
Metal-Involving Bifurcated Halogen Bonding with Iodide and Platinum(II) Center

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
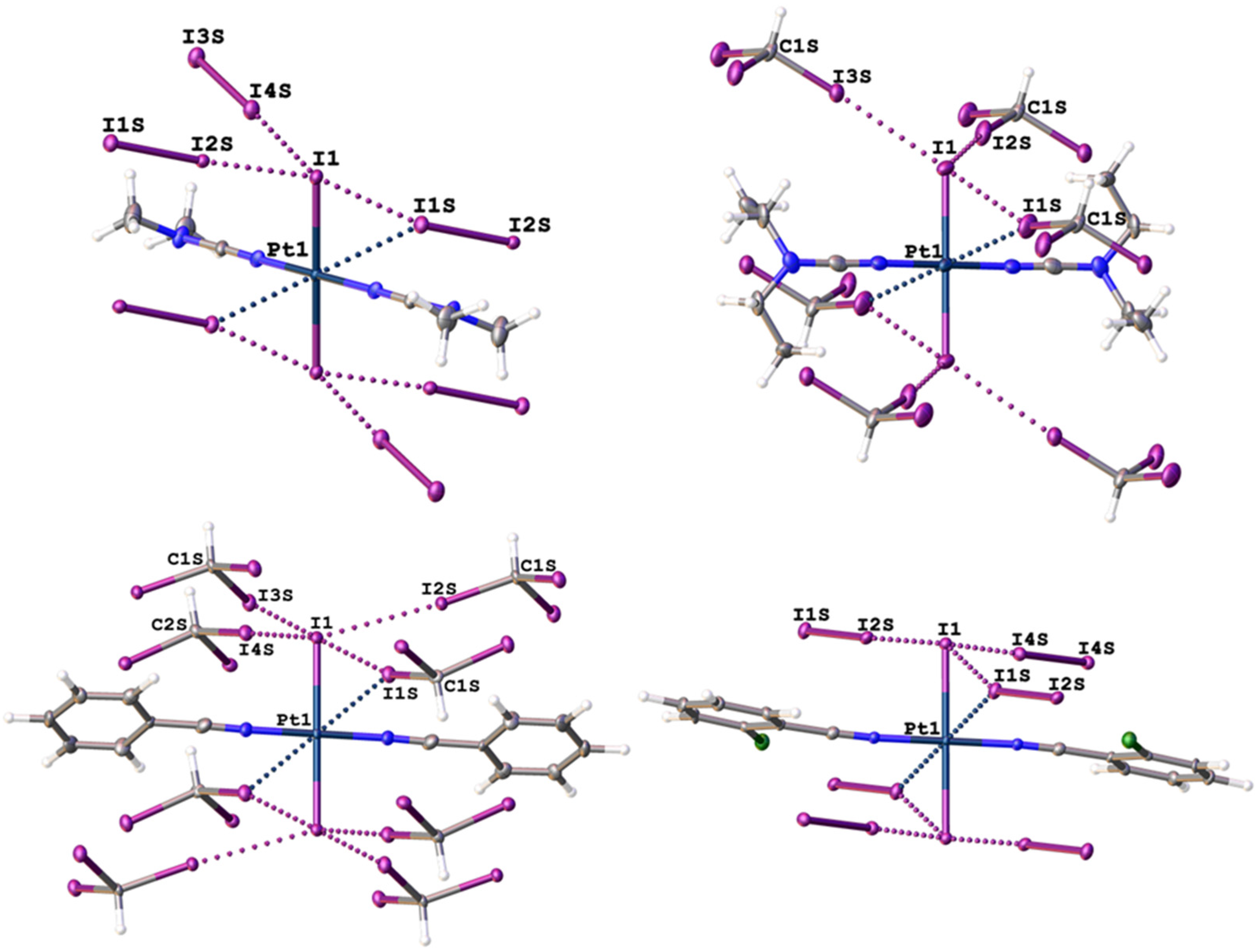
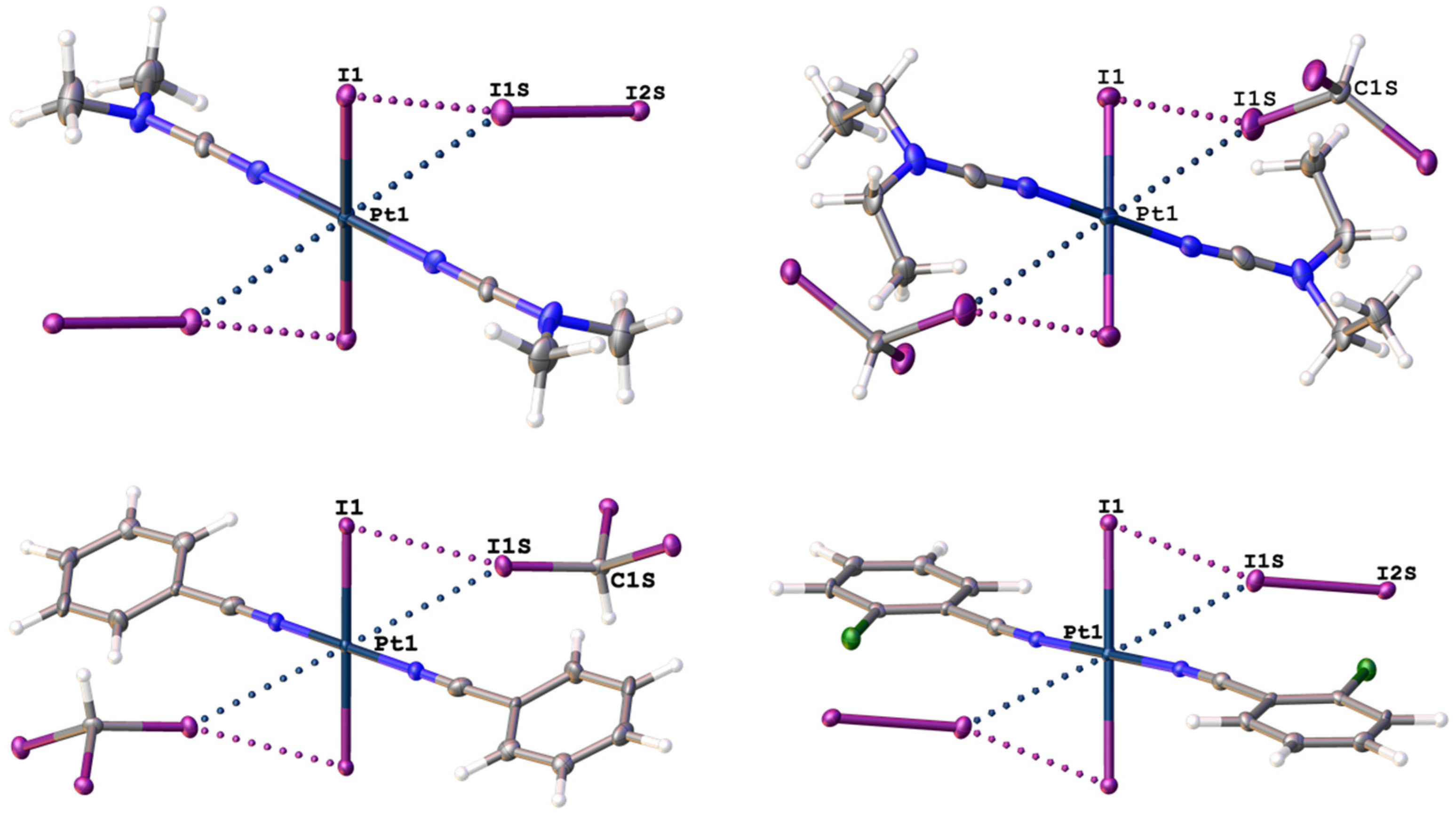
2.1. Single-Crystal X-Ray Diffraction Data
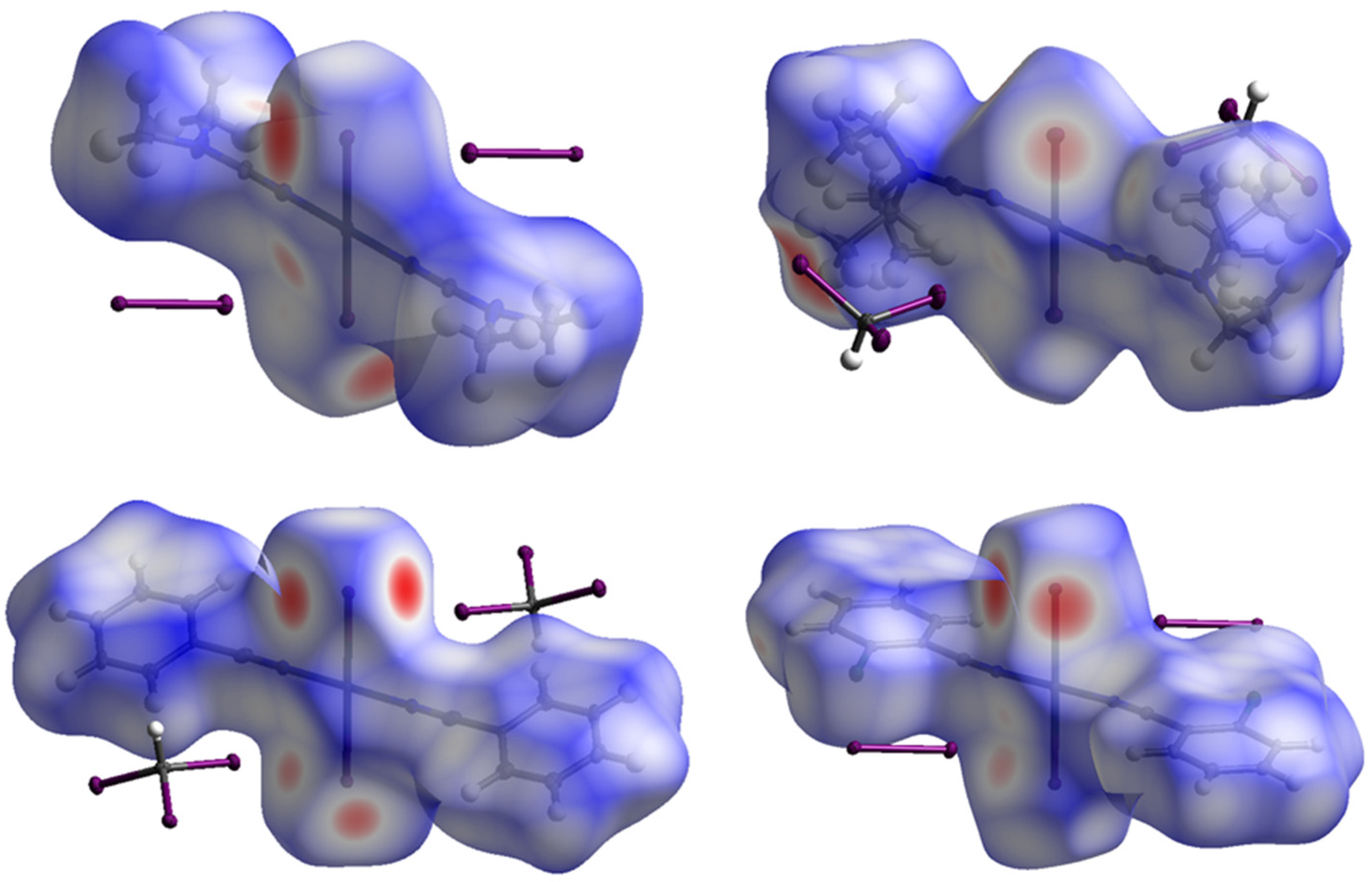
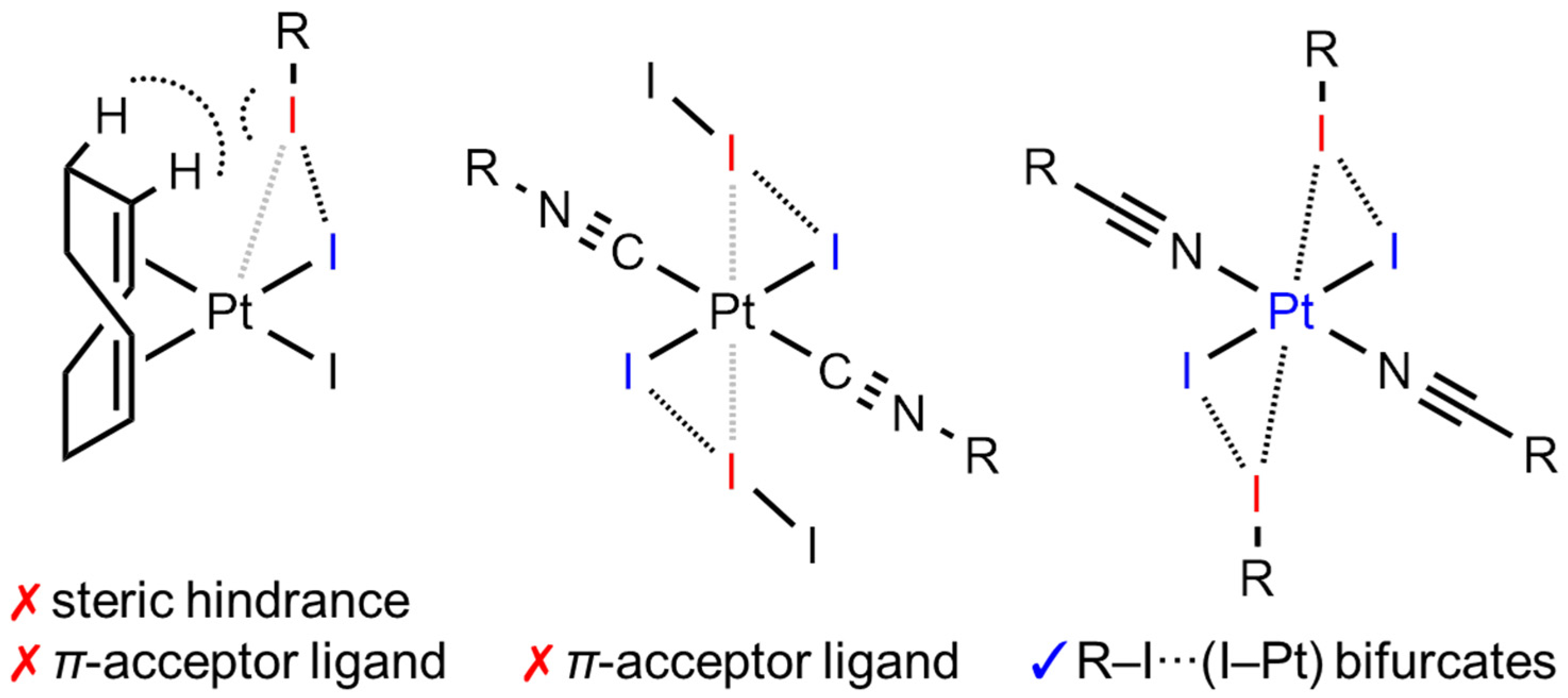
2.2. Theoretical Consideration
3. Materials and Methods
3.1. Synthesis of Trans-[PtI2(NCNR2)2] (R = Me 1, Et 2) Dialkylcyanamide Complexes
3.2. Synthesis of Trans-[PtI2(NCR)2] (R = Ph 3, 2-ClC6H4 4) Nitrile Complexes
3.3. Crystallizations of the Adducts
3.4. X-Ray Structure Determination and Refinement
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| XRD | X-ray diffraction |
| XB | halogen bonding, halogen bond |
| DFT | density functional theory |
| GAPW | Gaussian augmented plane waves |
| COD | 1,5-cycloocatadiene |
| FIB | 1,4-diiodotetrafluorobenzene |
| IUPAC | International Union of Pure and Applied Chemistry |
| RT | room temperature |
| DKH2 | Douglas–Kroll–Hess second-order scalar relativistic |
| QTAIM | quantum theory of atoms-in-molecules |
| BCP | bond critical point |
| RDG | reduced density gradient |
| NCI | noncovalent interaction (analysis) |
| ELF | electron localization function |
| sSAPT0 | scaled symmetry adapted perturbation theory (zero order) |
| NMR | nuclear magnetic resonance |
| TLC | thin-layer chromatography |
| HRESI+-MS | high-resolution electrospray ionization–mass spectroscopy (positive ions) |
References
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J. An Overview of Strengths and Directionalities of Noncovalent Interactions: σ-Holes and π-Holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef]
- Wang, H.; Bisoyi, H.K.; Urbas, A.M.; Bunning, T.J.; Li, Q. The Halogen Bond: An Emerging Supramolecular Tool in the Design of Functional Mesomorphic Materials. Chem.-A Eur. J. 2019, 25, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Sanz-Matias, A.; Velpula, G.; Waghray, D.; Ivasenko, O.; Bilbao, N.; Harvey, J.N.; Mali, K.S.; De Feyter, S. Halogenated building blocks for 2D crystal engineering on solid surfaces: Lessons from hydrogen bonding. Chem. Sci. 2019, 10, 3881–3891. [Google Scholar] [CrossRef]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Non-covalent interactions in the synthesis of coordination compounds: Recent advances. Coord. Chem. Rev. 2017, 345, 54–72. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Pavan, B.; Ferretti, V. Can pharmaceutical co-crystals provide an opportunity to modify the biological properties of drugs? Drug Discov. Today 2017, 22, 1134–1138. [Google Scholar] [CrossRef]
- Ho, P.S. Halogen bonding in medicinal chemistry: From observation to prediction. Future Med. Chem. 2017, 9, 637–640. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; Meyer, F. Halogen bonding in polymer science: From crystal engineering to functional supramolecular polymers and materials. Polym. Chem. 2015, 6, 3559–3580. [Google Scholar] [CrossRef]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem.-A Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent interactions in metal complex catalysis. Coord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef] [PubMed]
- Bennion, J.C.; Vogt, L.; Tuckerman, M.E.; Matzger, A.J. Isostructural Cocrystals of 1,3,5-Trinitrobenzene Assembled by Halogen Bonding. Cryst. Growth Des. 2016, 16, 4688–4693. [Google Scholar] [CrossRef]
- Landenberger, K.B.; Bolton, O.; Matzger, A.J. Energetic-energetic cocrystals of diacetone diperoxide (DADP): Dramatic and divergent sensitivity modifications via cocrystallization. J. Am. Chem. Soc. 2015, 137, 5074–5079. [Google Scholar] [CrossRef]
- Sivchik, V.V.; Solomatina, A.I.; Chen, Y.T.; Karttunen, A.J.; Tunik, S.P.; Chou, P.T.; Koshevoy, I.O. Halogen Bonding to Amplify Luminescence: A Case Study Using a Platinum Cyclometalated Complex. Angew. Chem. Int. Ed. 2015, 54, 14057–14060. [Google Scholar] [CrossRef] [PubMed]
- Sivchik, V.; Sarker, R.K.; Liu, Z.Y.; Chung, K.Y.; Grachova, E.V.; Karttunen, A.J.; Chou, P.T.; Koshevoy, I.O. Improvement of the photophysical performance of platinum-cyclometalated complexes in halogen-bonded adducts. Chem.-A Eur. J. 2018, 24, 11475–11484. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Kashina, M.V.; Mikherdov, A.S.; Mozheeva, E.A.; Novikov, A.S.; Smirnov, A.S.; Ivanov, D.M.; Kryukova, M.A.; Ivanov, A.Y.; Smirnov, S.N.; et al. Dramatically Enhanced Solubility of Halide-Containing Organometallic Species in Diiodomethane: The Role of Solvent⋯Complex Halogen Bonding. Angew. Chem. Int. Ed. 2018, 57, 12785–12789. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Novikov, A.S.; Boyarskiy, V.P.; Kukushkin, V.Y. The halogen bond with isocyano carbon reduces isocyanide odor. Nat. Commun. 2020, 11, 2921. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Type II halogen···halogen contacts are halogen bonds. IUCrJ 2014, 1, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Hassel, O. Structural aspects of interatomic charge-transfer bonding. Science 1970, 170, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.M.; Bokach, N.A.; Kukushkin, V.Y.; Frontera, A. Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering. Chem.-A Eur. J. 2022, 28, e202103173. [Google Scholar] [CrossRef] [PubMed]
- Eliseeva, A.A.; Ivanov, D.M.; Rozhkov, A.V.; Ananyev, I.V.; Frontera, A.; Kukushkin, V.Y. Bifurcated Halogen Bonding Involving Two Rhodium(I) Centers as an Integrated σ-Hole Acceptor. JACS Au 2021, 1, 354–361. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Kirina, Y.V.; Kukushkin, V.Y. Halogen Bonding Between Metal Center and Halocarbon. Chem. Commun. 2016, 52, 5565–5568. [Google Scholar] [CrossRef]
- Dabranskaya, U.; Ivanov, D.M.; Novikov, A.S.; Matveychuk, Y.V.; Bokach, N.A.; Kukushkin, V.Y. Metal-Involving Bifurcated Halogen Bonding C–Br···η2(Cl–Pt). Cryst. Growth Des. 2019, 19, 1364–1376. [Google Scholar] [CrossRef]
- Aliyarova, I.S.; Tupikina, E.Y.; Ivanov, D.M.; Kukushkin, V.Y. Metal-Involving Halogen Bonding Including Gold(I) as a Nucleophilic Partner. The Case of Isomorphic Dichloroaurate(I)·Halomethane Cocrystals. Inorg. Chem. 2022, 61, 2558–2567. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Haukka, M.; Levin, O.V.; Frontera, A.; Kukushkin, V.Y. Supramolecular Assembly of Metal Complexes by (Aryl)I⋯dz2[PtII] Halogen Bond. Chem.-A Eur. J. 2020, 26, 7692–7701. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Rozhkov, A.V.; Levin, O.V.; Haukka, M.; Kuznetsov, M.L.; Kukushkin, V.Y. Halogen Bonding Involving Palladium(II) as an XB Acceptor. Cryst. Growth Des. 2021, 21, 1159–1177. [Google Scholar] [CrossRef]
- Bulatova, M.; Ivanov, D.M.; Haukka, M. Classics Meet Classics: Theoretical and Experimental Studies of Halogen Bonding in Adducts of Platinum(II) 1,5-Cyclooctadiene Halide Complexes with Diiodine, Iodoform, and 1,4-Diiodotetrafluorobenzene. Cryst. Growth Des. 2021, 21, 974–987. [Google Scholar] [CrossRef]
- Bulatova, M.; Ivanov, D.M.; Rautiainen, J.M.; Kinzhalov, M.A.; Truong, K.N.; Lahtinen, M.; Haukka, M. Studies of Nature of Uncommon Bifurcated I-I···(I-M) Metal-Involving Noncovalent Interaction in Palladium(II) and Platinum(II) Isocyanide Cocrystals. Inorg. Chem. 2021, 60, 13200–13211. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Comas-Vilà, G.; Salvador, P. Quantification of the Donor-Acceptor Character of Ligands by the Effective Fragment Orbitals. ChemPhysChem 2024, 25, e202400582. [Google Scholar] [CrossRef]
- Kashina, M.V.; Kinzhalov, M.A.; Smirnov, A.S.; Ivanov, D.M.; Novikov, A.S.; Kukushkin, V.Y. Dihalomethanes as Bent Bifunctional XB/XB-Donating Building Blocks for Construction of Metal-involving Halogen Bonded Hexagons. Chem.-Asian J. 2019, 14, 3915–3920. [Google Scholar] [CrossRef] [PubMed]
- Cheranyova, A.M.; Zelenkov, L.E.; Baykov, S.V.; Izotova, Y.A.; Ivanov, D.M.; Bokach, N.A.; Kukushkin, V.Y. Intermolecular Metal-Involving Pnictogen Bonding: The Case of σ-(SbIII)-Hole···dz[PtII] Interaction. Inorg. Chem. 2024, 63, 14943–14957. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Khazanova, M.A.; Cheranyova, A.M.; Aliyarova, I.S.; Kravchuk, R.I.; Oganesyan, E.S.; Ryabykh, A.V.; Maslova, O.A.; Ivanov, D.M.; Beznosyuk, S.A. Metal-Involving Halogen Bonding Confirmed Using DFT Calculations with Periodic Boundary Conditions. Crystals 2023, 13, 712. [Google Scholar] [CrossRef]
- Bertolotti, F.; Shishkina, A.; Forni, A.; Gervasio, G.; Stash, A.; Tsirelson, V. Intermolecular Bonding Features in Solid Iodine. Cryst. Growth Des. 2014, 14, 3587–3595. [Google Scholar] [CrossRef]
- Johnson, M.T.; Džolić, Z.; Cetina, M.; Wendt, O.F.; Öhrström, L.; Rissanen, K. Neutral Organometallic Halogen Bond Acceptors: Halogen Bonding in Complexes of PCPPdX (X = Cl, Br, I) with Iodine (I2), 1,4-Diiodotetrafluorobenzene (F4DIBz), and 1,4-Diiodooctafluorobutane (F8DIBu). Cryst. Growth Des. 2012, 12, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 37, 3814–3816. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Jorge, F.E.; Canal Neto, A.; Camiletti, G.G.; MacHado, S.F. Contracted Gaussian basis sets for Douglas-Kroll-Hess calculations: Estimating scalar relativistic effects of some atomic and molecular properties. J. Chem. Phys. 2009, 130, 064108. [Google Scholar] [CrossRef]
- Barros, C.L.; De Oliveira, P.J.P.; Jorge, F.E.; Canal Neto, A.; Campos, M. Gaussian basis set of double zeta quality for atoms Rb through Xe: Application in non-relativistic and relativistic calculations of atomic and molecular properties. Mol. Phys. 2010, 108, 1965–1972. [Google Scholar] [CrossRef]
- de Berrêdo, R.C.; Jorge, F.E. All-electron double zeta basis sets for platinum: Estimating scalar relativistic effects on platinum(II) anticancer drugs. J. Mol. Struct. THEOCHEM 2010, 961, 107–112. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package -Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef]
- Frigo, M.; Johnson, S.G. The Design and Implementation of FFTW3. Proc. IEEE 2005, 93, 216–231. [Google Scholar] [CrossRef]
- Vandevondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Borštnik, U.; Vandevondele, J.; Weber, V.; Hutter, J. Sparse matrix multiplication: The distributed block-compressed sparse row library. Parallel Comput. 2014, 40, 47–58. [Google Scholar] [CrossRef]
- Schütt, O.; Messmer, P.; Hutter, J.; VandeVondele, J. GPU-Accelerated Sparse Matrix-Matrix Multiplication for Linear Scaling Density Functional Theory. In Electronic Structure Calculations on Graphics Processing Units; John Wiley & Sons, Ltd.: Chichester, UK, 2016; pp. 173–190. [Google Scholar] [CrossRef]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef]
- Lippert, G.; Hutter, J.; Parrinello, M. The Gaussian and augmented-plane-wave density functional method for ab initio molecular dynamics simulations. Theor. Chem. Acc. 1999, 103, 124–140. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X-H⋯F-Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chemie Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 94106. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Sarju, J.; Arbour, J.; Sayer, J.; Rohrmoser, B.; Scherer, W.; Wagner, G. Synthesis and characterisation of mixed ligand Pt(ii) and Pt(iv) oxadiazoline complexes. Dalt. Trans. 2008, 5302–5312. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Bugge, G. Vergleich der Nitrile und Isonitrile im Verhalten gegen Metallsalze, ein Beitrag zur Konstitution der Doppelcyanide. Berichte Der Dtsch. Chem. Ges. 1907, 40, 1772–1778. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X-H bond lengths from neutron diffraction data. Acta Crystallogr. Sect. B Struct. Sci. 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Barysz, M.; Sadlej, A.J. Two-component methods of relativistic quantum chemistry: From the Douglas-Kroll approximation to the exact two-component formalism. J. Mol. Struct. THEOCHEM 2001, 573, 181–200. [Google Scholar] [CrossRef]
- Reiher, M. Relativistic Douglas-Kroll-Hess theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 139–149. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Nguyen-Dang, T.T. Quantum Theory of Atoms in Molecules–Dalton Revisited. Adv. Quantum Chem. 1981, 14, 63–124. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. Psi4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef] [PubMed]
- Suslonov, V.V.; Soldatova, N.S.; Ivanov, D.M.; Galmés, B.; Frontera, A.; Resnati, G.; Postnikov, P.S.; Kukushkin, V.Y.; Bokach, N.A. Diaryliodonium Tetrachloroplatinates(II): Recognition of a Trifurcated Metal-Involving μ3-I···(Cl,Cl,Pt) Halogen Bond. Cryst. Growth Des. 2021, 21, 5360–5372. [Google Scholar] [CrossRef]
- Bokach, N.A.; Kukushkin, V.Y. Coordination chemistry of dialkylcyanamides: Binding properties, synthesis of metal complexes, and ligand reactivity. Coord. Chem. Rev. 2013, 257, 2293–2316. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Interaction | d(I⋯I), Å | RvdW * | ∠(R–I⋯I), ° | ∠(I⋯I–X), ° |
|---|---|---|---|---|---|
| 1∙4I2 | I1S ⋯I1–Pt1 | 3.9255(8) | 0.99 | 164.38(2) | 59.548(13) |
| I2S⋯I1–Pt1 | 3.2362(7) | 0.82 | 174.39(2) | 105.017(18) | |
| I3S⋯I1S–I2S | 3.4484(8) | 0.87 | 173.48(2) | 91.355(18) | |
| I4S⋯I1–Pt1 | 3.4568(8) | 0.87 | 177.88(2) | 103.462(18) | |
| 2∙2CHI3 | I1S ⋯I1–Pt1 | 3.9138(8) | 0.99 | 154.21(13) | 63.497(13) |
| I2S⋯I1–Pt1 | 3.5503(9) | 0.90 | 170.8(2) | 109.053(18) | |
| I3S⋯I1–Pt1 | 3.5687(6) | 0.90 | 173.6(2) | 124.317(16) | |
| 3∙2CHI3 | I1S ⋯I1–Pt1 | 3.7811(8) | 0.95 | 165.9(3) | 68.795(16) |
| I2S⋯I1–Pt1 | 3.5559(8) | 0.90 | 174.7(3) | 106.531(19) | |
| I3S⋯I1–Pt1 | 3.7176(8) | 0.94 | 167.6(3) | 98.484(18) | |
| I4S⋯I1–Pt1 | 3.5675(8) | 0.90 | 173.87(19) | 98.30(2) | |
| I5S⋯I1A–Pt1A | 3.5882(8) | 0.91 | 173.7(3) | 109.571(18) | |
| I6S⋯I1A–Pt1A | 3.6094(8) | 0.91 | 169.6(2) | 100.947(17) | |
| 4∙4I2 | I1S ⋯I1–Pt1 | 3.7517(5) | 0.95 | 158.298(13) | 66.909(8) |
| I2S⋯I1–Pt1 | 3.2980(4) | 0.83 | 177.373(15) | 95.769(10) | |
| I3S⋯I1S–I2S | 3.4385(4) | 0.87 | 178.450(17) | 99.193(13) | |
| I4S⋯I1–Pt1 | 3.4783(4) | 0.88 | 172.041(17) | 88.569(8) |
| Structure | Interaction | d(I⋯Pt), Å | RvdW * | ∠(R–I⋯Pt), ° |
|---|---|---|---|---|
| 1∙4I2 | I2S–I1S⋯Pt1 | 3.4414(6) | 0.92 | 148.77(2) |
| 2∙2CHI3 | C1S–I1S⋯Pt1 | 3.6065(7) | 0.97 | 165.46(14) |
| 3∙2CHI3 | C1S–I1S⋯Pt1 | 3.7362(6) | 1.00 | 152.8(3) |
| 4∙4I2 | I2S–I1S⋯Pt1 | 3.6338(3) | 0.97 | 150.58(1) |
| Structure | Contributions of Different Intermolecular Contacts to the Molecular Hirshfeld Surface * |
|---|---|
| 1∙4I2 | Pt⋯I 3.3%, I⋯I 19.6%, I⋯N 9.7%, I⋯C 3.8%, I⋯H 46.4%, C⋯H 1.7%, H⋯H 14.8% |
| 2∙2CHI3 | Pt⋯I 2.0%, I⋯I 7.4%, I⋯N 1.8%, I⋯H 44.5%, N⋯H 7.8%, C⋯H 2.9%, H⋯H 31.5% |
| 3∙2CHI3 | Pt⋯I 2.0%, Pt⋯H 1.6%, I⋯I 13.7%, I⋯N 5.3%, I⋯C 6.2%, I⋯H 38.6%, N⋯H 5.1%, C⋯C 3.1%, C⋯H 16.8%, H⋯H 7.0% |
| 4∙4I2 | Pt⋯I 2.8%, I⋯I 12.8%, I⋯Cl 10.1%, I⋯N 6.1%, I⋯C 2.1%, I⋯H 34.4%, Cl⋯C 8.9%, N⋯H 1.3%, C⋯C 6.9%, C⋯H 11.3%, H⋯H 2.4% |
| Structure | Interaction | d(I···Pt) | ρ | ∇2ρ | G | V | H | |V|/G |
|---|---|---|---|---|---|---|---|---|
| 1∙4I2 | I2S–I1S⋯Pt1 | 3.4414 | 0.018 | 0.039 | 0.010 | −0.010 | 0.000 | °1.003 |
| 2∙2CHI3 | C1S–I1S⋯Pt1 | 3.6065 | 0.012 | 0.029 | 0.006 | −0.006 | 0.000 | °0.872 |
| 3∙2CHI3 | C1S–I1S⋯Pt1 | 3.7362 | 0.010 | 0.025 | 0.005 | −0.004 | 0.001 | °0.798 |
| 4∙4I2 | I2S–I1S⋯Pt1 | 3.6338 | 0.012 | 0.030 | 0.007 | −0.006 | 0.001 | °0.884 |
| Structure | Interaction | d(I⋯I) | ρ | ∇2ρ | G | V | H | |V|/G |
|---|---|---|---|---|---|---|---|---|
| 1∙4I2 | I1S⋯I1–Pt1 | 3.9255 | 0.009 | 0.024 | 0.004 | −0.003 | 0.001 | 0.663 |
| I2S⋯I1–Pt1 | 3.2362 | 0.029 | 0.041 | 0.014 | −0.018 | −0.004 | 1.259 | |
| I3S⋯I1S–I2S | 3.4484 | 0.020 | 0.044 | 0.012 | −0.012 | 0.000 | 1.044 | |
| I4S⋯I1–Pt1 | 3.4568 | 0.019 | 0.045 | 0.012 | −0.012 | 0.000 | 1.031 | |
| 2∙2CHI3 | I1S⋯I1–Pt1 | 3.9138 | 0.008 | 0.029 | 0.006 | −0.004 | 0.002 | 0.768 |
| I2S⋯I1–Pt1 | 3.5503 | 0.015 | 0.043 | 0.010 | −0.010 | 0.000 | 0.952 | |
| I3S⋯I1–Pt1 | 3.5687 | 0.014 | 0.043 | 0.010 | −0.009 | 0.001 | 0.934 | |
| 3∙2CHI3 | I1S⋯I1–Pt1 | 3.7811 | 0.010 | 0.033 | 0.007 | −0.006 | 0.001 | 0.815 |
| I2S⋯I1–Pt1 | 3.5559 | 0.015 | 0.044 | 0.010 | −0.010 | 0.000 | 0.942 | |
| I3S⋯I1–Pt1 | 3.7176 | 0.011 | 0.036 | 0.008 | −0.007 | 0.001 | 0.852 | |
| I4S⋯I1–Pt1 | 3.5675 | 0.015 | 0.042 | 0.010 | −0.009 | 0.001 | 0.932 | |
| I5S⋯I1A–Pt1A | 3.5882 | 0.014 | 0.042 | 0.010 | −0.009 | 0.001 | 0.924 | |
| I6S⋯I1A–Pt1A | 3.6094 | 0.014 | 0.041 | 0.009 | −0.009 | 0.000 | 0.919 | |
| 4∙4I2 | I1S⋯I1–Pt1 | 3.7517 | 0.012 | 0.029 | 0.006 | −0.005 | 0.001 | 0.795 |
| I2S⋯I1–Pt1 | 3.2980 | 0.026 | 0.041 | 0.013 | −0.015 | −0.002 | 1.196 | |
| I3S⋯I1S–I2S | 3.4385 | 0.020 | 0.045 | 0.012 | –0.013 | –0.001 | 1.051 | |
| I4S⋯I1–Pt1 | 3.4783 | 0.017 | 0.046 | 0.011 | –0.011 | 0.000 | 0.991 |
| Cluster | Interaction | EsSAPT0 | Eelst | Eexch | Eind | Edisp | Edisp/Eelst |
|---|---|---|---|---|---|---|---|
| 1∙(I2)2 | I2S–I1S⋯(I1–Pt1) | –9.55 | –7.77 | 14.25 | –4.32 | –11.72 | 1.51 |
| 2∙(CHI3)2 | C1S–I1S⋯(I1–Pt1) | –8.58 | –5.97 | 12.58 | –2.93 | –12.27 | 2.06 |
| 3∙(CHI3)2 | C1S–I1S⋯(I1–Pt1) | –8.80 | –7.39 | 14.34 | –3.40 | –12.35 | 1.67 |
| 4∙(I2)2 | I2S–I1S⋯(I1–Pt1) | –9.47 | –7.26 | 14.07 | –4.17 | –12.11 | 1.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kryukova, M.A.; Kostareva, M.B.; Cheranyova, A.M.; Khazanova, M.A.; Rozhkov, A.V.; Ivanov, D.M. Metal-Involving Bifurcated Halogen Bonding with Iodide and Platinum(II) Center. Int. J. Mol. Sci. 2025, 26, 4555. https://doi.org/10.3390/ijms26104555
Kryukova MA, Kostareva MB, Cheranyova AM, Khazanova MA, Rozhkov AV, Ivanov DM. Metal-Involving Bifurcated Halogen Bonding with Iodide and Platinum(II) Center. International Journal of Molecular Sciences. 2025; 26(10):4555. https://doi.org/10.3390/ijms26104555
Chicago/Turabian StyleKryukova, Mariya A., Margarita B. Kostareva, Anna M. Cheranyova, Marina A. Khazanova, Anton V. Rozhkov, and Daniil M. Ivanov. 2025. "Metal-Involving Bifurcated Halogen Bonding with Iodide and Platinum(II) Center" International Journal of Molecular Sciences 26, no. 10: 4555. https://doi.org/10.3390/ijms26104555
APA StyleKryukova, M. A., Kostareva, M. B., Cheranyova, A. M., Khazanova, M. A., Rozhkov, A. V., & Ivanov, D. M. (2025). Metal-Involving Bifurcated Halogen Bonding with Iodide and Platinum(II) Center. International Journal of Molecular Sciences, 26(10), 4555. https://doi.org/10.3390/ijms26104555