
Signature Gene Mutations in Colorectal Cancer: Potential Neoantigens for Cancer Vaccines

Abstract

1. Introduction
2. Signaling Pathways and Characteristics of Signature Gene Mutations in CRC
2.1. MMR in CRC
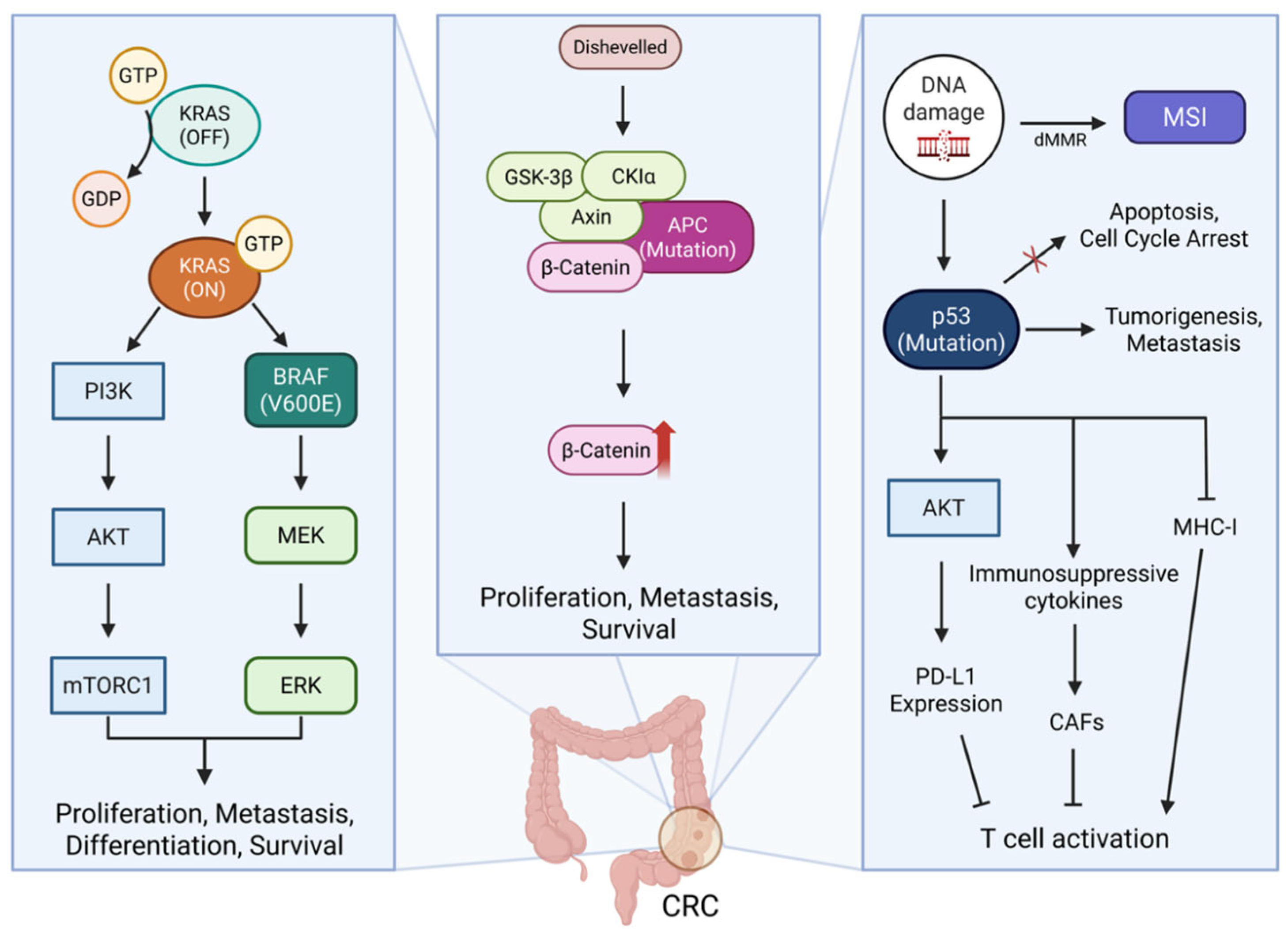
2.2. KRAS in CRC
2.3. BRAF in CRC
2.4. APC in CRC
2.5. TP53 in CRC
3. Immunological Function of TME in CRC
4. Applications of Cancer Vaccines Targeting Signature Gene Mutations in CRC
4.1. Types of Cancer Vaccines
4.1.1. Cell-Based Vaccine
4.1.2. Peptide-Based Vaccine
4.1.3. Nucleic Acid-Based Vaccine
4.1.4. Viral Vector-Based Vaccine
4.2. Advantages and Challenges of Cancer Vaccines Targeting Signature Genes
4.3. Development and Limitations of Signature Gene-Targeted Vaccines in CRC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| APC | Adenomatous polyposis coli |
| APCs | Antigens-presenting cells |
| Ascl2 | Achaete-scute family bHLH transcription factor 2 |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| CAFs | Cancer-associated fibroblasts |
| ChAd | Chimpanzee adenovirus |
| CK Iα | Casein kinase I alpha |
| CRC | Colorectal cancer |
| CTLs | Cytotoxic T lymphocytes |
| dMMR | Mismatch repair deficiency |
| DC | Dendritic cell |
| DTH | Delayed-type hypersensitivity |
| ECM | Extracellular matrix |
| EPCAM | Epithelial cell adhesion molecule |
| ERK | Extracellular signal-regulated kinase |
| FSP | Frameshift peptide |
| GaD | Gorilla adenovirus |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GSK-3β | Glycogen synthase kinase-3β |
| HIF-1α | Hypoxia-inducible factor-1 alpha |
| HLA | Human leukocyte antigen |
| ICIs | Immune checkpoint inhibitors |
| iCAFs | Inflammatory CAFs |
| ISC | Intestinal stem cell |
| KRAS | Kirsten rat sarcoma virus |
| LNP | Lipid nanoparticle |
| MEK | Mitogen-activated protein kinase |
| MHC | Major histocompatibility complex |
| MKI67 | Marker of proliferation Ki-67 |
| MLH1 | mutL homolog1 |
| MMR | Mismatch repair |
| MDSCs | Myeloid-derived suppressor cells |
| MSI | Microsatellite instability |
| MSI-H | Microsatellite instability-high |
| MSS | Microsatellite stable |
| mTORC1 | Mammalian target of rapamycin complex1 |
| myoCAFs | Myofibroblastic CAFs |
| MSH2 | muts homolog2 |
| MSH6 | muts homolog6 |
| NO | Nitric oxide |
| pDNA | Plasmid DNA |
| PD-L1 | Programmed cell death-ligand 1 |
| PCD | Programmed cell death |
| PI3K | Phosphatidylinositol-3-kinase |
| PMS2 | Postmeiotic segregation increased2 |
| PCD | Programmed cell death |
| ROS | Reactive oxygen species |
| samRNA | Self-amplifying mRNA |
| SLP | Synthetic long peptide |
| TAL | Tumor-associated lymphocyte |
| TAMs | Tumor-associated macrophages |
| TAAs | Tumor-associated antigens |
| TILs | Tumor-infiltrating lymphocytes |
| TMB | Tumor mutation burden |
| TME | Tumor microenvironment |
| Treg | Regulatory T cell |
| TSAs | Tumor-specific antigens |
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, H.; Xiang, T.; Wang, G. Clinical application of adaptive immune therapy in MSS colorectal cancer patients. Front. Immunol. 2021, 12, 762341. [Google Scholar] [CrossRef]
- Nunes, L.; Stenersen, J.M.; Kryeziu, K.; Sjöblom, T.; Glimelius, B.; Lothe, R.A.; Sveen, A. Co-occurring mutations identify prognostic subgroups of microsatellite stable colorectal cancer. Mol. Cancer 2024, 23, 264. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Song, I.H.; Lee, A.; Kang, J.; Lee, Y.S.; Lee, I.K.; Song, Y.S.; Lee, S.H. Enhancing the landscape of colorectal cancer using targeted deep sequencing. Sci. Rep. 2021, 11, 8154. [Google Scholar] [CrossRef]
- Poturnajova, M.; Furielova, T.; Balintova, S.; Schmidtova, S.; Kucerova, L.; Matuskova, M. Molecular features and gene expression signature of metastatic colorectal cancer. Oncol. Rep. 2021, 45, 10. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Goyal, S.; Pumpalova, Y.; Sonbol, M.B.; Das, S.; Haraldsdottir, S.; Ahn, D.; Ciombor, K.K.; Chen, Z.; Draper, A. Mismatch Repair (MMR) gene alteration and BRAF V600E mutation are potential predictive biomarkers of immune checkpoint inhibitors in MMR-deficient colorectal cancer. Oncologist 2021, 26, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Zhong, K.; Jiang, T.; Liu, Z.; Kwan, H.Y.; Su, T. The current understanding on the impact of KRAS on colorectal cancer. Biomed. Pharmacother. 2021, 140, 111717. [Google Scholar] [CrossRef]
- Fanelli, G.N.; Dal Pozzo, C.A.; Depetris, I.; Schirripa, M.; Brignola, S.; Biason, P.; Balistreri, M.; Dal Santo, L.; Lonardi, S.; Munari, G. The heterogeneous clinical and pathological landscapes of metastatic Braf-mutated colorectal cancer. Cancer Cell Int. 2020, 20, 30. [Google Scholar] [CrossRef]
- Feng, F.; Sun, H.; Zhao, Z.; Sun, C.; Zhao, Y.; Lin, H.; Yang, J.; Xiao, Y.; Wang, W.; Wu, D. Identification of APC mutation as a potential predictor for immunotherapy in colorectal cancer. J. Oncol. 2022, 2022, 6567998. [Google Scholar] [CrossRef]
- Wang, C.; Ouyang, C.; Cho, M.; Ji, J.; Sandhu, J.; Goel, A.; Kahn, M.; Fakih, M. Wild-type APC Is Associated with Poor Survival in Metastatic Microsatellite Stable Colorectal Cancer. Oncologist 2021, 26, 208–214. [Google Scholar] [CrossRef]
- Liebl, M.C.; Hofmann, T.G. The role of p53 signaling in colorectal cancer. Cancers 2021, 13, 2125. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, X.; Liu, H.; Wei, C.; Ru, H.; Qin, H.; Lai, H.; Meng, Y.; Wu, G.; Xie, W. Immune landscape and prognostic immune-related genes in KRAS-mutant colorectal cancer patients. J. Transl. Med. 2021, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Underwood, P.W.; Ruff, S.M.; Pawlik, T.M. Update on targeted therapy and immunotherapy for metastatic colorectal cancer. Cells 2024, 13, 245. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordāo, M.J.; Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Wang, L.; Geng, H.; Liu, Y.; Liu, L.; Chen, Y.; Wu, F.; Liu, Z.; Ling, S.; Wang, Y.; Zhou, L. Hot and cold tumors: Immunological features and the therapeutic strategies. MedComm 2023, 4, e343. [Google Scholar] [CrossRef]
- Ouyang, P.; Wang, L.; Wu, J.; Tian, Y.; Chen, C.; Li, D.; Yao, Z.; Chen, R.; Xiang, G.; Gong, J. Overcoming cold tumors: A combination strategy of immune checkpoint inhibitors. Front. Immunol. 2024, 15, 1344272. [Google Scholar] [CrossRef]
- Khosravi, G.R.; Mostafavi, S.; Bastan, S.; Ebrahimi, N.; Gharibvand, R.S.; Eskandari, N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun. 2024, 44, 521–553. [Google Scholar] [CrossRef]
- Benoit, A.; Vogin, G.; Duhem, C.; Berchem, G.; Janji, B. Lighting up the fire in the microenvironment of cold tumors: A major challenge to improve cancer immunotherapy. Cells 2023, 12, 1787. [Google Scholar] [CrossRef]
- Weng, J.; Li, S.; Zhu, Z.; Liu, Q.; Zhang, R.; Yang, Y.; Li, X. Exploring immunotherapy in colorectal cancer. J. Hematol. Oncol. 2022, 15, 95. [Google Scholar] [CrossRef]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Gwin, W.R., III; Mitchell, D.A. Vaccine therapies for cancer: Then and now. Target. Oncol. 2021, 16, 121–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, J.-A.; Zhang, H.-X.; Jiang, Y.-N.; Luo, W.-H. Cancer vaccines: Targeting KRAS-driven cancers. Expert Rev. Vaccines 2020, 19, 163–173. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef]
- Kumar, A.; Gautam, V.; Sandhu, A.; Rawat, K.; Sharma, A.; Saha, L. Current and emerging therapeutic approaches for colorectal cancer: A comprehensive review. World J. Gastrointest. Surg. 2023, 15, 495. [Google Scholar] [CrossRef]
- Kaczmarek, M.; Poznańska, J.; Fechner, F.; Michalska, N.; Paszkowska, S.; Napierała, A.; Mackiewicz, A. Cancer vaccine therapeutics: Limitations and effectiveness—A literature review. Cells 2023, 12, 2159. [Google Scholar] [CrossRef]
- Wan, Y.; Zhang, Y.; Wang, G.; Mwangi, P.M.; Cai, H.; Li, R. Recombinant KRAS G12D protein vaccines elicit significant anti-tumor effects in mouse CT26 tumor models. Front. Oncol. 2020, 10, 1326. [Google Scholar] [CrossRef]
- Ahmad, R.; Singh, J.K.; Wunnava, A.; Al-Obeed, O.; Abdulla, M.; Srivastava, S.K. Emerging trends in colorectal cancer: Dysregulated signaling pathways. Int. J. Mol. Med. 2021, 47, 14. [Google Scholar] [CrossRef]
- Wan, M.-l.; Wang, Y.; Zeng, Z.; Deng, B.; Zhu, B.-s.; Cao, T.; Li, Y.-k.; Xiao, J.; Han, Q.; Wu, Q. Colorectal cancer (CRC) as a multifactorial disease and its causal correlations with multiple signaling pathways. Biosci. Rep. 2020, 40, BSR20200265. [Google Scholar] [CrossRef]
- Zheng, Y.; Fu, Y.; Wang, P.-P.; Ding, Z.-Y. Neoantigen: A promising target for the immunotherapy of colorectal cancer. Dis. Markers 2022, 2022, 8270305. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.M.; Al Doghaither, H.A.; Al-Ghafari, A.B. General insight into cancer: An overview of colorectal cancer. Mol. Clin. Oncol. 2021, 15, 271. [Google Scholar] [CrossRef] [PubMed]
- Ijsselsteijn, R.; Jansen, J.G.; de Wind, N. DNA mismatch repair-dependent DNA damage responses and cancer. DNA Repair 2020, 93, 102923. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.-X.; Su, F.; Zhang, Y.; Zhang, X.-L.; Du, Y.-Y.; Gao, Y.-J.; Li, W.-L.; Hu, W.-Q.; Zhao, J. Oncological characteristics, treatments and prognostic outcomes in MMR-deficient colorectal cancer. Biomark. Res. 2024, 12, 89. [Google Scholar] [CrossRef]
- Dal Buono, A.; Gaiani, F.; Poliani, L.; Correale, C.; Laghi, L. Defects in MMR genes as a seminal example of personalized medicine: From diagnosis to therapy. J. Pers. Med. 2021, 11, 1333. [Google Scholar] [CrossRef]
- Venetis, K.; Frascarelli, C.; Bielo, L.B.; Cursano, G.; Adorisio, R.; Ivanova, M.; Mane, E.; Peruzzo, V.; Concardi, A.; Negrelli, M. Mismatch repair (MMR) and microsatellite instability (MSI) phenotypes across solid tumors: A comprehensive cBioPortal study on prevalence and prognostic impact. Eur. J. Cancer 2025, 217, 115233. [Google Scholar] [CrossRef]
- Taieb, J.; Svrcek, M.; Cohen, R.; Basile, D.; Tougeron, D.; Phelip, J.-M. Deficient mismatch repair/microsatellite unstable colorectal cancer: Diagnosis, prognosis and treatment. Eur. J. Cancer 2022, 175, 136–157. [Google Scholar] [CrossRef]
- Roudko, V.; Cimen Bozkus, C.; Greenbaum, B.; Lucas, A.; Samstein, R.; Bhardwaj, N. Lynch syndrome and MSI-H cancers: From mechanisms to “off-the-shelf” cancer vaccines. Front. Immunol. 2021, 12, 757804. [Google Scholar] [CrossRef]
- Patelli, G.; Tosi, F.; Amatu, A.; Mauri, G.; Curaba, A.; Patanè, D.; Pani, A.; Scaglione, F.; Siena, S.; Sartore-Bianchi, A. Strategies to tackle RAS-mutated metastatic colorectal cancer. ESMO Open 2021, 6, 100156. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Kilowski, K.A.; Dietrich, M.F.; Xiu, J.; Baca, Y.; Hinton, A.; Ahmad, S.; Herzog, T.J.; Thaker, P.; Holloway, R.W. KRAS mutations in endometrial cancers: Possible prognostic and treatment implications. Gynecol. Oncol. 2024, 191, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Ros, J.; Vaghi, C.; Baraibar, I.; Saoudi González, N.; Rodríguez-Castells, M.; García, A.; Alcaraz, A.; Salva, F.; Tabernero, J.; Elez, E. Targeting KRAS G12C Mutation in Colorectal Cancer, A Review: New Arrows in the Quiver. Int. J. Mol. Sci. 2024, 25, 3304. [Google Scholar] [CrossRef] [PubMed]
- Qunaj, L.; May, M.S.; Neugut, A.I.; Herzberg, B.O. Prognostic and therapeutic impact of the KRAS G12C mutation in colorectal cancer. Front. Oncol. 2023, 13, 1252516. [Google Scholar] [CrossRef]
- Poole, A.; Karuppiah, V.; Hartt, A.; Haidar, J.N.; Moureau, S.; Dobrzycki, T.; Hayes, C.; Rowley, C.; Dias, J.; Harper, S. Therapeutic high affinity T cell receptor targeting a KRASG12D cancer neoantigen. Nat. Commun. 2022, 13, 5333. [Google Scholar] [CrossRef]
- Cefalì, M.; Epistolio, S.; Palmarocchi, M.C.; Frattini, M.; De Dosso, S. Research progress on KRAS mutations in colorectal cancer. J. Cancer Metastasis Treat. 2021, 7, 26. [Google Scholar] [CrossRef]
- Negri, F.; Bottarelli, L.; de’Angelis, G.L.; Gnetti, L. KRAS: A druggable target in colon cancer patients. Int. J. Mol. Sci. 2022, 23, 4120. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.-i.; Restifo, N.P.; Yang, J.C. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef]
- Linette, G.P.; Bear, A.S.; Carreno, B.M. Facts and hopes in immunotherapy strategies targeting antigens derived from KRAS mutations. Clin. Cancer Res. 2024, 30, 2017–2024. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Li, X.; Chen, T.T.; Wang, J.X.; Zhou, Y.X.; Mu, X.L.; Du, Y.; Wang, J.L.; Tang, J.; Liu, J.Y. Personalised neoantigen-based therapy in colorectal cancer. Clin. Transl. Med. 2023, 13, e1461. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Fakih, M.; Tabernero, J. Management of BRAF-mutant metastatic colorectal cancer: A review of treatment options and evidence-based guidelines. Ann. Oncol. 2021, 32, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, R.M.; Labajos, V.A.; Ballena, S.L.; Macha, C.A.; Lezama, M.S.; Roman, C.P.; Beltran, P.M.; Torrejon, A.F. Targeting BRAF V600E in metastatic colorectal cancer: Where are we today? Ecancermedicalscience 2022, 16, 1489. [Google Scholar]
- Grassi, E.; Corbelli, J.; Papiani, G.; Barbera, M.A.; Gazzaneo, F.; Tamberi, S. Current therapeutic strategies in BRAF-mutant metastatic colorectal cancer. Front. Oncol. 2021, 11, 601722. [Google Scholar] [CrossRef]
- Liu, M.; Liu, Q.; Hu, K.; Dong, Y.; Sun, X.; Zou, Z.; Ji, D.; Liu, T.; Yu, Y. Colorectal cancer with BRAF V600E mutation: Trends in immune checkpoint inhibitor treatment. Crit. Rev. Oncol./Hematol. 2024, 204, 104497. [Google Scholar] [CrossRef]
- Shan, K.S.; Rehman, T.U.; Ivanov, S.; Domingo, G.; Raez, L.E. Molecular targeting of the BRAF proto-oncogene/mitogen-activated protein kinase (MAPK) pathway across cancers. Int. J. Mol. Sci. 2024, 25, 624. [Google Scholar] [CrossRef]
- Rodriquenz, M.G.; Ciardiello, D.; Latiano, T.P.; Maiorano, B.A.; Martinelli, E.; Silvestris, N.; Ciardiello, F.; Maiello, E. Exploring biological heterogeneity and implications on novel treatment paradigm in BRAF-mutant metastatic colorectal cancer. Crit. Rev. Oncol./Hematol. 2022, 173, 103657. [Google Scholar] [CrossRef]
- Tabernero, J.; Ros, J.; Élez, E. The Evolving Treatment Landscape in BRAF-V600E-Mutated Metastatic Colorectal Cancer. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2022; pp. 1–10. [Google Scholar]
- Abushukair, H.M.; Zaitoun, S.M.a.; Saeed, A. Insight on BRAFV600E mutated colorectal cancer immune microenvironment. World J. Gastrointest. Oncol. 2022, 14, 1213. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Xu, Y.; Huang, Z.; Cheng, G.; Xie, M.; Zhou, Z.; Yu, Y.; Xi, W.; Fan, Y. Tumor immune microenvironment and immunotherapy efficacy in BRAF mutation non-small-cell lung cancer. Cell Death Dis. 2022, 13, 1064. [Google Scholar] [CrossRef]
- Cho, S.M.; Esmail, A.; Abdelrahim, M. Triple-regimen of vemurafenib, irinotecan, and cetuximab for the treatment of BRAFV600E-mutant CRC: A case report and review. Front. Pharmacol. 2021, 12, 795381. [Google Scholar] [CrossRef] [PubMed]
- Ros, J.; Saoudi, N.; Baraibar, I.; Salva, F.; Tabernero, J.; Elez, E. Encorafenib plus cetuximab for the treatment of BRAF-V600E-mutated metastatic colorectal cancer. Ther. Adv. Gastroenterol. 2022, 15, 17562848221110644. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Wu, C.J. Cancer vaccines: Steering T cells down the right path to eradicate tumors. Cancer Discov. 2019, 9, 476–481. [Google Scholar] [CrossRef]
- Sotirov, S.; Dimitrov, I. Tumor-derived antigenic peptides as potential cancer vaccines. Int. J. Mol. Sci. 2024, 25, 4934. [Google Scholar] [CrossRef]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2020, 22, 130. [Google Scholar] [CrossRef]
- Tejeda-Muñoz, N.; Binder, G.; Mei, K.-C. Emerging therapeutic strategies for Wnt-dependent colon cancer targeting macropinocytosis. Cells Dev. 2024, 180, 203974. [Google Scholar] [CrossRef]
- Hankey, W.; Frankel, W.L.; Groden, J. Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: Implications for therapeutic targeting. Cancer Metastasis Rev. 2018, 37, 159–172. [Google Scholar] [CrossRef]
- Wang, T.; Fu, J.; Huang, Y.; Fu, C. Mechanism of APC truncation involved in colorectal cancer tumorigenesis. Oncol. Lett. 2024, 29, 2. [Google Scholar] [CrossRef]
- Song, P.; Gao, Z.; Bao, Y.; Chen, L.; Huang, Y.; Liu, Y.; Dong, Q.; Wei, X. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J. Hematol. Oncol. 2024, 17, 46. [Google Scholar] [CrossRef]
- Xi, Y.; Cui, W.; Tan, Y.; Zhang, Q.; Duan, Q.; Chen, D.-s. Impact of APC mutations on prognosis and tumor microenvironment in colorectal signet ring cell carcinoma. J. Clin. Oncol. 2024, 42, e15500. [Google Scholar] [CrossRef]
- Aghabozorgi, A.S.; Bahreyni, A.; Soleimani, A.; Bahrami, A.; Khazaei, M.; Ferns, G.A.; Avan, A.; Hassanian, S.M. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie 2019, 157, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Kelson, C.O.; Zaytseva, Y.Y. Altered lipid metabolism in APC-driven colorectal cancer: The potential for therapeutic intervention. Front. Oncol. 2024, 14, 1343061. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Sai, Y.; Zhou, L.; Liu, Z.; Du, P.; Wu, J.; Zhang, J. Current insights into the regulation of programmed cell death by TP53 mutation in cancer. Front. Oncol. 2022, 12, 1023427. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Lei, L.; Lu, Q.; Ma, G.; Li, T.; Deng, J.; Li, W. P53 protein and the diseases in central nervous system. Front. Genet. 2023, 13, 1051395. [Google Scholar] [CrossRef]
- Kim, J.Y.; Jung, J.; Kim, K.M.; Lee, J.; Im, Y.H. TP53 mutations predict poor response to immunotherapy in patients with metastatic solid tumors. Cancer Med. 2023, 12, 12438–12451. [Google Scholar] [CrossRef]
- Hassin, O.; Nataraj, N.B.; Shreberk-Shaked, M.; Aylon, Y.; Yaeger, R.; Fontemaggi, G.; Mukherjee, S.; Maddalena, M.; Avioz, A.; Iancu, O.; et al. Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nat. Commun. 2022, 13, 2800. [Google Scholar] [CrossRef]
- Ho, V.; Chung, L.; Lim, S.H.; Ma, Y.; Wang, B.; Lea, V.; Abubakar, A.; Ng, W.; Lee, M.; Roberts, T.L. Prognostic impact of TP53 mutations and tumor mutational load in colorectal cancer. Gastrointest. Disord. 2022, 4, 165–179. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Sheng, J.; Wu, F.; Li, K.; Huang, R.; Wang, X.; Jiao, T.; Guan, X.; Lu, Y.; et al. P53-R273H mutation enhances colorectal cancer stemness through regulating specific lncRNAs. J. Exp. Clin. Cancer Res. 2019, 38, 379. [Google Scholar] [CrossRef]
- Liu, N.; Jiang, X.; Guo, L.; Zhang, C.; Jiang, M.; Sun, Z.; Zhang, Y.; Mi, W.; Li, J.; Fu, Y. Mutant p53 achieved Gain-of-Function by promoting tumor growth and immune escape through PHLPP2/AKT/PD-L1 pathway. Int. J. Biol. Sci. 2022, 18, 2419. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, T.; Zhu, W.; Xie, S.; Zhao, Z.; Feng, B.; Guo, H.; Yang, R. Targeting MDSC for immune-checkpoint blockade in cancer immunotherapy: Current progress and new prospects. Clin. Med. Insights Oncol. 2021, 15, 11795549211035540. [Google Scholar] [CrossRef] [PubMed]
- Makaremi, S.; Asadzadeh, Z.; Hemmat, N.; Baghbanzadeh, A.; Sgambato, A.; Ghorbaninezhad, F.; Safarpour, H.; Argentiero, A.; Brunetti, O.; Bernardini, R. Immune checkpoint inhibitors in colorectal cancer: Challenges and future prospects. Biomedicines 2021, 9, 1075. [Google Scholar] [CrossRef] [PubMed]
- Zafari, N.; Khosravi, F.; Rezaee, Z.; Esfandyari, S.; Bahiraei, M.; Bahramy, A.; Ferns, G.A.; Avan, A. The role of the tumor microenvironment in colorectal cancer and the potential therapeutic approaches. J. Clin. Lab. Anal. 2022, 36, e24585. [Google Scholar] [CrossRef]
- Li, N.; Zhu, Q.; Tian, Y.; Ahn, K.J.; Wang, X.; Cramer, Z.; Jou, J.; Folkert, I.W.; Yu, P.; Adams-Tzivelekidis, S. Mapping and modeling human colorectal carcinoma interactions with the tumor microenvironment. Nat. Commun. 2023, 14, 7915. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, H.; Jiang, D.; Li, Y.; Feng, L.; Tian, C.; Pu, M.; Wang, X.; Zhang, J.; Hu, Y. Multi gene mutation signatures in colorectal cancer patients: Predict for the diagnosis, pathological classification, staging and prognosis. BMC Cancer 2021, 21, 380. [Google Scholar] [CrossRef]
- Park, K.; Veena, M.S.; Shin, D.S. Key players of the immunosuppressive tumor microenvironment and emerging therapeutic strategies. Front. Cell Dev. Biol. 2022, 10, 830208. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L.; Zhang, Z.; Wu, P.; Zhang, Y.; Chen, X. Advances in targeting tumor microenvironment for immunotherapy. Front. Immunol. 2024, 15, 1472772. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365. [Google Scholar] [CrossRef]
- Wang, J.X.; Choi, S.Y.; Niu, X.; Kang, N.; Xue, H.; Killam, J.; Wang, Y. Lactic acid and an acidic tumor microenvironment suppress anticancer immunity. Int. J. Mol. Sci. 2020, 21, 8363. [Google Scholar] [CrossRef] [PubMed]
- Sieminska, I.; Baran, J. Myeloid-derived suppressor cells in colorectal cancer. Front. Immunol. 2020, 11, 1526. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Hidalgo, I.; Sorolla, A.; Montal, R.; Pallisé, O.; Salud, A.; Parisi, E. Microenvironmental reactive oxygen species in colorectal cancer: Involved processes and therapeutic opportunities. Cancers 2021, 13, 5037. [Google Scholar] [CrossRef]
- Wang, H.; Tian, T.; Zhang, J. Tumor-associated macrophages (TAMs) in colorectal cancer (CRC): From mechanism to therapy and prognosis. Int. J. Mol. Sci. 2021, 22, 8470. [Google Scholar] [CrossRef]
- Janssen, J.B.; Medema, J.P.; Gootjes, E.C.; Tauriello, D.V.; Verheul, H.M. Mutant RAS and the tumor microenvironment as dual therapeutic targets for advanced colorectal cancer. Cancer Treat. Rev. 2022, 109, 102433. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Kumagai, S.; Momoi, Y.; Nishikawa, H. Immunogenomic cancer evolution: A framework to understand cancer immunosuppression. Sci. Immunol. 2025, 10, eabo5570. [Google Scholar] [CrossRef]
- Dzobo, K.; Dandara, C. Architecture of cancer-associated fibroblasts in tumor microenvironment: Mapping their origins, heterogeneity, and role in cancer therapy resistance. Omics A J. Integr. Biol. 2020, 24, 314–339. [Google Scholar] [CrossRef]
- Shasha, T.; Gruijs, M.; van Egmond, M. Mechanisms of colorectal liver metastasis development. Cell. Mol. Life Sci. 2022, 79, 607. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, H.; Mao, Y.; Jiang, S.; Yi, H.; Zhang, Z.; Wang, M.; Zong, Z. Myeloid-derived suppressor cells: Key immunosuppressive regulators and therapeutic targets in colorectal cancer. Int. J. Oncol. 2024, 65, 154711. [Google Scholar] [CrossRef]
- Le, I.; Dhandayuthapani, S.; Chacon, J.; Eiring, A.M.; Gadad, S.S. Harnessing the immune system with cancer vaccines: From prevention to therapeutics. Vaccines 2022, 10, 816. [Google Scholar] [CrossRef] [PubMed]
- Ros, J.; Balconi, F.; Baraibar, I.; Saoudi Gonzalez, N.; Salva, F.; Tabernero, J.; Elez, E. Advances in immune checkpoint inhibitor combination strategies for microsatellite stable colorectal cancer. Front. Oncol. 2023, 13, 1112276. [Google Scholar] [CrossRef] [PubMed]
- Najafi, S.; Mortezaee, K. Advances in dendritic cell vaccination therapy of cancer. Biomed. Pharmacother. 2023, 164, 114954. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; Scaggiante, B.; Morris, R.; Chai, D.; Catalano, M.; Tardiel-Cyril, D.R.; Neeli, P.; Roviello, G.; Mondani, G.; Li, Y. Therapeutic cancer vaccines: From biological mechanisms and engineering to ongoing clinical trials. Cancer Treat. Rev. 2022, 109, 102429. [Google Scholar] [CrossRef]
- Yu, J.; Sun, H.; Cao, W.; Song, Y.; Jiang, Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp. Hematol. Oncol. 2022, 11, 3. [Google Scholar] [CrossRef]
- Filin, I.Y.; Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Recent advances in experimental dendritic cell vaccines for cancer. Front. Oncol. 2021, 11, 730824. [Google Scholar] [CrossRef]
- Lee, K.-W.; Yam, J.W.P.; Mao, X. Dendritic cell vaccines: A shift from conventional approach to new generations. Cells 2023, 12, 2147. [Google Scholar] [CrossRef]
- Kim, V.M.; Pan, X.; Soares, K.C.; Azad, N.S.; Ahuja, N.; Gamper, C.J.; Blair, A.B.; Muth, S.; Ding, D.; Ladle, B.H. Neoantigen-based EpiGVAX vaccine initiates antitumor immunity in colorectal cancer. JCI Insight 2020, 5, e136368. [Google Scholar] [CrossRef]
- Abd-Aziz, N.; Poh, C.L. Development of peptide-based vaccines for cancer. J. Oncol. 2022, 2022, 9749363. [Google Scholar] [CrossRef]
- Buonaguro, L.; Tagliamonte, M. Peptide-based vaccine for cancer therapies. Front. Immunol. 2023, 14, 1210044. [Google Scholar] [CrossRef]
- Hamley, I.W. Peptides for vaccine development. ACS Appl. Bio Mater. 2022, 5, 905–944. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.J.; Burgess-Brown, N.A.; Jiang, S. Beyond just peptide antigens: The complex world of peptide-based cancer vaccines. Front. Immunol. 2021, 12, 696791. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Wainberg, Z.A.; Weekes, C.D.; Furqan, M.; Kasi, P.M.; Devoe, C.E.; Leal, A.D.; Chung, V.; Perry, J.; Seenappa, L. AMPLIFY-201, a first-in-human safety and efficacy trial of adjuvant ELI-002 2P immunotherapy for patients with high-relapse risk with KRAS G12D-or G12R-mutated pancreatic and colorectal cancer. J. Clin. Oncol. 2023, 41, 2528. [Google Scholar] [CrossRef]
- Pant, S.; Wainberg, Z.A.; Weekes, C.D.; Furqan, M.; Kasi, P.M.; Devoe, C.E.; Leal, A.D.; Chung, V.; Basturk, O.; VanWyk, H. Lymph-node-targeted, mKRAS-specific amphiphile vaccine in pancreatic and colorectal cancer: The phase 1 AMPLIFY-201 trial. Nat. Med. 2024, 30, 531–542. [Google Scholar] [CrossRef]
- Haldar, S.D.; Heumann, T.R.; Berg, M.; Ferguson, A.; Lim, S.J.; Wang, H.; Nauroth, J.; Laheru, D.; Jaffee, E.M.; Azad, N.S. A phase I study of a mutant KRAS-targeted long peptide vaccine combined with ipilimumab/nivolumab in resected pancreatic cancer and MMR-proficient metastatic colorectal cancer. J. Clin. Oncol. 2023, 41, TPS814. [Google Scholar] [CrossRef]
- Nusrat, M.; Yaeger, R. KRAS inhibition in metastatic colorectal cancer: An update. Curr. Opin. Pharmacol. 2023, 68, 102343. [Google Scholar] [CrossRef]
- Hager, S.; Fittler, F.J.; Wagner, E.; Bros, M. Nucleic acid-based approaches for tumor therapy. Cells 2020, 9, 2061. [Google Scholar] [CrossRef]
- Liao, H.-C.; Liu, S.-J. Advances in nucleic acid-based cancer vaccines. J. Biomed. Sci. 2025, 32, 10. [Google Scholar] [CrossRef]
- Chi, W.-Y.; Hu, Y.; Huang, H.-C.; Kuo, H.-H.; Lin, S.-H.; Kuo, C.-T.J.; Tao, J.; Fan, D.; Huang, Y.-M.; Wu, A.A. Molecular targets and strategies in the development of nucleic acid cancer vaccines: From shared to personalized antigens. J. Biomed. Sci. 2024, 31, 94. [Google Scholar] [CrossRef]
- Mollé, L.M.; Smyth, C.H.; Yuen, D.; Johnston, A.P. Nanoparticles for vaccine and gene therapy: Overcoming the barriers to nucleic acid delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1809. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, Y.; Zhang, J.; Kuo, J.C.-T.; Zhang, Z.; Xie, H.; Zhu, J.; Liu, T. Modification of lipid-based nanoparticles: An efficient delivery system for nucleic acid-based immunotherapy. Molecules 2022, 27, 1943. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.E.; Maurel, J.; Morris, V.; Kopetz, S.; Galligan, B.; Ali, S.; Derhovanessian, E.; Unsal-Kacmaz, K.; Manning, L.; Henn, H. 29P Characterization of T cell responses induced by the individualized mRNA neoantigen vaccine autogene cevumeran in adjuvant stage II (high risk)/stage III colorectal cancer (CRC) patients (pts) from the biomarker cohort of the phase II BNT122-01 trial. Ann. Oncol. 2024, 35, S14–S15. [Google Scholar] [CrossRef]
- Shanmugaraj, B.; Priya, L.B.; Mahalakshmi, B.; Subbiah, S.; Hu, R.-M.; Velmurugan, B.K.; Baskaran, R. Bacterial and viral vectors as vaccine delivery vehicles for breast cancer therapy. Life Sci. 2020, 250, 117550. [Google Scholar] [CrossRef] [PubMed]
- Travieso, T.; Li, J.; Mahesh, S.; Mello, J.D.F.R.E.; Blasi, M. The use of viral vectors in vaccine development. NPJ Vaccines 2022, 7, 75. [Google Scholar] [CrossRef]
- bin Umair, M.; Akusa, F.N.; Kashif, H.; Butt, F.; Azhar, M.; Munir, I.; Ahmed, M.; Khalil, W.; Sharyar, H.; Rafique, S. Viruses as tools in gene therapy, vaccine development, and cancer treatment. Arch. Virol. 2022, 167, 1387–1404. [Google Scholar] [CrossRef]
- Bezeljak, U. Cancer gene therapy goes viral: Viral vector platforms come of age. Radiol. Oncol. 2022, 56, 1. [Google Scholar] [CrossRef]
- Apolonio, J.S.; de Souza Gonçalves, V.L.; Santos, M.L.C.; Luz, M.S.; Souza, J.V.S.; Pinheiro, S.L.R.; de Souza, W.R.; Loureiro, M.S.; de Melo, F.F. Oncolytic virus therapy in cancer: A current review. World J. Virol. 2021, 10, 229. [Google Scholar] [CrossRef]
- Mondal, M.; Guo, J.; He, P.; Zhou, D. Recent advances of oncolytic virus in cancer therapy. Hum. Vaccines Immunother. 2020, 16, 2389–2402. [Google Scholar] [CrossRef]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Monge Bonilla, M.C.; Xie, C.; Steinberg, S.M.; Fioraventi, S.; Walker, M.; Mabry-Hrones, D.; Wood, B.J.; Kleiner, D.E.; Greten, T.F. A phase I/II study of Pexa-Vec oncolytic virus in combination with immune checkpoint inhibition in refractory colorectal cancer. J. Clin. Oncol. 2020, 38, 117. [Google Scholar] [CrossRef]
- Monge, C.; Xie, C.; Myojin, Y.; Coffman, K.; Hrones, D.M.; Wang, S.; Hernandez, J.M.; Wood, B.J.; Levy, E.B.; Juburi, I. Phase I/II study of PexaVec in combination with immune checkpoint inhibition in refractory metastatic colorectal cancer. J. Immunother. Cancer 2023, 11, e005640. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, A.R.; Kyi, C.; Lane, M.; Hart, M.G.; Johnson, M.L.; Henick, B.S.; Liao, C.-Y.; Mahipal, A.; Shergill, A.; Spira, A.I. A shared neoantigen vaccine combined with immune checkpoint blockade for advanced metastatic solid tumors: Phase 1 trial interim results. Nat. Med. 2024, 30, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Huang, C.Y.; Zhu, Q.; Ferguson, A.K.; Durham, J.N.; Anders, R.A.; Thompson, E.D.; Rozich, N.S.; Thomas, D.L.; Nauroth, J.M. A phase 2 study of GVAX colon vaccine with cyclophosphamide and pembrolizumab in patients with mismatch repair proficient advanced colorectal cancer. Cancer Med. 2020, 9, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Monsen, H.; Norseth, H.M.; Remen, N.; Abrahamsen, I.; Lysén, A.; Schjesvold, F. PB2118: TG01-Study: A phase 1/2 study with TG01 immunotherapy vaccination in patients with confirmed NRAS/KRAS mutation on codon 12/13 and multiple myeloma or high-risk smoldering myeloma. HemaSphere 2023, 7, e51467aa. [Google Scholar] [CrossRef]
- Seenappa, L.M.; Jakubowski, A.; Steinbuck, M.P.; Palmer, E.; Haqq, C.M.; Carter, C.; Fontenot, J.; Villinger, F.; McNeil, L.K.; DeMuth, P.C. Amphiphile-CpG vaccination induces potent lymph node activation and COVID-19 immunity in mice and non-human primates. NPJ Vaccines 2022, 7, 128. [Google Scholar] [CrossRef]
- Brown, T.A.; Byrd, K.; Vreeland, T.J.; Clifton, G.T.; Jackson, D.O.; Hale, D.F.; Herbert, G.S.; Myers, J.W.; Greene, J.M.; Berry, J.S. Final analysis of a phase I/IIa trial of the folate-binding protein-derived E39 peptide vaccine to prevent recurrence in ovarian and endometrial cancer patients. Cancer Med. 2019, 8, 4678–4687. [Google Scholar] [CrossRef]
- Belnoue, E.; Leystra, A.A.; Carboni, S.; Cooper, H.S.; Macedo, R.T.; Harvey, K.N.; Colby, K.B.; Campbell, K.S.; Vanderveer, L.A.; Clapper, M.L. Novel protein-based vaccine against self-antigen reduces the formation of sporadic colon adenomas in mice. Cancers 2021, 13, 845. [Google Scholar] [CrossRef]
- Kopetz, S.; Morris, V.K.; Alonso-Orduña, V.; Garcia-Alfonso, P.; Reboredo, M.; Fernandez Montes, A.; Maurel, J.; Paez, D.; Reinacher-Schick, A.C.; Höhler, T. A phase 2 multicenter, open-label, randomized, controlled trial in patients with stage II/III colorectal cancer who are ctDNA positive following resection to compare efficacy of autogene cevumeran versus watchful waiting. J. Clin. Oncol. 2022, 40, TPS3641. [Google Scholar] [CrossRef]
- Wei, J.; Hui, A.-M. The paradigm shift in treatment from COVID-19 to oncology with mRNA vaccines. Cancer Treat. Rev. 2022, 107, 102405. [Google Scholar] [CrossRef]
- D’Alise, M.; Willis, J.; Leoni, G.; Cruz-Correa, M.; Hall, M.J.; Idos, G.E.; Garzia, I.; Antonucci, L.; Cotugno, G.; Siani, L. 1526 Nous-209 genetic vaccine encoding shared cancer neoantigens is safe and elicits robust immune response in healthy Lynch syndrome carriers: Interim results from Phase 1 cancer interception trial. J. ImmunoTher. Cancer 2023, 11. [Google Scholar] [CrossRef]
- Pant, S.; Furqan, M.; Abdul-Karim, R.M.; Chung, V.; Devoe, C.E.; Johnson, M.L.; Leal, A.D.; Park, H.; Wainberg, Z.A.; Welkowsky, E. First-in-human phase 1 trial of ELI-002 immunotherapy as treatment for subjects with Kirsten rat sarcoma (KRAS)-mutated pancreatic ductal adenocarcinoma and other solid tumors. J. Clin. Oncol. 2022, 40, TPS2701. [Google Scholar] [CrossRef]
- Jou, J.; Harrington, K.J.; Zocca, M.-B.; Ehrnrooth, E.; Cohen, E.E. The changing landscape of therapeutic cancer vaccines—Novel platforms and neoantigen identification. Clin. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Maurel, J.; Oberstein, P.E.; Roselló-Keränen, S.; Le, D.T.; Pedersen, K.S.; Mukherjee, S.; D’Alise, A.M.; Leoni, G.; Siani, L. Results of phase I-II bridging study for Nous-209, a neoantigen cancer immunotherapy, in combination with pembrolizumab as first line treatment in patients with advanced dMMR/MSI-h colorectal cancer. J. Clin. Oncol. 2023, 41, e14665. [Google Scholar] [CrossRef]
- Formica, V.; Sera, F.; Cremolini, C.; Riondino, S.; Morelli, C.; Arkenau, H.-T.; Roselli, M. KRAS and BRAF mutations in stage II and III colon cancer: A systematic review and meta-analysis. JNCI J. Natl. Cancer Inst. 2022, 114, 517–527. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. Colorectal cancer: Genetic abnormalities, tumor progression, tumor heterogeneity, clonal evolution and tumor-initiating cells. Med. Sci. 2018, 6, 31. [Google Scholar] [CrossRef]
- Quandt, J.; Schlude, C.; Bartoschek, M.; Will, R.; Cid-Arregui, A.; Schölch, S.; Reissfelder, C.; Weitz, J.; Schneider, M.; Wiemann, S. Long-peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation-specific effector as well as regulatory T cell responses. Oncoimmunology 2018, 7, e1500671. [Google Scholar] [CrossRef]
- Zhou, S.; Fan, C.; Zeng, Z.; Young, K.H.; Li, Y. Clinical and immunological effects of p53-targeting vaccines. Front. Cell Dev. Biol. 2021, 9, 762796. [Google Scholar] [CrossRef]
- Chiang, Y.-T.; Chien, Y.-C.; Lin, Y.-H.; Wu, H.-H.; Lee, D.-F.; Yu, Y.-L. The function of the mutant p53-R175H in cancer. Cancers 2021, 13, 4088. [Google Scholar] [CrossRef]
- Chen, C.; Liu, S.; Qu, R.; Li, B. Recurrent neoantigens in colorectal cancer as potential immunotherapy targets. BioMed Res. Int. 2020, 2020, 2861240. [Google Scholar] [CrossRef]
- Wang, Z.; Burigotto, M.; Ghetti, S.; Vaillant, F.; Tan, T.; Capaldo, B.D.; Palmieri, M.; Hirokawa, Y.; Tai, L.; Simpson, D.S. Loss-of-function but not gain-of-function properties of mutant TP53 are critical for the proliferation, survival, and metastasis of a broad range of cancer cells. Cancer Discov. 2024, 14, 362–379. [Google Scholar] [CrossRef]
- Madden-Hennessey, K.; Gupta, D.; Radecki, A.A.; Guild, C.; Rath, A.; Heinen, C.D. Loss of mismatch repair promotes a direct selective advantage in human stem cells. Stem Cell Rep. 2022, 17, 2661–2673. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Ying, J.; Zang, J.; Lu, H.; Zhao, X.; Yang, P.; Wang, X.; Li, J.; Gong, Z.; Zhang, D. Specific mutations in APC, with prognostic implications in metastatic colorectal cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2023, 55, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Capietto, A.-H.; Hoshyar, R.; Delamarre, L. Sources of cancer neoantigens beyond single-nucleotide variants. Int. J. Mol. Sci. 2022, 23, 10131. [Google Scholar] [CrossRef]
- Corulli, L.R.; Cecil, D.L.; Gad, E.; Koehnlein, M.; Coveler, A.L.; Childs, J.S.; Lubet, R.A.; Disis, M.L. Multi-Epitope-Based vaccines for colon cancer treatment and prevention. Front. Immunol. 2021, 12, 729809. [Google Scholar] [CrossRef]
- Zhang, D.; Ni, Q.-Q.; Liang, Q.-Y.; He, L.-L.; Qiu, B.-W.; Zhang, L.-J.; Mou, T.-Y.; Le, C.-C.; Huang, Y.; Li, T.-T. ASCL2 induces an immune excluded microenvironment by activating cancer-associated fibroblasts in microsatellite stable colorectal cancer. Oncogene 2023, 42, 2841–2853. [Google Scholar] [CrossRef]
- Mestrallet, G.; Brown, M.; Bozkus, C.C.; Bhardwaj, N. Immune escape and resistance to immunotherapy in mismatch repair deficient tumors. Front. Immunol. 2023, 14, 1210164. [Google Scholar] [CrossRef]
- Li, X.; You, J.; Hong, L.; Liu, W.; Guo, P.; Hao, X. Neoantigen cancer vaccines: A new star on the horizon. Cancer Biol. Med. 2024, 21, 274–311. [Google Scholar] [CrossRef]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges and prospects. Signal Transduct. Target. Ther. 2023, 8, 450. [Google Scholar] [CrossRef]
- Yu, Y.J.; Shan, N.; Li, L.Y.; Zhu, Y.S.; Lin, L.M.; Mao, C.C.; Hu, T.T.; Xue, X.Y.; Su, X.P.; Shen, X.; et al. Preliminary clinical study of personalized neoantigen vaccine therapy for microsatellite stability (MSS)-advanced colorectal cancer. Cancer Immunol. Immunother. 2023, 72, 2045–2056. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Hundal, J.; Kiwala, S.; McMichael, J.; Miller, C.A.; Xia, H.; Wollam, A.T.; Liu, C.J.; Zhao, S.; Feng, Y.-Y.; Graubert, A.P. pVACtools: A computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 2020, 8, 409–420. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Drug/Formulation | Related Signature Genes | Mechanism | Clinical Trial Phase | Combination/ Adjuvant | Reference |
|---|---|---|---|---|---|
| GVAX/Cell-based | KRAS | Secretes GM-CSF to activate DC | II | Pembrolizumab | [134] |
| ELI-002/Lipopeptide-based | KRAS | AMP-CpG activates immune system, AMP-mKRAS peptide targets KRAS | Ib | - | [114,142] |
| -/Long peptide-based | KRAS | Increases diversity of T-cell responses by long peptide | I | Nivolumab, Ipilimumab | [116] |
| TG01/Peptide-based | KRAS | Enhances DTH response and T cell proliferation | I/II | QS-21 | [135] |
| E-39/Peptide-based | NRAS/KRAS | TALs from E39-stimulated induce cytolysis in tumor | I/II | - | [137] |
| KISIMA-Ascl2 vaccine/Peptide-based | APC | Utilizing Ascl2 upregulated in ISCs with KISIMA vaccine platform | - | Anti-PD-1/AS15 | [138] |
| BNT122/mRNA-based | KRAS/BRAF | Upregulates tumor-specific cytotoxic immune response | II | Atezolizumab | [139] |
| mRNA5681(V941)/ mRNA-based | KRAS | Administered mRNA induces antigen expression | II | Pembrolizumab | [140] |
| GRT-R904/saRNA-based, Vector-based | KRAS | Prime-boost strategy with ChAdV and saRNA-LNP combined | I/II | Keytruda, Pembrolizumab | [133,143] |
| Pexa-Vec(JX-594)/ Vector-based | KRAS/TP53 | Oncolytic virus-based that directly destroys tumors and promotes immune response | I/II | - | [131,132] |
| Nous-209/Vector-based | MMR | Prime-boost strategy with GAd20-209-FSP eliciting robust T cell response | Ib/II | Pembrolizumab | [141,144] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, J.; Moon, H.; Jeon, Y.; Choe, S.; Yoon, H. Signature Gene Mutations in Colorectal Cancer: Potential Neoantigens for Cancer Vaccines. Int. J. Mol. Sci. 2025, 26, 4559. https://doi.org/10.3390/ijms26104559
Yoon J, Moon H, Jeon Y, Choe S, Yoon H. Signature Gene Mutations in Colorectal Cancer: Potential Neoantigens for Cancer Vaccines. International Journal of Molecular Sciences. 2025; 26(10):4559. https://doi.org/10.3390/ijms26104559
Chicago/Turabian StyleYoon, Jaegoo, Haeun Moon, Yuna Jeon, Soohyun Choe, and Hyunho Yoon. 2025. "Signature Gene Mutations in Colorectal Cancer: Potential Neoantigens for Cancer Vaccines" International Journal of Molecular Sciences 26, no. 10: 4559. https://doi.org/10.3390/ijms26104559
APA StyleYoon, J., Moon, H., Jeon, Y., Choe, S., & Yoon, H. (2025). Signature Gene Mutations in Colorectal Cancer: Potential Neoantigens for Cancer Vaccines. International Journal of Molecular Sciences, 26(10), 4559. https://doi.org/10.3390/ijms26104559