
Molecular Mechanisms of Type 2 Diabetes-Related Heart Disease and Therapeutic Insights

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Type 2 Diabetes Mellitus and the Cardiomyocyte
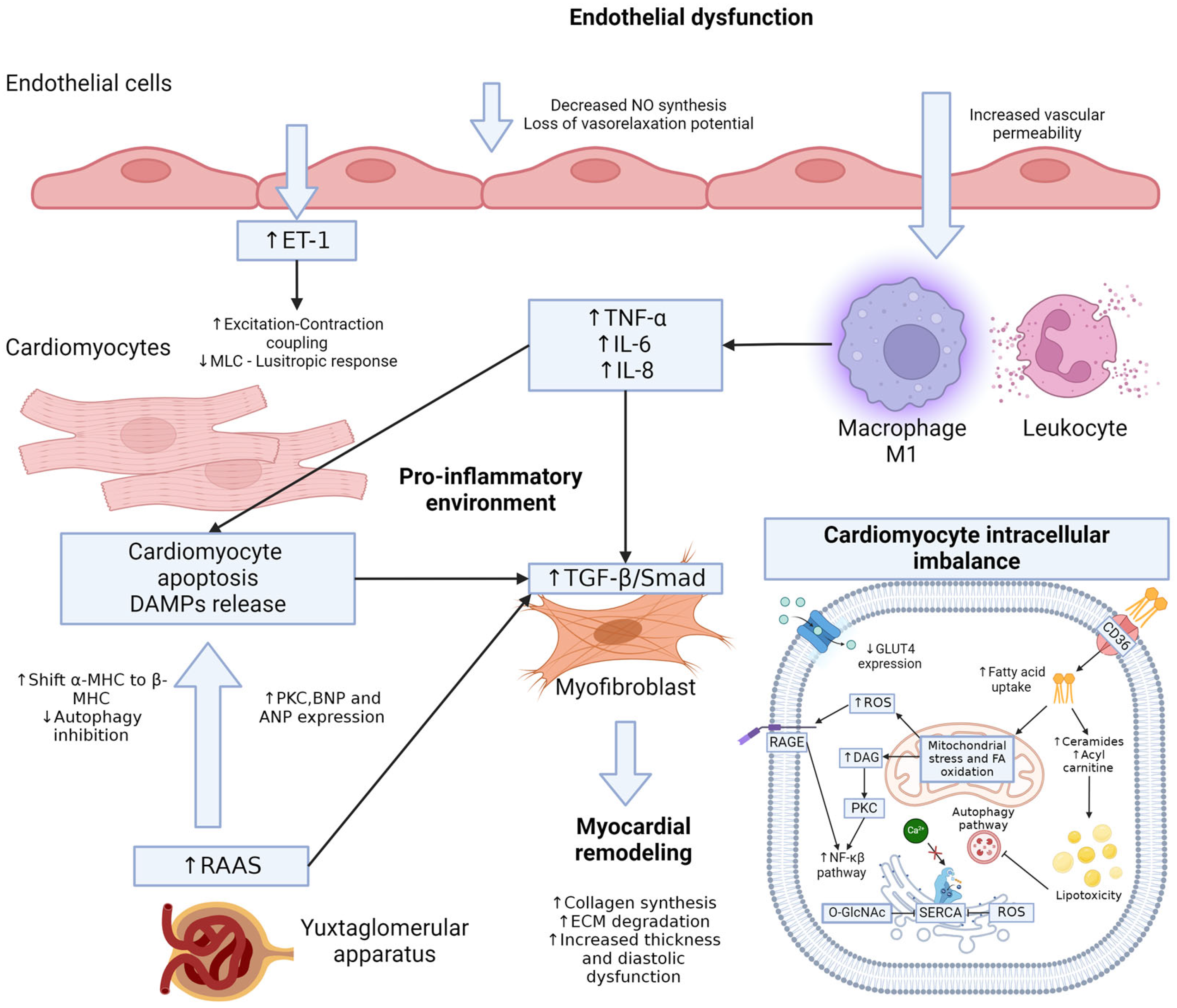
3. Type 2 Diabetes Mellitus and the Endothelium
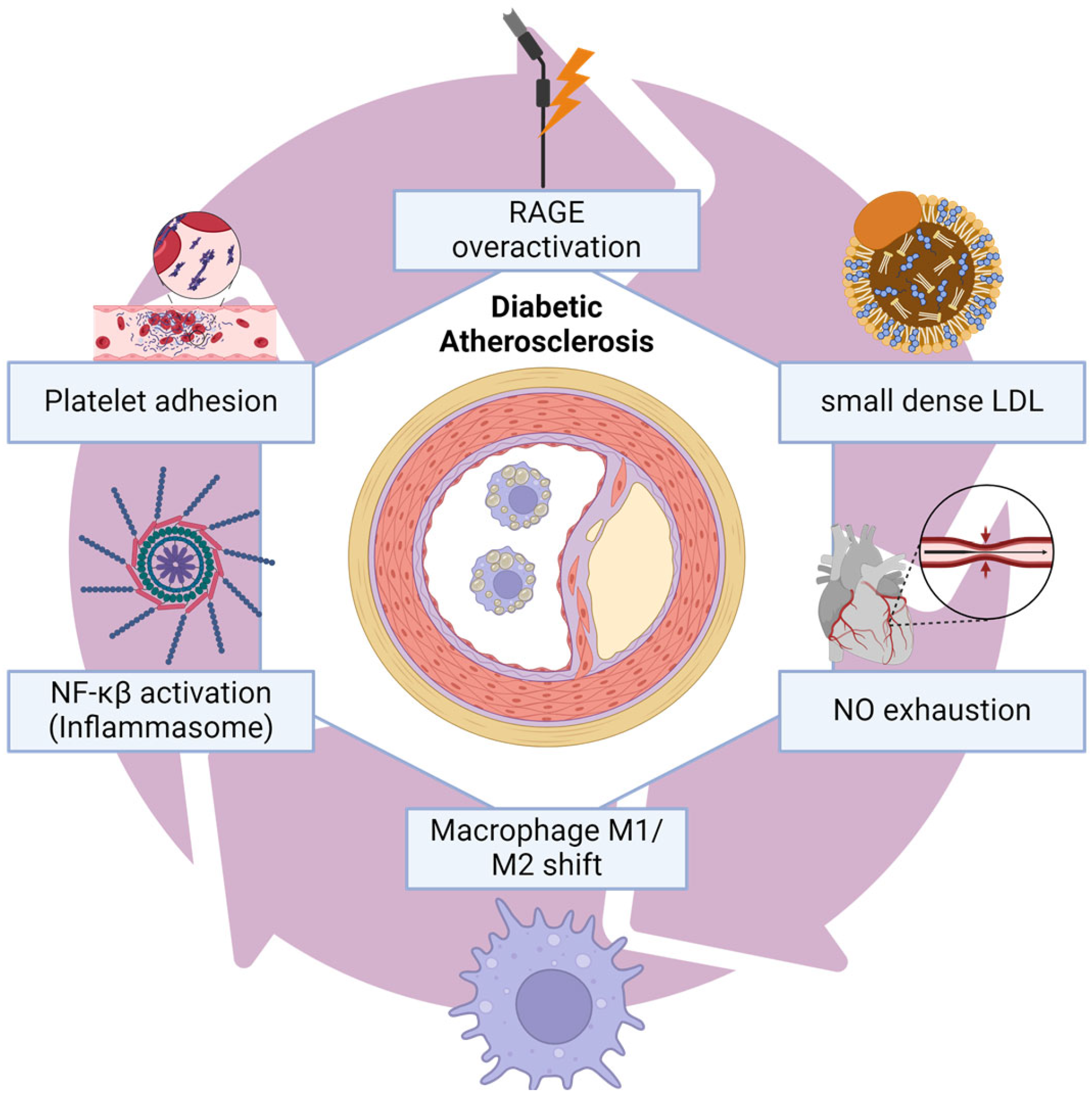
4. Coronary Artery Disease
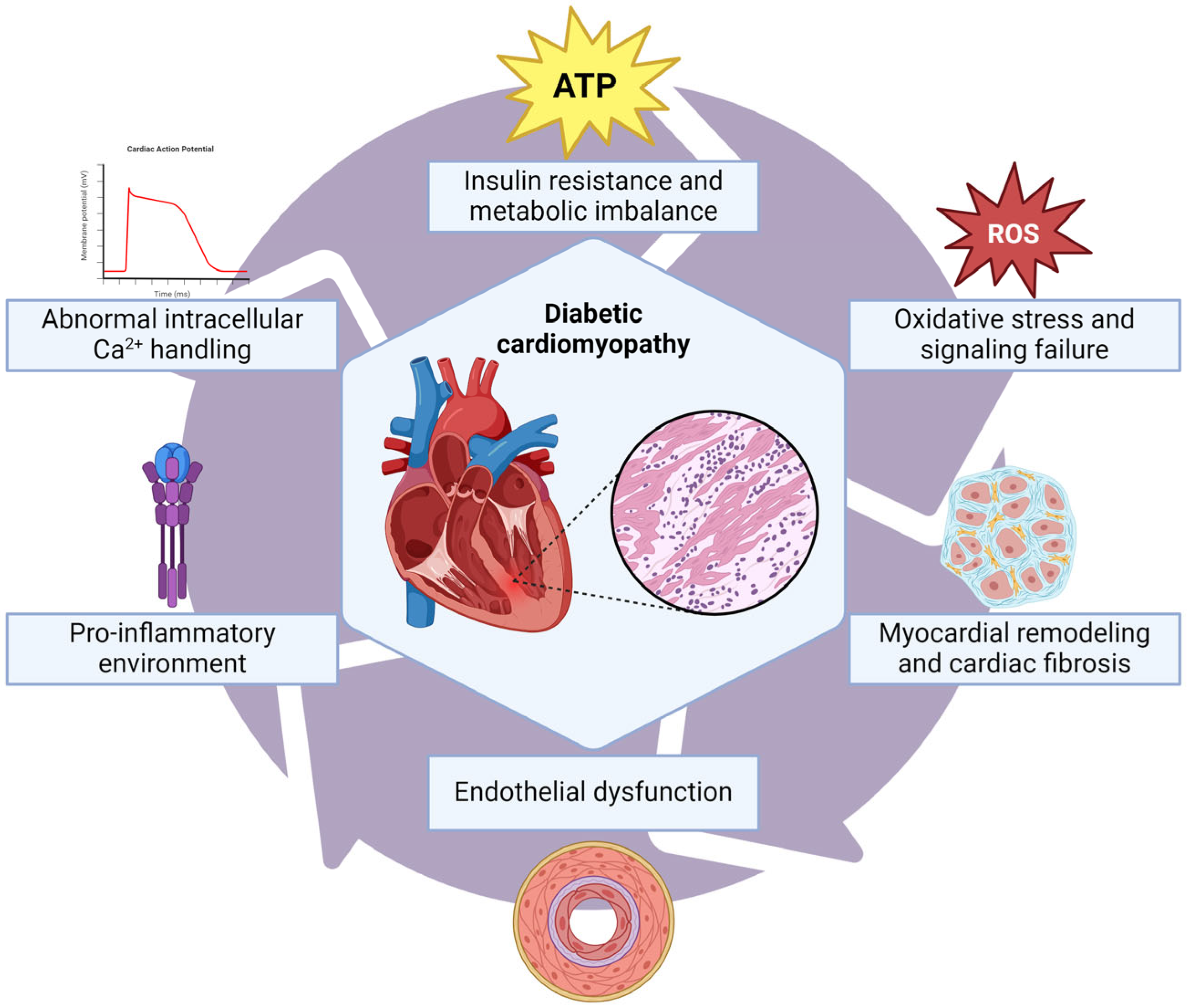
5. Diabetic Cardiomyopathy
6. Heart Failure
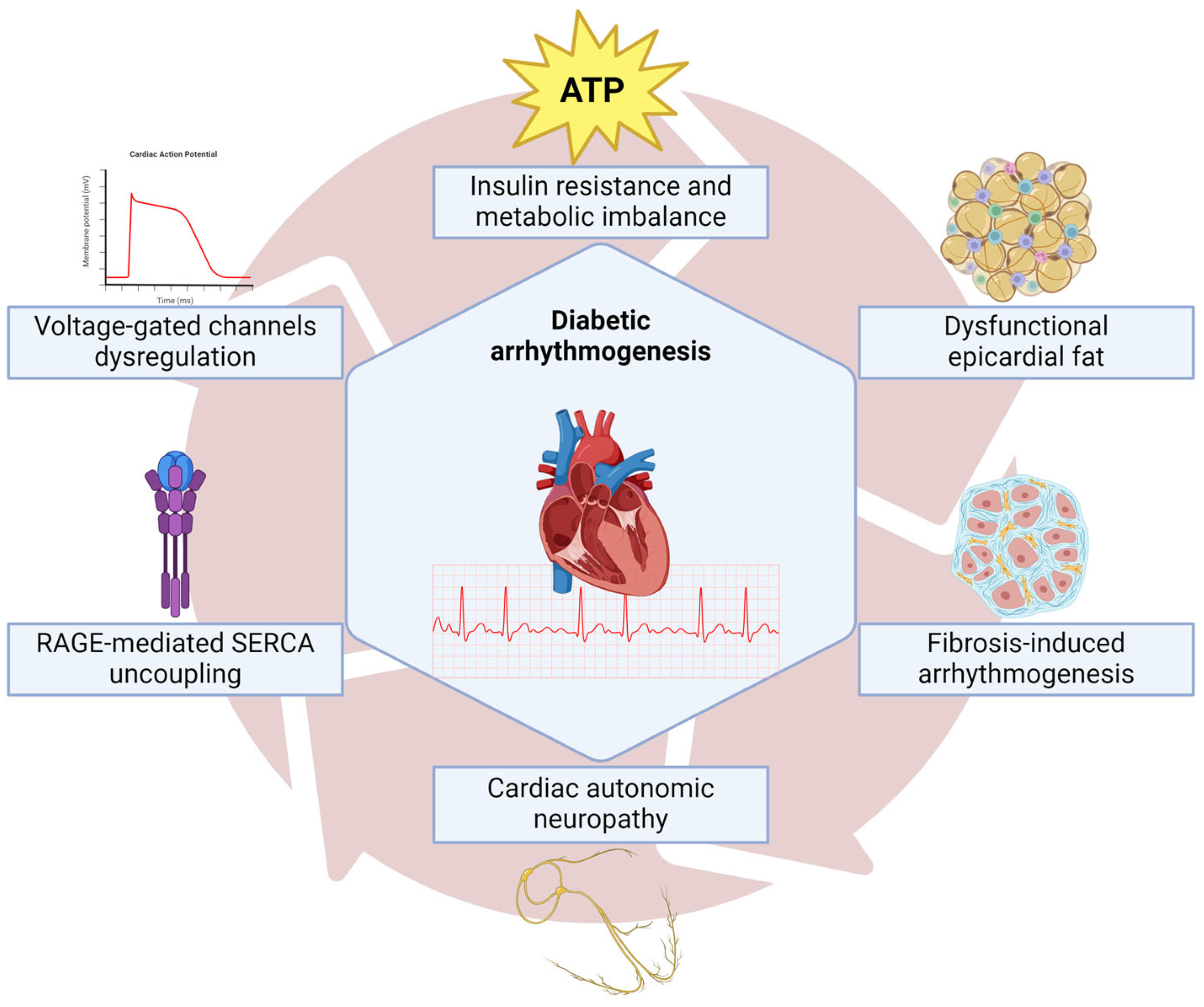
7. Arrhythmias
8. Therapeutic Insights
9. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular Disease and Risk Management: Standards of Care in Diabetes—2024. Diabetes Care 2024, 47, S179–S218. [Google Scholar] [CrossRef]
- Avagimyan, A.; Fogacci, F.; Pogosova, N.; Kakrurskiy, L.; Kogan, E.; Urazova, O.; Kobalava, Z.; Mikhaleva, L.; Vandysheva, R.; Zarina, G.; et al. Diabetic Cardiomyopathy: 2023 Update by the International Multidisciplinary Board of Experts. Curr. Probl. Cardiol. 2024, 49, 102052. [Google Scholar] [CrossRef]
- Tune, J.D.; Goodwill, A.G.; Sassoon, D.J.; Mather, K.J. Cardiovascular consequences of metabolic syndrome. Transl. Res. 2017, 183, 57–70. [Google Scholar] [CrossRef]
- Hsuan, C.F.; Teng, S.I.F.; Hsu, C.N.; Liao, D.; Chang, A.J.W.; Lee, H.L.; Hee, S.-W.; Chang, Y.-C.; Chuang, L.-M. Emerging Therapy for Diabetic Cardiomyopathy: From Molecular Mechanism to Clinical Practice. Biomedicines 2023, 11, 662. [Google Scholar] [CrossRef]
- Kolka, C.M.; Bergman, R.N. The endothelium in diabetes: Its role in insulin access and diabetic complications. Rev. Endocr. Metab. Disord. 2013, 14, 13–19. [Google Scholar] [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef]
- Henning, R.J. Type-2 diabetes mellitus and cardiovascular disease. Future Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef]
- Rajbhandari, J.; Fernandez, C.J.; Agarwal, M.; Yeap, B.X.Y.; Pappachan, J.M. Diabetic heart disease: A clinical update. World J. Diabetes 2021, 12, 383–406. [Google Scholar] [CrossRef]
- Wende, A.R.; Schell, J.C.; Ha, C.M.; Pepin, M.E.; Khalimonchuk, O.; Schwertz, H.; Pereira, R.O.; Brahma, M.K.; Tuinei, J.; Contreras-Ferrat, A.; et al. Maintaining myocardial glucose utilization in diabetic cardiomyopathy accelerates mitochondrial dysfunction. Diabetes 2020, 69, 2094–2111. [Google Scholar] [CrossRef]
- Ma, C.X.; Ma, X.N.; Guan, C.H.; Li, Y.D.; Mauricio, D.; Fu, S.B. Cardiovascular disease in type 2 diabetes mellitus: Progress toward personalized management. Cardiovasc. Diabetol. 2022, 21, 74. [Google Scholar] [CrossRef]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e22. [Google Scholar] [CrossRef]
- Nesci, A.; Carnuccio, C.; Ruggieri, V.; D’Alessandro, A.; Di Giorgio, A.; Santoro, L.; Gasbarrini, A.; Santoliquido, A.; Ponziani, F.R. Gut Microbiota and Cardiovascular Disease: Evidence on the Metabolic and Inflammatory Background of a Complex Relationship. Int. J. Mol. Sci. 2023, 24, 9087. [Google Scholar] [CrossRef]
- Lei, X.; Qiu, S.; Yang, G.; Wu, Q. Adiponectin and metabolic cardiovascular diseases: Therapeutic opportunities and challenges. Genes Dis. 2023, 10, 1525–1536. [Google Scholar] [CrossRef]
- Horáková, D.; Azeem, K.; Benešová, R.; Pastucha, D.; Horák, V.; Dumbrovská, L.; Martínek, A.; Novotný, D.; Švagera, Z.; Hobzová, M.; et al. Total and High Molecular Weight Adiponectin Levels and Prediction of Cardiovascular Risk in Diabetic Patients. Int. J. Endocrinol. 2015, 2015, 545068. [Google Scholar] [CrossRef]
- Lindberg, S.; Jensen, J.S.; Bjerre, M.; Pedersen, S.H.; Frystyk, J.; Flyvbjerg, A.; Galatius, S.; Jeppesen, J.; Mogelvang, R. Adiponectin, type 2 diabetes and cardiovascular risk. Eur. J. Prev. Cardiol. 2015, 22, 276–283. [Google Scholar] [CrossRef]
- Faramoushi, M.; Amir Sasan, R.; Sari Sarraf, V.; Karimi, P. Cardiac fibrosis and down regulation of GLUT4 in experimental diabetic cardiomyopathy are ameliorated by chronic exposures to intermittent altitude. J. Cardiovasc. Thorac. Res. 2016, 8, 26–33. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef]
- Jaishy, B.; Abel, E.D. Thematic review series: Lipotoxicity: Many roads to cell dysfunction and cell death lipids, lysosomes, and autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef]
- Arow, M.; Waldman, M.; Yadin, D.; Nudelman, V.; Shainberg, A.; Abraham, N.G.; Freimark, D.; Kornowski, R.; Aravot, D.; Hochhauser, E.; et al. Sodium-glucose cotransporter 2 inhibitor Dapagliflozin attenuates diabetic cardiomyopathy. Cardiovasc. Diabetol. 2020, 19, 7. [Google Scholar] [CrossRef]
- Peng, M.L.; Fu, Y.; Wu, C.W.; Zhang, Y.; Ren, H.; Zhou, S.S. Signaling Pathways Related to Oxidative Stress in Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 907757. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, Y.; Yin, R.; Xu, Y.; Zhang, L.; Zhang, Y.; Yang, L.; Zhao, D. Central role of cardiac fibroblasts in myocardial fibrosis of diabetic cardiomyopathy. Front. Endocrinol. 2023, 14, 1162754. [Google Scholar] [CrossRef]
- Pan, K.L.; Hsu, Y.C.; Chang, S.T.; Chung, C.M.; Lin, C.L. The Role of Cardiac Fibrosis in Diabetic Cardiomyopathy: From Pathophysiology to Clinical Diagnostic Tools. Int. J. Mol. Sci. 2023, 24, 8604. [Google Scholar] [CrossRef]
- Gupta, M.P. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2007, 43, 388–403. [Google Scholar] [CrossRef]
- Garcia, A.G.; Wilson, R.M.; Heo, J.; Murthy, N.R.; Baid, S.; Ouchi, N.; Sam, F. Interferon-γ ablation Exacerbates Myocardial Hypertrophy in Diastolic Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, 587–596. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial Dysfunction and Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 851941. [Google Scholar] [CrossRef]
- Huo, J.L.; Feng, Q.; Pan, S.; Fu, W.J.; Liu, Z.; Liu, Z. Diabetic cardiomyopathy: Early diagnostic biomarkers, pathogenetic mechanisms, and therapeutic interventions. Cell Death Discov. 2023, 9, 256. [Google Scholar] [CrossRef]
- Fernandes, R.; Hosoya, K.I.; Pereira, P. Reactive oxygen species downregulate glucose transport system in retinal endothelial cells. J. Physiol. Cell Physiol. 2011, 300, 927–936. [Google Scholar] [CrossRef]
- Molinaro, C.; Salerno, L.; Marino, F.; Scalise, M.; Salerno, N.; Pagano, L.; De Angelis, A.; Cianflone, E.; Torella, D.; Urbanek, K. Unraveling and Targeting Myocardial Regeneration Deficit in Diabetes. Antioxidants 2022, 11, 208. [Google Scholar] [CrossRef]
- Ke, J.; Pan, J.; Lin, H.; Gu, J. Diabetic cardiomyopathy: A brief summary on lipid toxicity. ESC Heart Fail. 2023, 10, 776–790. [Google Scholar] [CrossRef]
- Gaens, K.H.J.; Goossens, G.H.; Niessen, P.M.; Van Greevenbroek, M.M.; Van Der Kallen, C.J.H.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.M.; Blaak, E.E.; et al. Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arter. Thromb. Vasc. Biol. 2014, 34, 1199–1208. [Google Scholar] [CrossRef]
- Camarena, V.; Sant, D.; Mohseni, M.; Salerno, T.; Zaleski, M.L.; Wang, G.; Iacobellis, G. Novel atherogenic pathways from the differential transcriptome analysis of diabetic epicardial adipose tissue. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Ann. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, H.; Fan, X.; Wang, J.; Yin, Y.; Zhang, Y.; Shi, K.; Yu, F. The Role of Gut Microbiota and Trimethylamine N-oxide in Cardiovascular Diseases. J. Cardiovasc. Transl. Res. 2023, 16, 581–589. [Google Scholar] [CrossRef]
- Jing, L.; Zhang, H.; Xiang, Q.; Shen, L.; Guo, X.; Zhai, C.; Hu, H. Targeting Trimethylamine N-Oxide: A New Therapeutic Strategy for Alleviating Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 864600. [Google Scholar] [CrossRef]
- Al Kury, L.T. Calcium Homeostasis in Ventricular Myocytes of Diabetic Cardiomyopathy. J. Diabetes Res. 2020, 2020, 1942086. [Google Scholar]
- He, X.; Liu, S.; Zhang, Z.; Liu, Q.; Dong, J.; Lin, Z.; Chen, J.; Li, L.; Liu, W.; Liu, S.; et al. M1 macrophage-derived exosomes inhibit cardiomyocyte proliferation through delivering miR-155. BMC Cardiovasc. Disord. 2024, 24, 365. [Google Scholar] [CrossRef]
- Fredersdorf, S.; Thumann, C.; Zimmermann, W.H.; Vetter, R.; Graf, T.; Luchner, A.; Riegger, G.A.; Schunkert, H.; Eschenhagen, T.; Weil, J. Increased myocardial SERCA expression in early type 2 diabetes mellitus is insulin dependent: In vivo and in vitro data. Cardiovasc. Diabetol. 2012, 11, 57. [Google Scholar]
- Niggli, E.; Ullrich, N.D.; Gutierrez, D.; Kyrychenko, S.; Poláková, E.; Shirokova, N. Posttranslational modifications of cardiac ryanodine receptors: Ca2+ signaling and EC-coupling. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 866–875. [Google Scholar] [CrossRef]
- Macvanin, M.T.; Gluvic, Z.; Radovanovic, J.; Essack, M.; Gao, X.; Isenovic, E.R. Diabetic cardiomyopathy: The role of microRNAs and long non-coding RNAs. Front. Endocrinol. 2023, 14, 1124613. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Frati, G.; Schirone, L.; Chimenti, I.; Yee, D.; Biondi-Zoccai, G.; Volpe, M.; Sciarretta, S. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc. Res. 2017, 113, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Sewastianik, T.; Szydlowski, M.; Jablonska, E.; Bialopiotrowicz, E.; Kiliszek, P.; Gorniak, P.; Polak, A.; Prochorec-Sobieszek, M.; Szumera-Cieckiewicz, A.; Kaminski, T.S.; et al. FOXO1 is a TXN-and p300-dependent sensor and effector of oxidative stress in diffuse large B-cell lymphomas characterized by increased oxidative metabolism. Oncogene 2016, 35, 5989–6000. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.J.; Jiang, N.; Blagg, A.; Johnstone, J.L.; Gondalia, R.; Oh, M.; Luo, X.; Yang, K.; Shelton, J.M.; Rothermel, B.A.; et al. Mechanical unloading activates FoxO3 to trigger Bnip3-dependent cardiomyocyte atrophy. J. Am. Heart Assoc. 2013, 2, e000016. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.C.; Abel, E.D. Heart Failure in Type 2 Diabetes Mellitus: Impact of Glucose-Lowering Agents, Heart Failure Therapies, and Novel Therapeutic Strategies. Circ. Res. 2019, 124, 121–141. [Google Scholar] [CrossRef]
- Shiojima, I.; Sato, K.; Izumiya, Y.; Schiekofer, S.; Ito, M.; Liao, R.; Colucci, W.S.; Walsh, K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Investig. 2005, 115, 2108–2118. [Google Scholar] [CrossRef]
- Mengstie, M.A.; Abebe, E.C.; Teklemariam, A.B.; Mulu, A.T.; Teshome, A.A.; Zewde, E.A.; Muche, Z.T.; Azezew, M.T. Molecular and cellular mechanisms in diabetic heart failure: Potential therapeutic targets. Front. Endocrinol. 2022, 13, 947294. [Google Scholar] [CrossRef]
- Pan, G.; Roy, B.; Giri, S.; Lanfear, D.E.; Thandavarayan, R.A.; Guha, A.; Ortiz, P.A.; Palaniyandi, S.S. Aldehyde Dehydrogenase 2 Activator Augments the Beneficial Effects of Empagliflozin in Mice with Diabetes-Associated HFpEF. Int. J. Mol. Sci. 2022, 23, 10439. [Google Scholar] [CrossRef]
- Mali, V.R.; Palaniyandi, S.S. Regulation and therapeutic strategies of 4-hydroxy-2-nonenal metabolism in heart disease. Free Radic. Res. 2014, 48, 251–263. [Google Scholar] [CrossRef]
- Bipradas, R.; Guodong, P.; Shailendra, G.; Rajarajan, T.; Suresh, S. Aldehyde dehydrogenase 2 augments adiponectin signaling in coronary angiogenesis in HFpEF associated with diabetes. FASEB J. 2022, 36, e22440–e22465. [Google Scholar]
- Ramesh Iyer, V. Ventricular dysfunction: Tachycardia induced cardiomyopathy. Indian Pacing Electrophysiol. J. 2008, 8 (Suppl. S1), S122–S129. [Google Scholar]
- Tu, C.; Caudal, A.; Liu, Y.; Gorgodze, N.; Zhang, H.; Lam, C.K.; Dai, Y.; Zhang, A.; Wnorowski, A.; Wu, M.A.; et al. Tachycardia-induced metabolic rewiring as a driver of contractile dysfunction. Nat. Biomed. Eng. 2024, 8, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Huizar, J.F.; Ellenbogen, K.A.; Tan, A.Y.; Kaszala, K. Arrhythmia-Induced Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2328–2344. [Google Scholar] [CrossRef] [PubMed]
- Donghua, Z.; Jian, P.; Zhongbo, X.; Feifei, Z.; Xinhui, P.; Hao, Y.; Fuqiang, L.; Yan, L.; Yong, X.; Xinfu, H.; et al. Reversal of cardiomyopathy in patients with congestive heart failure secondary to tachycardia. J. Interv. Card. Electrophysiol. 2013, 36, 27–32. [Google Scholar] [CrossRef]
- Bergonti, M.; Ascione, C.; Marcon, L.; Pambrun, T.; Della Rocca, D.G.; Ferrero, T.G.; Pannone, L.; Kühne, M.; Compagnucci, P.; Bonomi, A.; et al. Left ventricular functional recovery after atrial fibrillation catheter ablation in heart failure: A prediction model. Eur. Heart J. 2023, 44, 3327–3335. [Google Scholar] [CrossRef]
- Redfield, M.M.; Neal Kay, G.; Jenkins, L.S.; Mianulli, M.; Nick Jensen, D.; Ellenbogen, K.A. Tachycardia-related cardiomyopathy: A common cause of ventricular dysfunction in patients with atrial fibrillation referred for atrioventricular ablation. Mayo Clin. Proc. 2000, 75, 790–795. [Google Scholar] [CrossRef]
- Rodriguez, L.M.; Smeets, J.L.R.M.; Xie, B.; De Chillou, C.; Cheriex, E.; Pieters, F.; Metzger, J.; den Dulk, K.; Wellens, H.J.J. Arrhythmias and conduction disturbances Improvement in Left Ventricular Function by Ablation of Atrioventricular Nodal Conduction in Selected Patients with Lone Atrial Fibrillation. Am. J. Cardiol. 1993, 72, 1137–1141. [Google Scholar] [CrossRef]
- Mueller, K.A.L.; Heinzmann, D.; Klingel, K.; Fallier-Becker, P.; Kandolf, R.; Kilias, A.; Walker-Allgaier, B.; Borst, O.; Kumbrink, J.; Kirchner, T.; et al. Histopathological and Immunological Characteristics of Tachycardia-Induced Cardiomyopathy. J. Am. Coll. Cardoil. 2017, 69, 2160–2172. [Google Scholar] [CrossRef]
- Nerheim, P.; Birger-Botkin, S.; Piracha, L.; Olshansky, B. Heart failure and sudden death in patients with tachycardia-induced cardiomyopathy and recurrent tachycardia. Circulation 2004, 110, 247–252. [Google Scholar] [CrossRef]
- Ellis, E.R.; Josephson, M.R. What About Tachycardia-induced Cardiomyopathy? Arrhythm. Electrophysiol. Rev. 2013, 2, 82–90. [Google Scholar] [CrossRef]
- Reddy, R.K.; Ardissino, M.; Ng, F.S. Type 2 Diabetes and Atrial Fibrillation: Evaluating Causal and Pleiotropic Pathways Using Mendelian Randomization. J. Am. Heart Assoc. 2023, 12, e030298. [Google Scholar] [CrossRef]
- Kaze, A.D.; Fonarow, G.C.; Echouffo-Tcheugui, J.B. Cardiac Autonomic Dysfunction and Risk of Silent Myocardial Infarction Among Adults With Type 2 Diabetes. J. Am. Heart Assoc. 2023, 12, e029814. [Google Scholar] [CrossRef] [PubMed]
- Duque, A.; Mediano, M.F.F.; De Lorenzo, A.; Rodrigues, L.F., Jr. Cardiovascular autonomic neuropathy in diabetes: Pathophysiology, clinical assessment and implications. World J. Diabetes 2021, 12, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Sloan, G.; Ye, Y.; Wang, S.; Duan, B.; Tesfaye, S.; Gao, L. New Perspective in Diabetic Neuropathy: From the Periphery to the Brain, a Call for Early Detection, and Precision Medicine. Front. Endocrinol. 2020, 10, 929. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk between oxidative stress and endoplasmic reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: A focus on the protective roles of heme oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef]
- Fiaschi, T.; Magherini, F.; Gamberi, T.; Lucchese, G.; Faggian, G.; Modesti, A.; Modesti, P.A. Hyperglycemia and angiotensin II cooperate to enhance collagen I deposition by cardiac fibroblasts through a ROS-STAT3-dependent mechanism. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2603–2610. [Google Scholar] [CrossRef]
- Kato, T.; Yamashita, T.; Sekiguchi, A.; Tsuneda, T.; Sagara, K.; Takamura, M.; Kaneko, S.; Aizawa, T.; Fu, L. AGEs-RAGE system mediates atrial structural remodeling in the diabetic rat. J. Cardiovasc. Electrophysiol. 2008, 19, 415–420. [Google Scholar] [CrossRef]
- Bohne, L.J.; Johnson, D.; Rose, R.A.; Wilton, S.B.; Gillis, A.M. The association between diabetes mellitus and atrial fibrillation: Clinical and mechanistic insights. Front. Physiol. 2019, 10, 135. [Google Scholar] [CrossRef]
- Ma, T.; Huang, X.; Zheng, H.; Huang, G.; Li, W.; Liu, X.; Liang, J.; Cao, Y.; Hu, Y.; Huang, Y. SFRP2 Improves Mitochondrial Dynamics and Mitochondrial Biogenesis, Oxidative Stress, and Apoptosis in Diabetic Cardiomyopathy. Oxid. Med. Cell Longev. 2021, 2021, 9265016. [Google Scholar] [CrossRef]
- Nakamura, K.; Miyoshi, T.; Yoshida, M.; Akagi, S.; Saito, Y.; Ejiri, K.; Matsuo, N.; Ichikawa, K.; Iwasaki, K.; Naito, T.; et al. Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus. Int. J. Mol. Sci. 2022, 23, 3587. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, W.R.; Kaul, S.; Smith, R.J. The Cardiovascular Safety of Diabetes Drugs—Insights from the Rosiglitazone Experience. N. Engl. J. Med. 2013, 369, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Wada, H.; Satoh-Asahara, N.; Inoue, H.; Uehara, K.; Funada, J.; Ogo, A.; Horie, T.; Fujita, M.; Shimatsu, A.; et al. Effects of Metformin on Left Ventricular Size and Function in Hypertensive Patients with Type 2 Diabetes Mellitus: Results of a Randomized, Controlled, Multicenter, Phase IV Trial. Am. J. Cardiovasc. Drugs 2020, 20, 283–293. [Google Scholar] [CrossRef]
- Li, J.; Minćzuk, K.; Massey, J.C.; Howell, N.L.; Jack Roy, R.; Paul, S.; Patrie, J.T.; Kramer, C.M.; Epstein, F.H.; Care, R.M.; et al. Metformin improves cardiac metabolism and function, and prevents left ventricular hypertrophy in spontaneously hypertensive rats. J. Am. Heart Assoc. 2020, 9, e015154. [Google Scholar] [CrossRef]
- Spigoni, V.; Fantuzzi, F.; Carubbi, C.; Pozzi, G.; Masselli, E.; Gobbi, G.; Solini, A.; Bonadonna, R.C.; Cas, A.D. Sodium-glucose cotransporter 2 inhibitors antagonize lipotoxicity in human myeloid angiogenic cells and ADP-dependent activation in human platelets: Potential relevance to prevention of cardiovascular events. Cardiovasc. Diabetol. 2020, 19, 46. [Google Scholar] [CrossRef]
- Matthews, V.B.; Elliot, R.H.; Rudnicka, C.; Hricova, J.; Herat, L.; Schlaich, M.P. Role of the sympathetic nervous system in regulation of the sodium glucose cotransporter 2. J. Hypertens. 2017, 35, 2059–2068. [Google Scholar] [CrossRef]
- Longo, M.; Scappaticcio, L.; Cirillo, P.; Maio, A.; Carotenuto, R.; Maiorino, M.I.; Bellastella, G.; Esposito, K. Glycemic Control and the Heart: The Tale of Diabetic Cardiomyopathy Continues. Biomolecules 2022, 12, 272. [Google Scholar] [CrossRef]
- Ghosh, N.; Chacko, L.; Bhattacharya, H.; Vallamkondu, J.; Nag, S.; Dey, A.; Karmakar, T.; Reddy, P.H.; Kandimalla, R.; Dewanjee, S. Exploring the Complex Relationship between Diabetes and Cardiovascular Complications: Understanding Diabetic Cardiomyopathy and Promising Therapies. Biomedicines 2023, 11, 1126. [Google Scholar] [CrossRef]
- Kim, M.; Platt, M.J.; Shibasaki, T.; Quaggin, S.E.; Backx, P.H.; Seino, S.; Simpson, J.A.; Drucker, D.J. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat. Med. 2013, 19, 567–575. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Maleki, M.; Jamialahmadi, T.; Sahebkar, A. Anti-inflammatory benefits of semaglutide: State of the art. J. Clin. Transl. Endocrinol. 2024, 36, 100340. [Google Scholar] [CrossRef]
- Ndumele, C.E.; Rangaswami, J.; Chow, S.L.; Neeland, I.J.; Tuttle, K.R.; Khan, S.S.; Coresh, J.; Mathew, R.O.; Baker-Smith, C.M.; Carnethon, M.R.; et al. Cardiovascular-Kidney-Metabolic Health: A Presidential Advisory from the American Heart Association. Circulation 2023, 148, 1606–1635. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giraldo-Gonzalez, G.C.; Roman-Gonzalez, A.; Cañas, F.; Garcia, A. Molecular Mechanisms of Type 2 Diabetes-Related Heart Disease and Therapeutic Insights. Int. J. Mol. Sci. 2025, 26, 4548. https://doi.org/10.3390/ijms26104548
Giraldo-Gonzalez GC, Roman-Gonzalez A, Cañas F, Garcia A. Molecular Mechanisms of Type 2 Diabetes-Related Heart Disease and Therapeutic Insights. International Journal of Molecular Sciences. 2025; 26(10):4548. https://doi.org/10.3390/ijms26104548
Chicago/Turabian StyleGiraldo-Gonzalez, German Camilo, Alejandro Roman-Gonzalez, Felipe Cañas, and Andres Garcia. 2025. "Molecular Mechanisms of Type 2 Diabetes-Related Heart Disease and Therapeutic Insights" International Journal of Molecular Sciences 26, no. 10: 4548. https://doi.org/10.3390/ijms26104548
APA StyleGiraldo-Gonzalez, G. C., Roman-Gonzalez, A., Cañas, F., & Garcia, A. (2025). Molecular Mechanisms of Type 2 Diabetes-Related Heart Disease and Therapeutic Insights. International Journal of Molecular Sciences, 26(10), 4548. https://doi.org/10.3390/ijms26104548