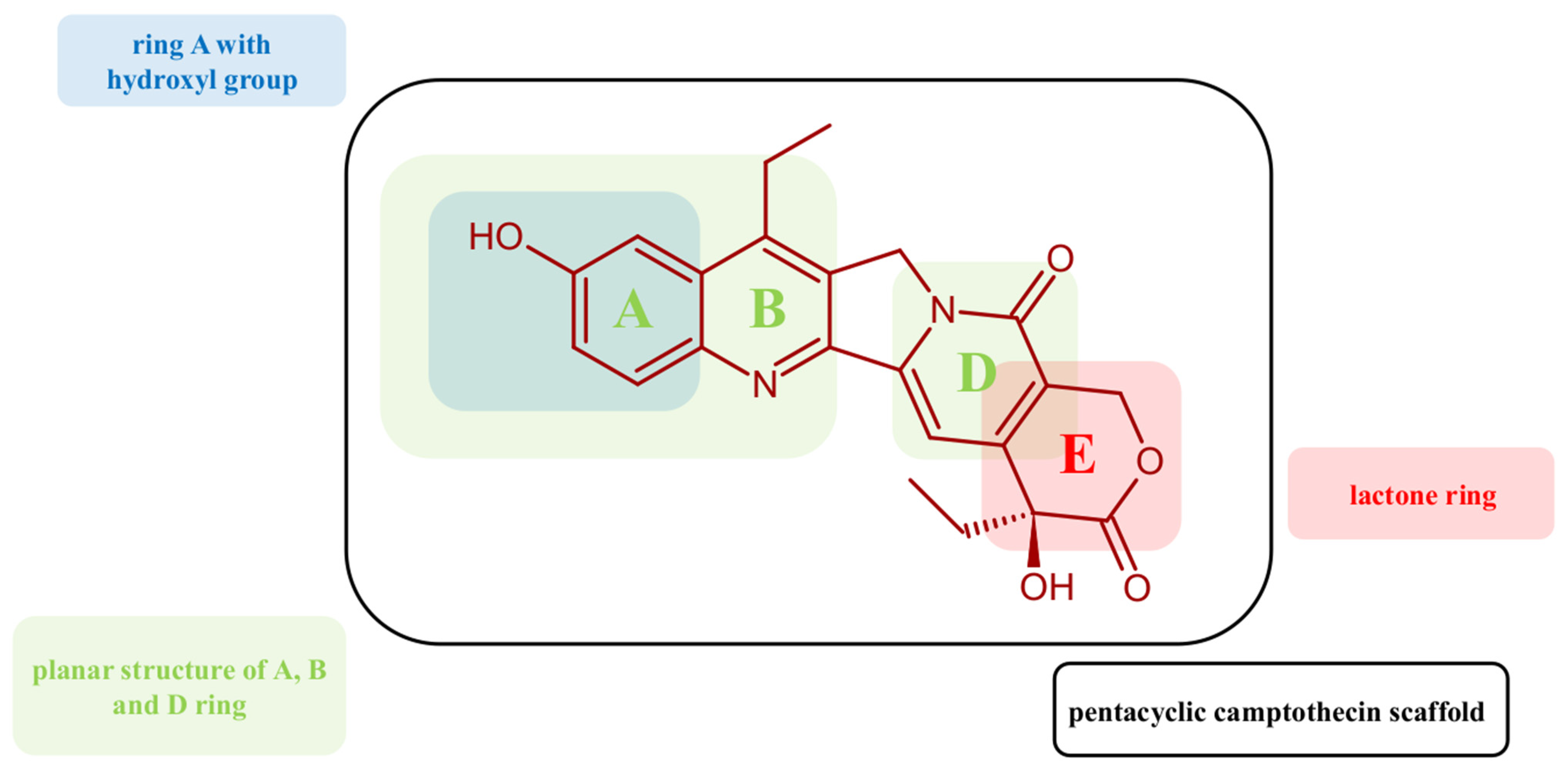
2.1. Designing Strategy
2.1.1. Molecular Targets
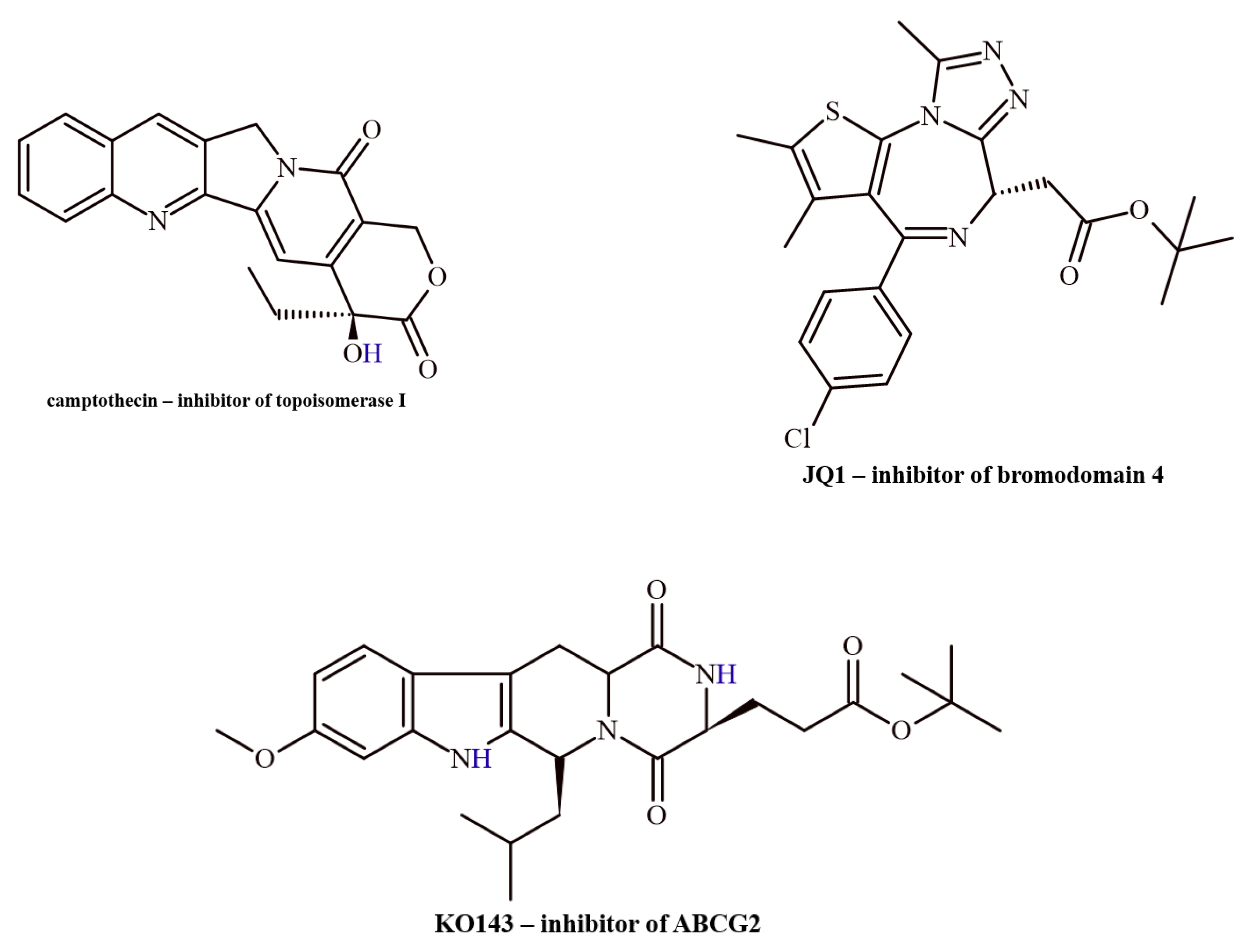
2.1.2. Reference Compounds
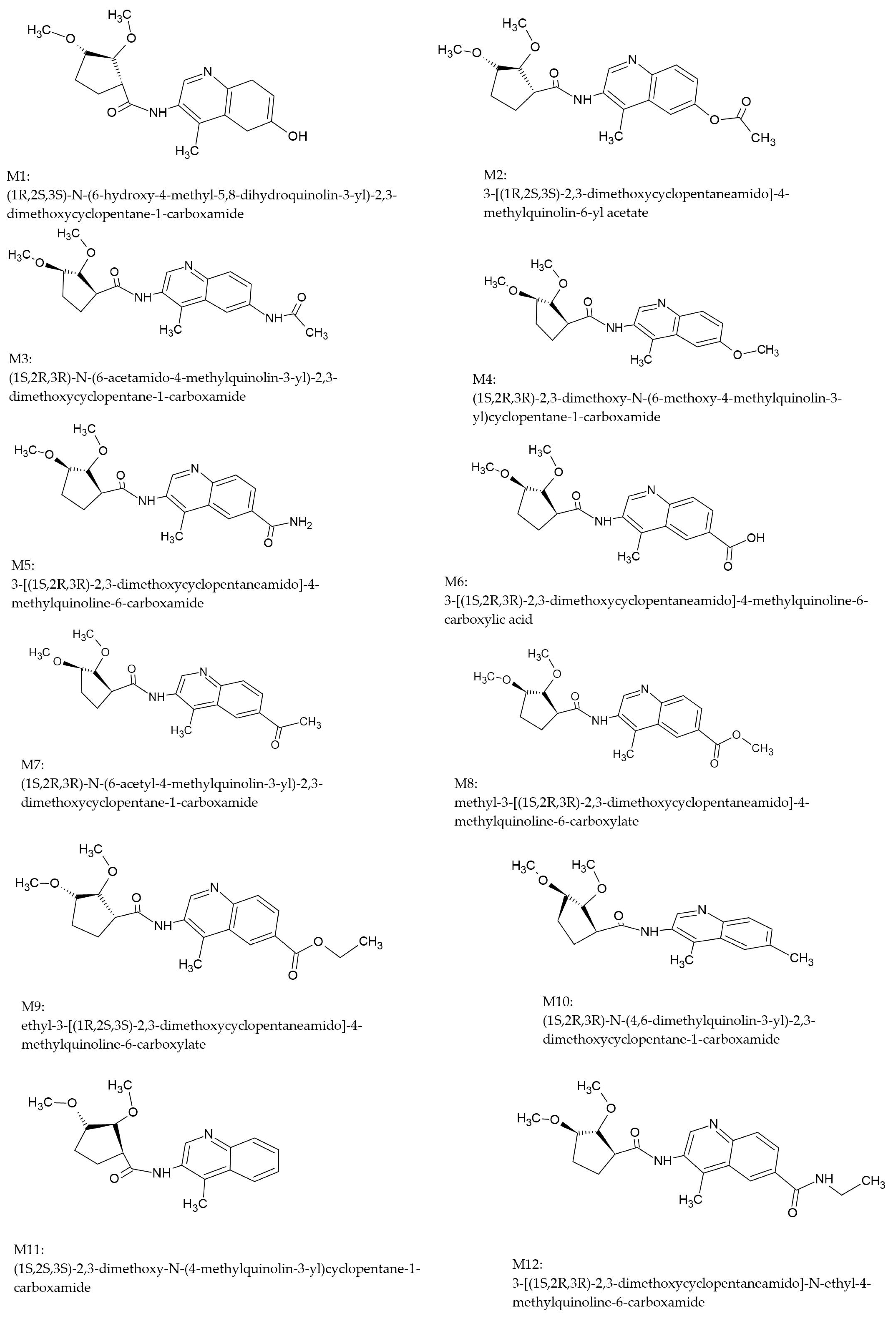
2.1.3. Designed Compounds
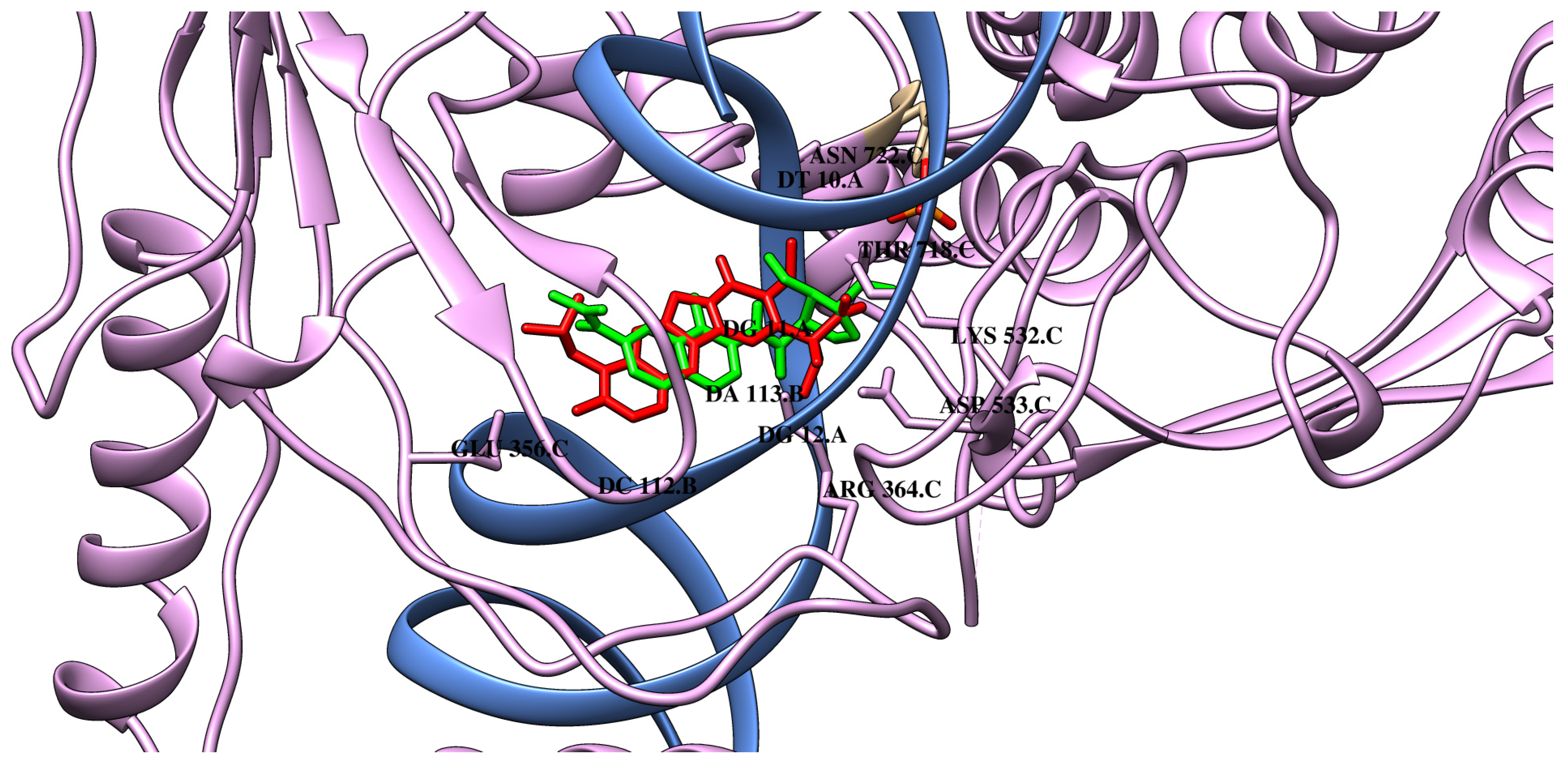
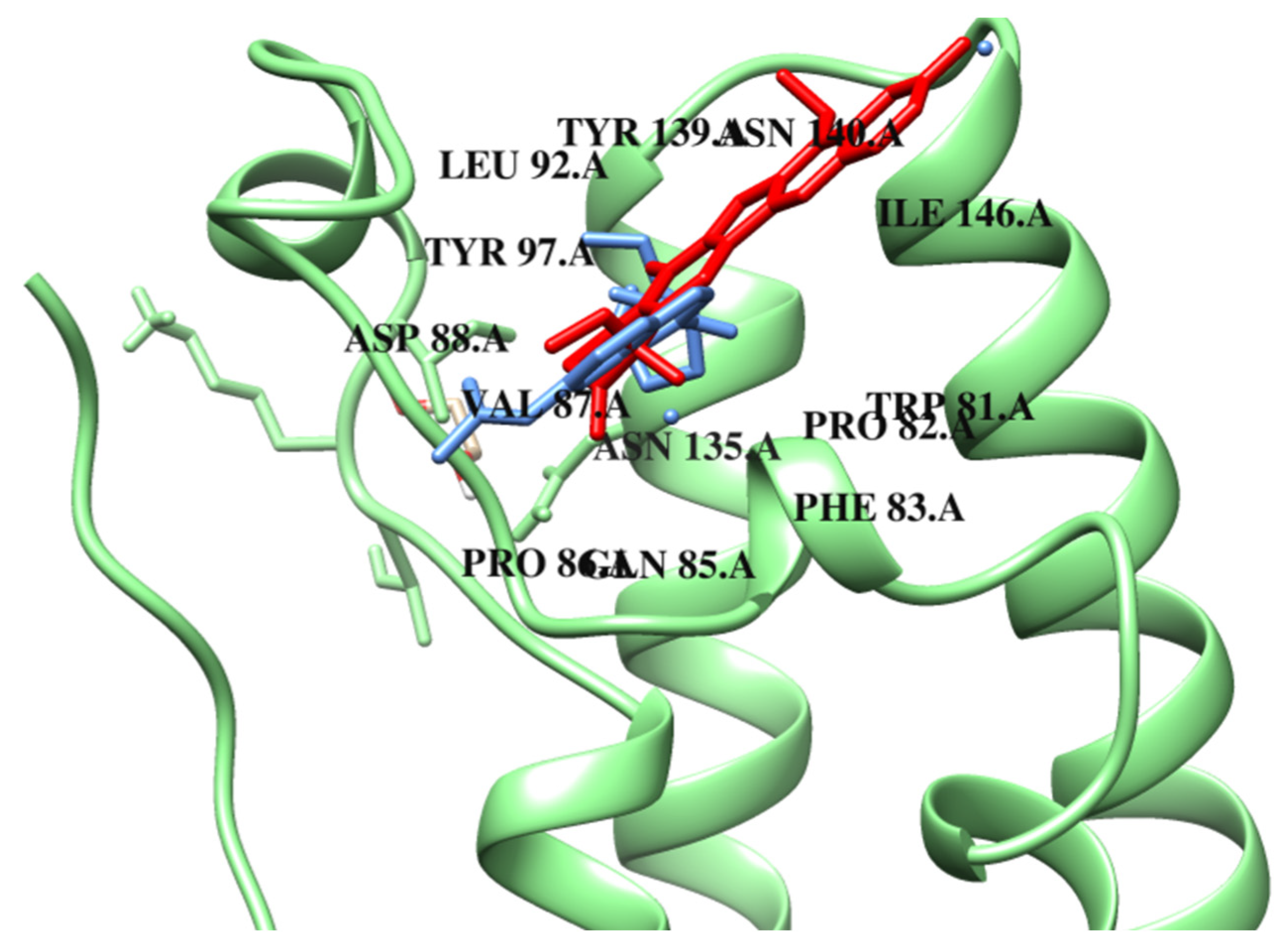
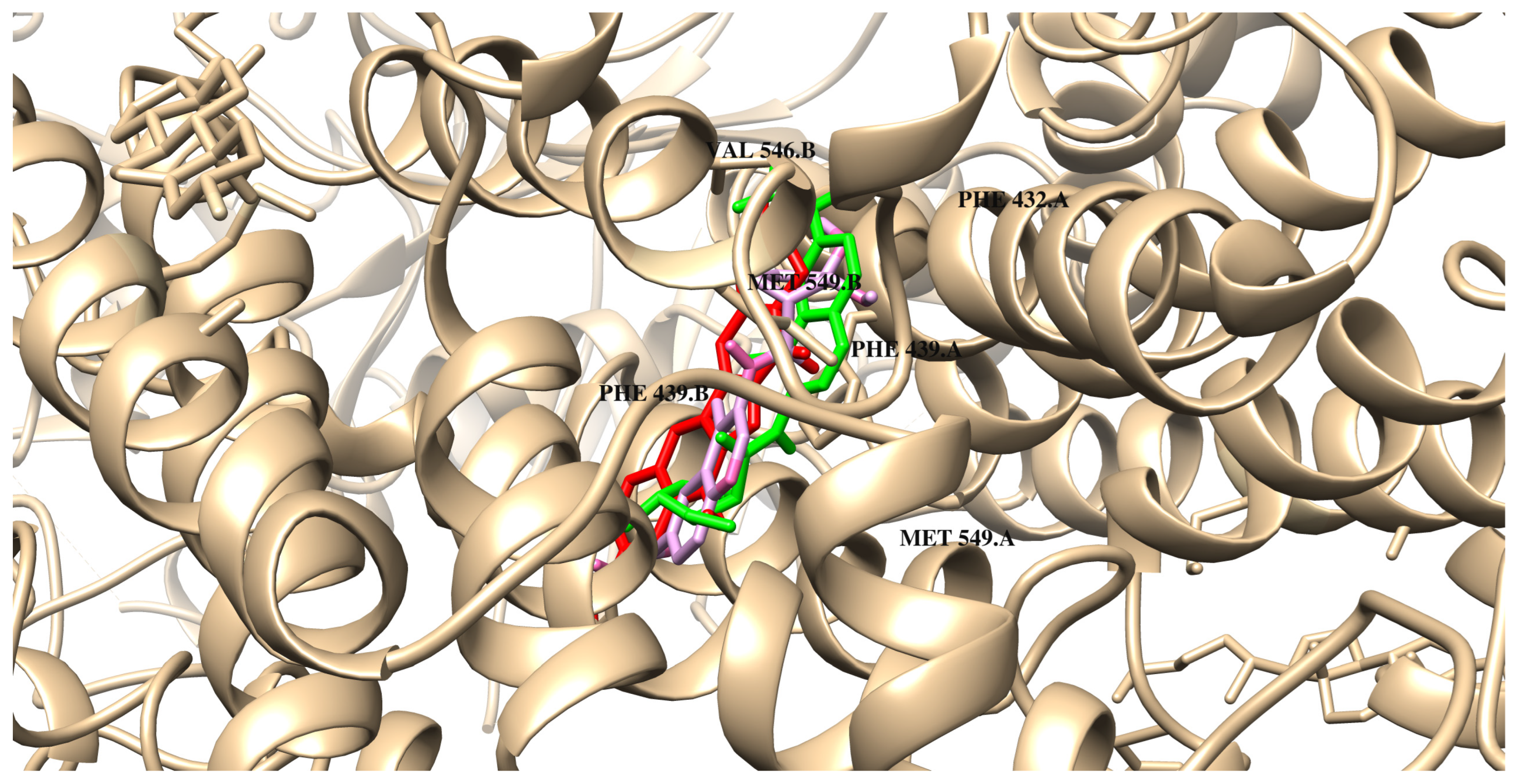
2.2. Binding Properties of Designed Compounds Based on Molecular Docking
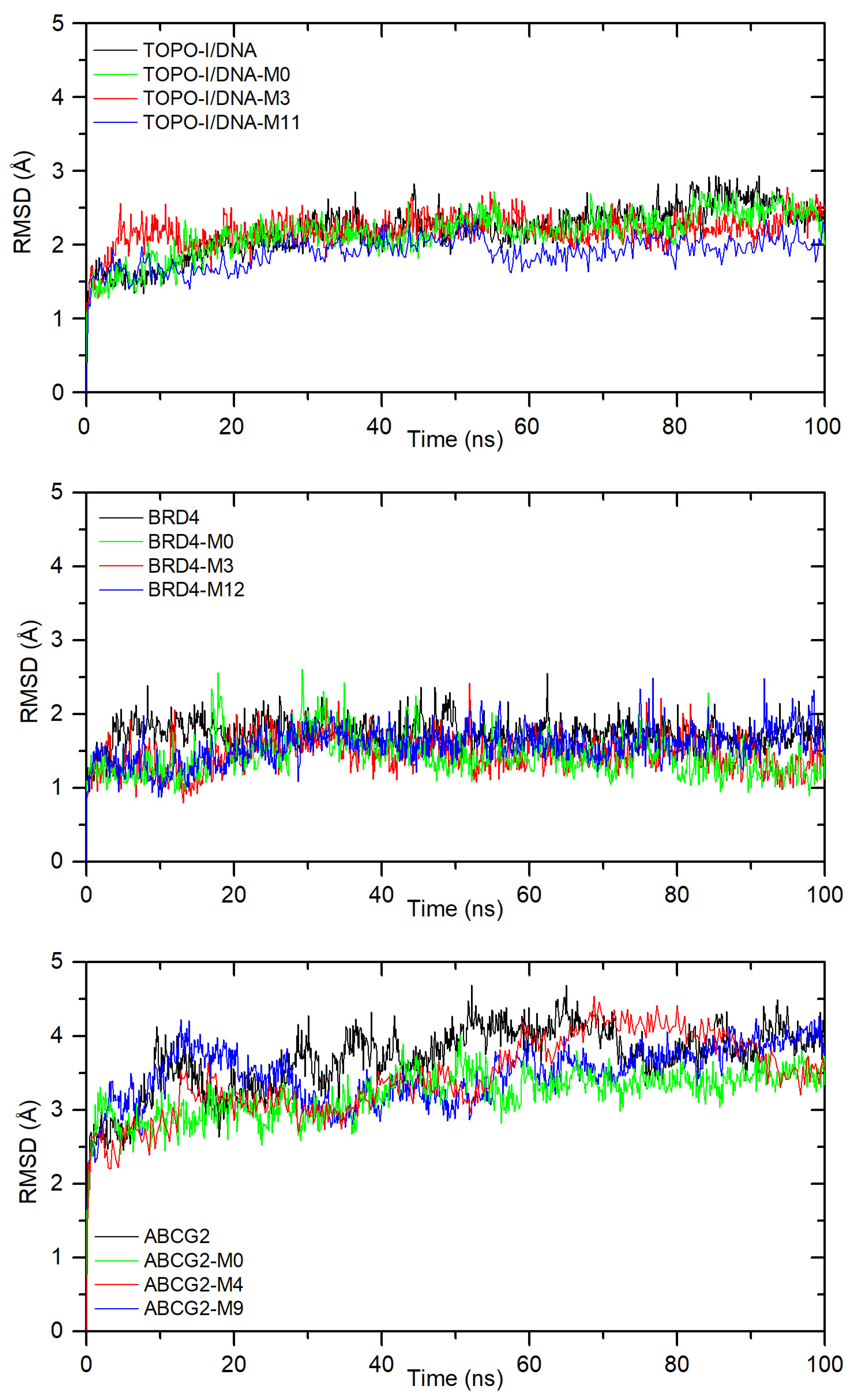
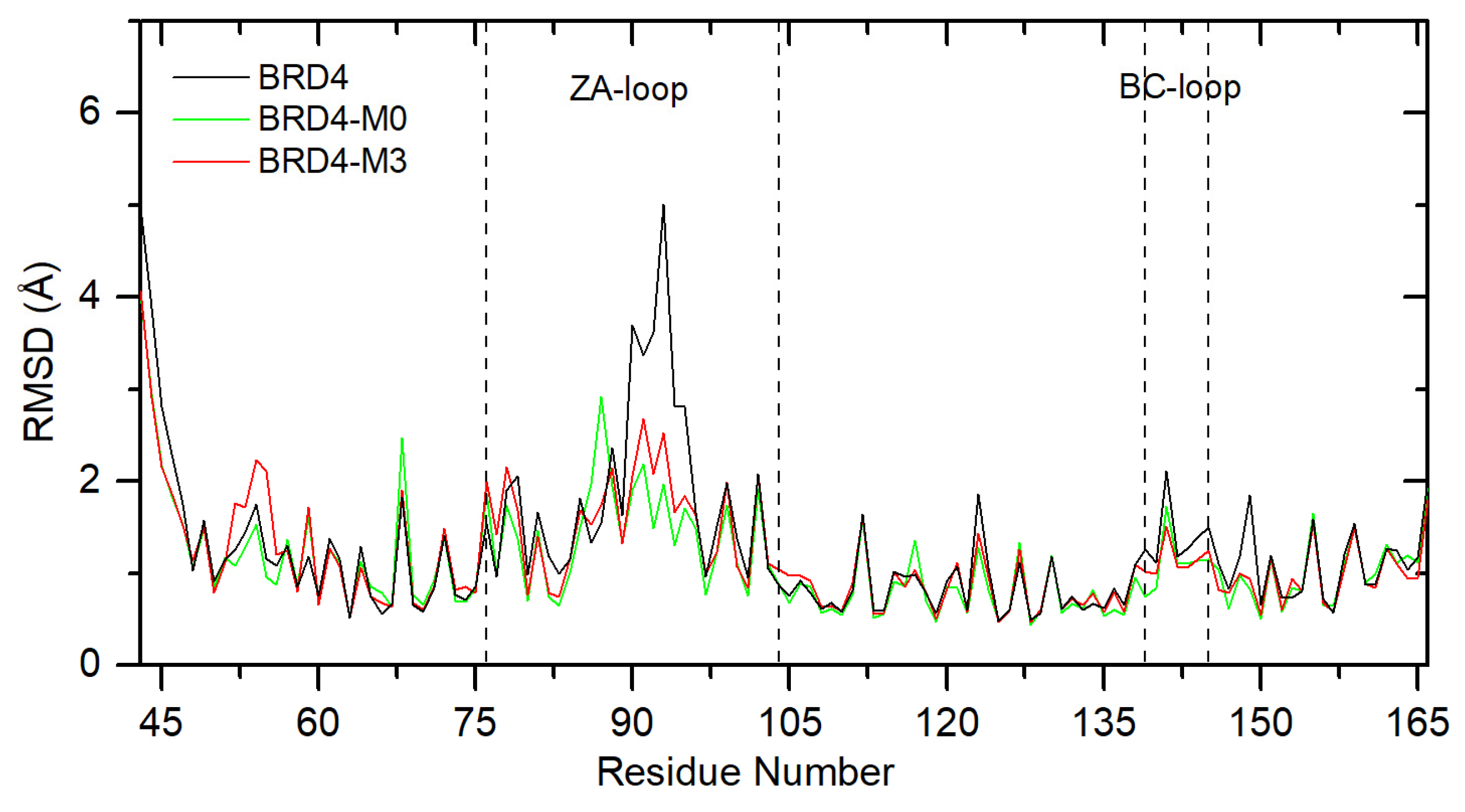
2.3. Molecular Dynamics Simulation Results
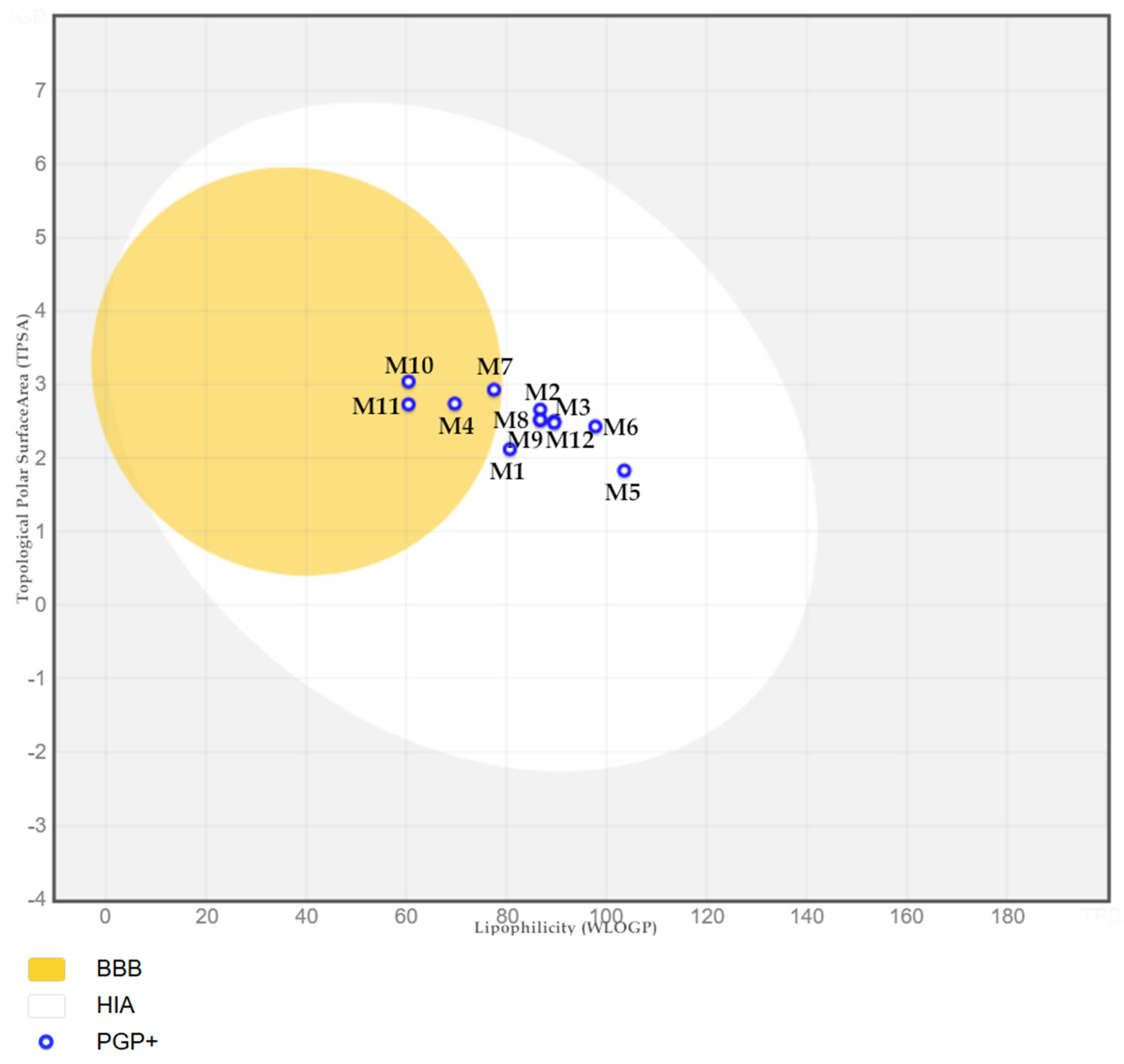
2.4. ADMET Properties of the Tested Compounds
3. Materials and Methods
3.1. Molecular Docking
3.2. Molecular Dynamics Simulation Details
3.3. ADMET Evaluation
4. Conclusions and Summary
- -
- All designed compounds bind to the active site of the three tested proteins with negative binding affinity;
- -
- Molecular dynamics simulation confirmed the stability of the formed complexes, and the calculated binding free energies for all derivatives were found to be negative (Table 7);
- -
- Reducing the size of the molecule does not affect key structural elements that determine binding to selected molecular targets;
- -
- The introduced modifications had a positive impact on the ADMET parameter profile (Table 9);
- -
- -
- Potential disadvantages, such as a short biological half life or effects on metabolic enzymes, can be overcome, for example, by developing a suitable formulation of the new drug.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orlando, B.J.; Liao, M. ABCG2 Transports Anticancer Drugs via a Closed-to-Open Switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, L.; Li, M.; Wang, S.; Wang, C. Case Report: 7-Ethyl-10-Hydroxycamptothecin, a DNA Topoisomerase I Inhibitor, Performs BRD4 Inhibitory Activity and Inhibits Human Leukemic Cell Growth. Front. Pharmacol. 2021, 12, 664176. [Google Scholar] [CrossRef]
- Bailly, C. Irinotecan: 25 Years of Cancer Treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef]
- Mathijssen, R.H.J.; Verweij, J.; Loos, W.J.; De Bruijn, P.; Nooter, K.; Sparreboom, A. Irinotecan Pharmacokinetics-Pharmacodynamics: The Clinical Relevance of Prolonged Exposure to SN-38. Br. J. Cancer 2002, 87, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Sharkey, R.M. Antibody-Drug Conjugates Targeting TROP-2 and Incorporating SN-38: A Case Study of Anti-TROP-2 Sacituzumab Govitecan. MAbs 2019, 11, 987–995. [Google Scholar] [CrossRef]
- Nascimento, I.J.d.S.; de Aquino, T.M.; da Silva-Júnior, E.F. The New Era of Drug Discovery: The Power of Computer-Aided Drug Design (CADD). Lett. Drug Des. Discov. 2022, 19, 951–955. [Google Scholar] [CrossRef]
- Tandon, H.; Chakraborty, T.; Suhag, V. A Concise Review on the Significance of QSAR in Drug Design. Chem. Biomol. Eng. 2019, 4, 45. [Google Scholar] [CrossRef]
- Dong, J.; Hao, X. Docking-based Virtual Screening of BRD4 (BD1) Inhibitors: Assessment of Docking Methods, Scoring Functions and in Silico Molecular Properties. BMC Chem. 2024, 18, 247. [Google Scholar] [CrossRef]
- Ahmad, S.; Hassan, M.I.; Gupta, D.; Dwivedi, N.; Islam, A. Design and Evaluation of Pyrimidine Derivatives as Potent Inhibitors of ABCG2, a Breast Cancer Resistance Protein. 3 Biotech 2022, 12, 182. [Google Scholar] [CrossRef]
- Redinbo, M.R.; Stewart, L.; Kuhn, P.; Champoux, J.J.; Hol, W.G.J. Crystal Structures of Human Topoisomerase I in Covalent and Noncovalent Complexes with DNA. Science 1998, 279, 1504–1513. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I−DNA Covalent Complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the Human Multidrug Transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the Breast Cancer Resistance Protein (BCRP/ABCG2) in Drug Transport—An Update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET Modeling Approaches in Drug Discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Andrushko, V.; Andrushko, N. Principles, Concepts and Strategies of Stereoselective Synthesis; Wiley: Hoboken, NJ, USA, 2013; ISBN 9781118596784. [Google Scholar]
- Parr, B.T.; Davies, H.M.L. Highly Stereoselective Synthesis of Cyclopentanes Bearing Four Stereocentres by a Rhodium Carbene-Initiated Domino Sequence. Nat. Commun. 2014, 5, 4455. [Google Scholar] [CrossRef] [PubMed]
- Caddick, S.; Cheung, S.; Doyle, V.E.; Frost, L.M.; Soscia, M.G.; Delisser, V.M.; Williams, M.R.V.; Etheridge, Z.C.; Khan, S.; Hitchcock, P.B.; et al. Stereoselective Synthesis of Polyfunctionalised Hydroxylated Cyclopentanes from Dihydroxylated 2-Cyclopentenone Derivatives. Tetrahedron 2001, 57, 6295–6303. [Google Scholar] [CrossRef]
- Abdel-Jalil, R.J.; Steinbrecher, T.; Al-Harthy, T.; Mahal, A.; Abou-Zied, O.K.; Voelter, W. Stereoselective Synthesis and Molecular Modeling of Chiral Cyclopentanes. Carbohydr. Res. 2015, 415, 12–16. [Google Scholar] [CrossRef]
- Kaeobamrung, J.; Bode, J.W. Stereodivergency of Triazolium and Imidazolium-Derived NHcs for Catalytic, Enantioselective Cyclopentane Synthesis. Org. Lett. 2009, 11, 677–680. [Google Scholar] [CrossRef]
- Grandvuinet, A.S.; Steffansen, B. Interactions between Organic Anions on Multiple Transporters in Caco-2 Cells. J. Pharm. Sci. 2011, 100, 3817–3830. [Google Scholar] [CrossRef] [PubMed]
- Volpe, D.A. Variability in Caco-2 and MDCK Cell-Based Intestinal Permeability Assays. J. Pharm. Sci. 2008, 97, 712–725. [Google Scholar] [CrossRef]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby Canine Kidney) Cells: A Tool for Membrane Permeability Screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Zhao, Q.; Peng, H.; Hou, T. ADME Evaluation in Drug Discovery. 10. Predictions of P-Glycoprotein Inhibitors Using Recursive Partitioning and Naive Bayesian Classification Techniques. Mol. Pharm. 2011, 8, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, L.; Li, Y.; Tian, S.; Sun, H.; Hou, T. ADMET Evaluation in Drug Discovery. 13. Development of in Silico Prediction Models for p-Glycoprotein Substrates. Mol. Pharm. 2014, 11, 716–726. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Staker, B.L.; Burgin, A.B.; Pourquier, P.; Pommier, Y.; Stewart, L.; Redinbo, M.R. Mechanisms of Camptothecin Resistance by Human Topoisomerase I Mutations. J. Mol. Biol. 2004, 339, 773–784. [Google Scholar] [CrossRef]
- Wang, J.; Erazo, T.; Ferguson, F.M.; Buckley, D.L.; Gomez, N.; Muñoz-Guardiola, P.; Diéguez-Martínez, N.; Deng, X.; Hao, M.; Massefski, W.; et al. Structural and Atropisomeric Factors Governing the Selectivity of Pyrimido-Benzodiazipinones as Inhibitors of Kinases and Bromodomains. ACS Chem. Biol. 2018, 13, 2438–2448. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Citation; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Velázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A Web Server for Automatic Structure Matching and RMSD Calculations among Identical and Similar Compounds in Protein-Ligand Docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Leppard, J.B.; Champoux, J.J. Human DNA Topoisomerase I: Relaxation, Roles, and Damage Control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef]
- Corbett, K.D.; Berger, J.M. Structure, Molecular Mechanisms, and Evolutionary Relationships in DNA Topoisomerases. Annu. Rev. Biophys. 2004, 33, 95–118. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Salerno, S.; Da Settimo, F.; Taliani, S.; Simorini, F.; La Motta, C.; Fornaciari, G.; Marini, A.M. Recent Advances in the Development of Dual Topoisomerase I and II Inhibitors as Anticancer Drugs. Curr. Med. Chem. 2010, 17, 4270–4290. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Kim, C.; Zhou, M.M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- White, M.E.; Fenger, J.M.; Carson, W.E. Emerging Roles of and Therapeutic Strategies Targeting BRD4 in Cancer. Cell Immunol. 2019, 337, 48–53. [Google Scholar] [CrossRef]
- Polgar, O.; Robey, R.W.; Bates, S.E. ABCG2: Structure, Function and Role in Drug Response. Expert. Opin. Drug Metab. Toxicol. 2008, 4, 1–15. [Google Scholar] [CrossRef]
- Moinul, M.; Amin, S.A.; Jha, T.; Gayen, S. Updated Chemical Scaffolds of ABCG2 Inhibitors and Their Structure-Inhibition Relationships for Future Development. Eur. J. Med. Chem. 2022, 241, 114628. [Google Scholar] [CrossRef]
- Nielsen, D.L.; Palshof, J.A.; Brünner, N.; Stenvang, J.; Viuff, B.M. Implications of ABCG2 Expression on Irinotecan Treatment of Colorectal Cancer Patients: A Review. Int. J. Mol. Sci. 2017, 18, 1926. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M0 | M1 | M2 | M3 | M4 | M5 | M6 | |
|---|---|---|---|---|---|---|---|
| van der Waals interactions | DG A:12 ARG C:488 PTR C:723 ASN C:722 THR C:718 | ARG C:488 PTR C:723 ASN C:722 ILE C:535 | ARG C:364 DG A:12 ARG C:488 PTR C:723 THR C:718 ASP C:533 ILE C:535 | ARG C:364 DG A:12 ARG C:488 ASN C:722 THR C:718 LYS C:452 | ARG C:364 DG A:12 ARG C:488 PTR C:723 ASN C:722 THR C:718 | ARG C:364 DG A:12 ARG C:488 PTR C:723 ASN C:722 THR C:718 | ARG C:364 DG A:12 ARG C:488 PTR C:723 ASN C:722 THR C:718 HIS C:632 |
| Hydrogen bonds | ARG C:364 LYS C:532 GLU C:356 ASP C:533 | DG A:11 ARG C:364 THR C:718 | DG A:11 DA B:113 LYS C:532 | DG A:11 DA B:113 LYS C:532 | DG A:11 LYS C:532 | DG A:11 LYS C:532 | DG A:11 DA B:113 LYS C:532 |
| Carbon–hydrogen interactions | DT A:10 | DG A:11 ASP C:533 HIS C:632 DG A:12 | DT A:10 | DT A:10 DG A:11 | DT A:10 DG A:11 ASN C:532 | DT A:10 DG A:11 ASP C:533 | DT A:10 DG A:11 |
| π-type interactions | DA B:113 DG A:11 DC B:112 | DT A:10 DG A:11 DA B:113 DC B:112 | DT A:10 DG A:11 DA B:113 DC B:112 | DT A:10 DG A:11 DA B:113 DC B:112 | DG A:11 DA B:113 DC B:112 DT A:10 | DT A:10 DG A:11 DA B:113 DC B:112 | DT A:10 DG A:11 DA B:113 DC B:112 |
| Alkyl/π–alkyl interactions | DG A:11 | DA B:113 | DT A:10 DG A:11 DA B:113 | DT A:10 | DT A:10 | DT A:10 | DT A:10 DA B:113 |
| M7 | M8 | M9 | M10 | M11 | M12 | |
|---|---|---|---|---|---|---|
| van der Waals interactions | ARG C:364 DG A:12 ARG C:488 PTR C:723 ASN C:722 THR C:718 LYS C:532 | ARG C:364 DG A:12 ARG C:488 PTR C:723 ASN C:722 THR C:718 ASP C:533 HIS C:632 | DG A:12 ARG C:488 PTR C:723 ASN C:722 THR C:718 | ARG C:364 DG A:12 ARG C:488 PTR C:723 ASN C:722 THR C:718 ASP C:533 | ARG C:488 ASN C:722 THR C:718 | ARG C:488 PTR C:723 ASN C:722 THR C:718 ASP C:533 ASN C:532 GLU C:356 |
| Hydrogen bonds | DG A:11 DA B:113 | DG A:11 DA B:113 LYS C:532 | DG A:11 LYS C:532 LYS C:751 DT A:10 | DG A:11 LYS C:532 | ARG C:364 LYS C:532 | DG A:11 LYS C:532 |
| Carbon–hydrogen interactions | DT A:10 ASP C:533 | DT A:10 DG A:11 GLU C:356 | ASP C:533 DA B:113 DA B:114 | DT A:10 | PTR C:723 | DT A:10 DG A:11 |
| π-type interactions | DG A:11 DA B:113 DC B:112 | DT A:10 DG A:11 DA B:113 DC B:112 | DT A:11 DT A:10 | DG A:11 DA B:113 DC B:112 | DA B:113 DG A:11 DC B:112 | DA B:113 DG A:11 DC B:112 |
| π–alkyl/alkyl | DT A:10 DA B:113 | DT A:10 DA B:113 | LEU C:721 | DT A:10 DA B:113 | DT A:10 DG A:11 DA B:113 | DT A:10 DA B:113 |
| M0 | M1 | M2 | M3 | M4 | M5 | M6 | |
|---|---|---|---|---|---|---|---|
| van der Waals interactions | ASP A:88 ASP A:144 | TRP A:81 LYS A:91 ASP A:88 GLN A:85 PRO A:86 LEU A:96 PHE A:83 TYR A:139 CYS A:136 | TRP A:81 GLN A:85 PRO A:86 PHE A:83 TYR A:139 LEU A:94 ASN A:140 | TRP A:81 PHE A:83 TYR A:139 LEU A:94 ASN A:140 TYR A:97 ASN A:135 LEU A:92 ASP A:144 | PRO A:86 PHE A:83 TYR A:139 CYS A:136 TYR A:97 | ASP A:88 GLN A:85 PRO A:86 PHE A:83 TYR A:139 ASN A:140 ASN A:135 | TRP A:81 PHE A:83 TYR A:139 LEU A:94 TYR A:97 LEU A:92 ASP A:144 |
| Hydrogen bond | ASP A:145 GLN A:85 TRP A:81 | PRO A: 82 ASN A:140 | ASP A:88 TYR A:97 | ILE A:146 ASP A:145 | PRO A:82 ASN A:140 | TYR A:97 | |
| Carbon–hydrogen interactions | VAL A:87 | ASN A:140 | ASN A:135 | PRO A:82 GLN A:85 | MET A:132 | ||
| π-type interactions | PRO A:86 ILE A:146 | LEU A:92 ILE A:146 | ILE A:146 | LEU A:92 | ILE A:146 | LEU A:92 ILE A:146 | ILE A:146 |
| π–alkyl/alkyl | LEU A:92 | VAL A:87 TYR A:97 LEU A:92 | LEU A:92 VAL A:87 CYS A:136 PRO A:82 | VAL A:87 CYS A:136 PRO A:82 | VAL A:87 PRO A:82 TRP A:81 | VAL A:87 CYS A:136 PRO A:82 TRP A:81 | VAL A:87 CYS A:136 PRO A:82 |
| M7 | M8 | M9 | M10 | M11 | M12 | |
|---|---|---|---|---|---|---|
| van der Waals interactions | TRP A:81 PHE A:83 TYR A:139 LEU A:94 TYR A:97 LEU A:92 ASP A:144 | ASP A:88 GLN A:85 PRO A:86 PHE A:83 TYR A:139 ASN A:140 | ASP A:88 GLN A:85 PRO A:86 PHE A:83 TYR A:139 LEU A:94 TYR A:97 | ASP A:88 PRO A:86 PHE A:83 TYR A:139 ASN A:140 MET A:132 TRP A:81 | ASP A:88 TYR A:139 ASN A:140 ASN A:135 LEU A:92 | PRO A:86 PHE A:83 TYR A:139 LEU A:94 ASN A:140 GLN A:84 |
| Hydrogen bonds | ASN A:140 ASP A:145 | TYR A:97 | PRO A:82 TYR A:97 | GLN A:85 | ASP A:88 TYR A:97 TRP A:81 | |
| Carbon–hydrogen interactions | ASN A:135 MET A:132 TRP A:81 | ASN A:140 VAL A:87 | ASN A:135 GLN A:85 | TRP A:81 TYR A:97 PRO A:86 | ASN A:135 | |
| π-type interactions | ILE A:146 | PHE A:83 CYS A:136 | LEU A:92 | |||
| π–alkyl/alkyl | ILE A:146 VAL A:87 CYS A:136 PRO A:82 | ILE A:146 LEU A:92 VAL A:87 CYS A:136 PRO A:82 TRP A:81 | ILE A:146 LEU A:92 VAL A:87 CYS A:136 PRO A:82 TRP A:81 | ILE A:146 LEU A:92 VAL A:87 CYS A:136 PRO A:82 | ILE A:146 VAL A:87 CYS A:136 PRO A:82 | ILE A:146 VAL A:87 CYS A:136 PRO A:82 |
| M0 | M1 | M2 | M3 | M4 | M5 | M6 | |
|---|---|---|---|---|---|---|---|
| van der Waals interactions | THR B:435 SER A:440 PHE A:432 SER B:440 THR B:542 ASN A:436 MET B:549 | LEU B:405 PHE B:432 THR B:435 SER A:440 THR A:435 PHE A:432 SER B:440 | LEU B:405 PHE B:432 THR B:435 SER A:440 THR A:435 PHE A:432 SER B:440 ASN B:436 | PHE B:432 THR B:435 SER A:440 THR A:435 PHE A::432 SER B:440 THR B:542 ILE B:543 | PHE B:432 THR B:435 SER A:440 THR A:435 PHE A:432 SER B:440 | PHE B:432 THR B:435 SER A:440 THR A:435 PHE A:432 SER B:440 ASN B:436 | SER A:440 THR A:435 PHE A:432 THR B:542 ASN B:436 LEU A:405 VAL B:546 |
| Hydrogen bonds | ASN B:436 | ASN B:436 | MET B:549 | ASN A:436 | MET A:549 | MET B:549 | ASN A:436 |
| Carbon–hydrgen interactions | THR A:435 | ASN A:436 | ASN A:436 | ASN B:436 | ASN A:436 ASN B:436 | ASN A:436 | |
| π-type interactions | PHE A:439 PHE B:439 | PHE A:439 PHE B:439 | MET A:549 PHE B:439 PHE A:439 | PHE A:439 PHE B:439 | PHE A:439 PHE B:439 | PHE B:439 PHE B:439 | PHE B:439 PHE A:439 |
| π–alkyl/alkyl | VAL A:546 VAL B:546 MET A:549 PHE B:432 | VAL A:546 VAL B:546 MET A:549 PHE A:439 | VAL A:546 VAL B:546 PHE A:439 | VAL A:546 VAL B:546 MET B:549 PHE B:439 | VAL A:546 VAL B:546 MET B:549 PHE B:439 | VAL A:546 VAL B:546 MET A:549 PHE A:439 | VAL A:546 MET B:549 PHE A:439 |
| Sulfur interactions | MET B:549 | MET B:549 | MET B:549 | MET B:549 MET A:549 | MET B:549 | MET B:549 | MET A:549 |
| M7 | M8 | M9 | M10 | M11 | M12 | |
|---|---|---|---|---|---|---|
| van der Waals interactions | PHE B:432 THR B:435 SER A:440 THR A:435 PHE A:432 SER B:440 ASN A:436 | PHE B:432 THR B:435 SER A:440 THR A:435 SER B:440 ASN A:436 ASN B:436 ILE B:543 | PHE B:432 THR A:435 PHE A:432 SER B:440 ASN A:436 ASN B:436 LEU A:405 THR A:435 | LEU B:405 THR B:435 THR A:435 SER B:440 ASN A:436 | PHE B:432 THR B:432 THR A:435 PHE A:432 ASN A:436 ASN B:436 | LEU B:405 PHE B:432 THR B:435 SER A:440 THR A:435 PHE A:432 SER B:440 ILE A:543 VAL B:401 |
| Hydrogen bonds | MET B:549 ASN B:436 | |||||
| Carbon–hydrogen interactions | ASN B:436 | THR B:542 | PHE A:439 | ASN A:436 | ||
| π-type interactions | PHE A:439 PHE A:439 | PHE A:439 PHE A:439 PHE B:439 | PHE A:439 PHE A:439 PHE B:439 | PHE A:439 PHE B:439 | PHE A:439 PHE B:439 | PHE A:439 PHE B:439 |
| π–alkyl/alkyl | VAL A:546 VAL B:546 MET B:549 PHE B:439 | VAL A:546 VAL B:546 | VAL B:546 MET A:549 PHE B:439 | VAL A:546 MET A:549 MET B:549 PHE A:432 PHE B:432 | VAL A:546 VAL B:546 MET A:549 MET B:549 PHE A:439 PHE B:439 | VAL A:546 VAL B:546 PHE A:439 |
| Sulfur interactions | MET A:549 | MET B:549 MET A:549 | MET B:549 | MET A:549 | MET B:549 MET A:549 |
| Compound | Binding Free Energy (MD) (kcal/mol) | ||
|---|---|---|---|
| TOPO-I/DNA | BRD4 | ABCG2 | |
| M0 | −18.19 ± 2.53 | −22.21 ± 2.69 | −25.42 ± 2.93 |
| M1 | −19.57 ± 2.52 | −21.60 ± 1.78 | −30.41 ± 1.93 |
| M2 | −15.01 ± 2.67 | −15.48 ± 1.87 | −35.00 ± 3.06 |
| M3 | −24.98 ± 2.65 | −25.92 ± 3.71 | −37.62 ± 3.73 |
| M4 | −21.13 ± 3.23 | −17.60 ± 1.86 | −38.99 ± 2.22 |
| M5 | −22.74 ± 1.29 | −21.02 ± 2.45 | −36.24 ± 1.80 |
| M6 | −18.97 ± 1.38 | −24.67 ± 2.53 | −36.05 ± 2.05 |
| M7 | −24.09 ± 1.05 | −23.84 ± 2.63 | −37.36 ± 2.11 |
| M8 | −23.87 ± 1.39 | −19.93 ± 2.15 | −37.76 ± 1.51 |
| M9 | −23.63 ± 3.25 | −21.28 ± 2.77 | −43.97 ± 1.83 |
| M10 | −19.50 ± 2.24 | −22.01 ± 1.94 | −34.06 ± 3.06 |
| M11 | −28.73 ± 1.26 | −23.76 ± 2.23 | −28.10 ± 2.40 |
| M12 | −22.67 ± 2.96 | −25.06 ± 2.61 | −38.89 ± 2.09 |
| Property | MW [Da] | HBA | HBD | Nrot | TPSA [Å2] | logP | logS | SAscore |
|---|---|---|---|---|---|---|---|---|
| M0 | 392.14 | 7 | 2 | 2 | 101.65 | 1.045 | −3.581 | - |
| M1 | 332.17 | 6 | 2 | 5 | 80.68 | 1.027 | −2.407 | 4.27 |
| M2 | 372.17 | 7 | 1 | 7 | 86.75 | 1.530 | −2.786 | 3.83 |
| M3 | 371.18 | 7 | 2 | 7 | 89.55 | 1.551 | −2.775 | 3.86 |
| M4 | 344.17 | 6 | 1 | 6 | 69.68 | 1.935 | −3.077 | 3.70 |
| M5 | 357.17 | 7 | 3 | 6 | 103.54 | 0.963 | −2.608 | 3.70 |
| M6 | 358.15 | 7 | 2 | 6 | 97.75 | 1.491 | −3.109 | 3.69 |
| M7 | 356.17 | 6 | 1 | 6 | 77.52 | 1.813 | −3.069 | 3.73 |
| M8 | 372.17 | 7 | 1 | 7 | 86.75 | 2.073 | −3.247 | 3.83 |
| M9 | 386.18 | 7 | 1 | 8 | 86.75 | 2.631 | −3.528 | 3.97 |
| M10 | 328.18 | 5 | 1 | 5 | 60.45 | 2.587 | −3.418 | 3.67 |
| M11 | 314.16 | 5 | 1 | 5 | 60.45 | 1.857 | −2.910 | 3.56 |
| M12 | 385.20 | 7 | 2 | 8 | 89.55 | 1.394 | −2.496 | 3.89 |
| Property | Caco-2 | MDCK | Pgp Inhibitor | Pgp Substrate | Plasma Protein-Binding Parameter |
|---|---|---|---|---|---|
| M0 | −5.154 | −4.794 | --- | +++ | 98.89% |
| M1 | −5.030 | −4.858 | --- | - | 61.40% |
| M2 | −4.958 | −4.652 | - | + | 83.10% |
| M3 | −4.987 | −4.633 | --- | -- | 80.30% |
| M4 | −4.925 | −4.671 | + | - | 89.80% |
| M5 | −5.117 | −4.623 | --- | - | 74.90% |
| M6 | −5.117 | −4.984 | --- | --- | 91.20% |
| M7 | −4.987 | −4.764 | + | --- | 91.00% |
| M8 | −5.008 | −4.716 | ++ | -- | 97.30% |
| M9 | −4.990 | −4.698 | + | --- | 96.50% |
| M10 | −4.934 | −4.612 | ++ | --- | 95.20% |
| M11 | −4.902 | −4.611 | - | + | 95.60% |
| M12 | −4.923 | −4.615 | --- | +++ | 82.50% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czyżnikowska, Ż.; Mysłek, M.; Marciniak, A.; Płaczek, R.; Kotynia, A.; Krzyżak, E. In Silico Approach to Design of New Multi-Targeted Inhibitors Based on Quinoline Ring with Potential Anticancer Properties. Int. J. Mol. Sci. 2025, 26, 4620. https://doi.org/10.3390/ijms26104620
Czyżnikowska Ż, Mysłek M, Marciniak A, Płaczek R, Kotynia A, Krzyżak E. In Silico Approach to Design of New Multi-Targeted Inhibitors Based on Quinoline Ring with Potential Anticancer Properties. International Journal of Molecular Sciences. 2025; 26(10):4620. https://doi.org/10.3390/ijms26104620
Chicago/Turabian StyleCzyżnikowska, Żaneta, Martyna Mysłek, Aleksandra Marciniak, Remigiusz Płaczek, Aleksandra Kotynia, and Edward Krzyżak. 2025. "In Silico Approach to Design of New Multi-Targeted Inhibitors Based on Quinoline Ring with Potential Anticancer Properties" International Journal of Molecular Sciences 26, no. 10: 4620. https://doi.org/10.3390/ijms26104620
APA StyleCzyżnikowska, Ż., Mysłek, M., Marciniak, A., Płaczek, R., Kotynia, A., & Krzyżak, E. (2025). In Silico Approach to Design of New Multi-Targeted Inhibitors Based on Quinoline Ring with Potential Anticancer Properties. International Journal of Molecular Sciences, 26(10), 4620. https://doi.org/10.3390/ijms26104620