
New Dimethoxyaryl-Sesquiterpene Derivatives with Cytotoxic Activity Against MCF-7 Breast Cancer Cells: From Synthesis to Topoisomerase I/II Inhibition and Cell Death Mechanism Studies

 ,
,  , , ,
, , ,  , ,
, ,  , and
, and
Abstract

1. Introduction
2. Results and Discussion
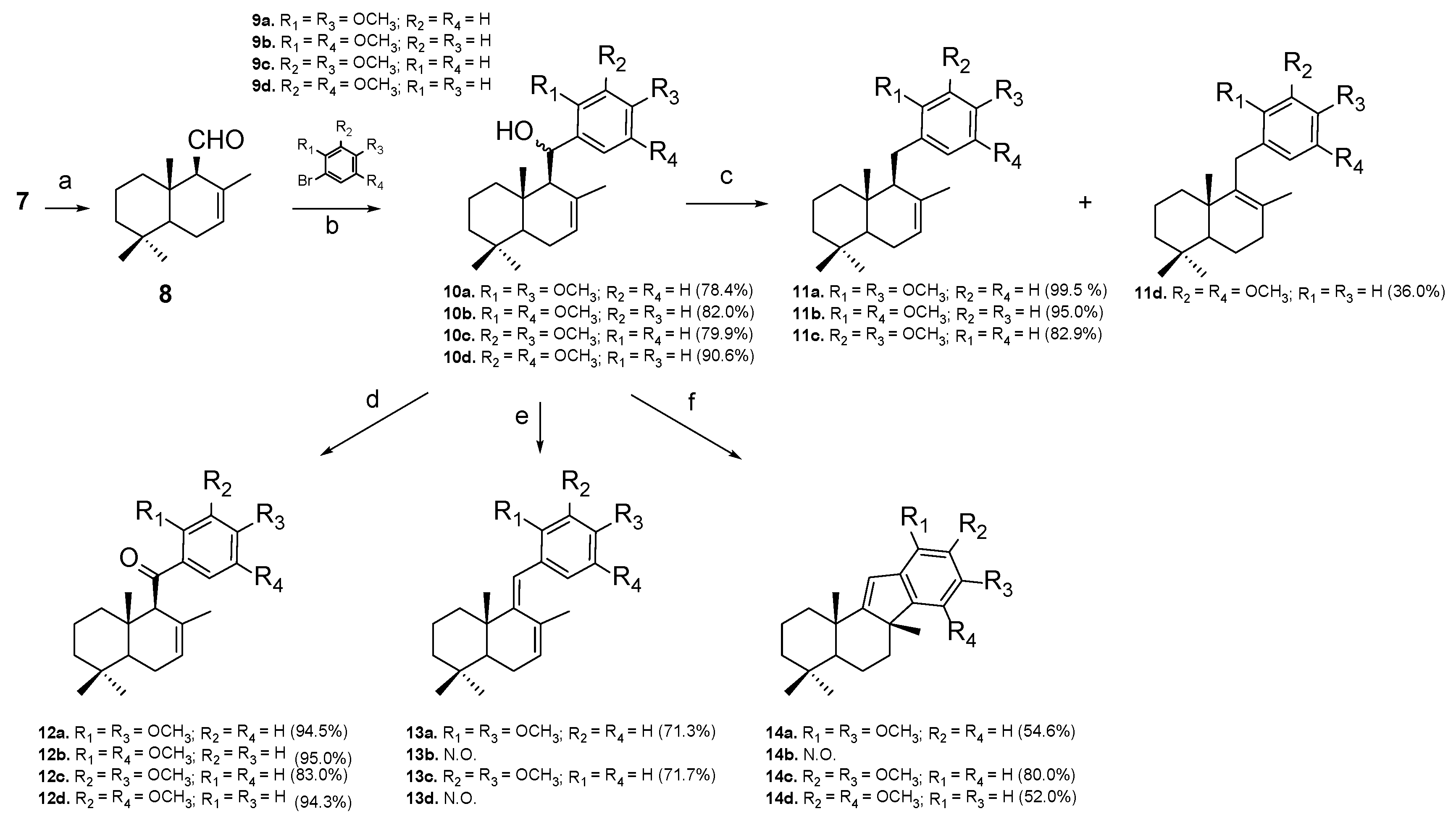
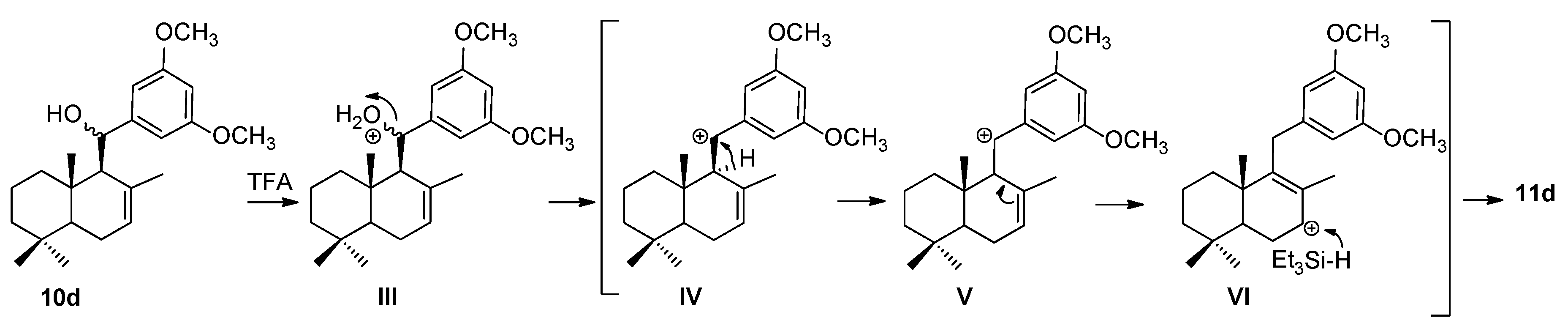
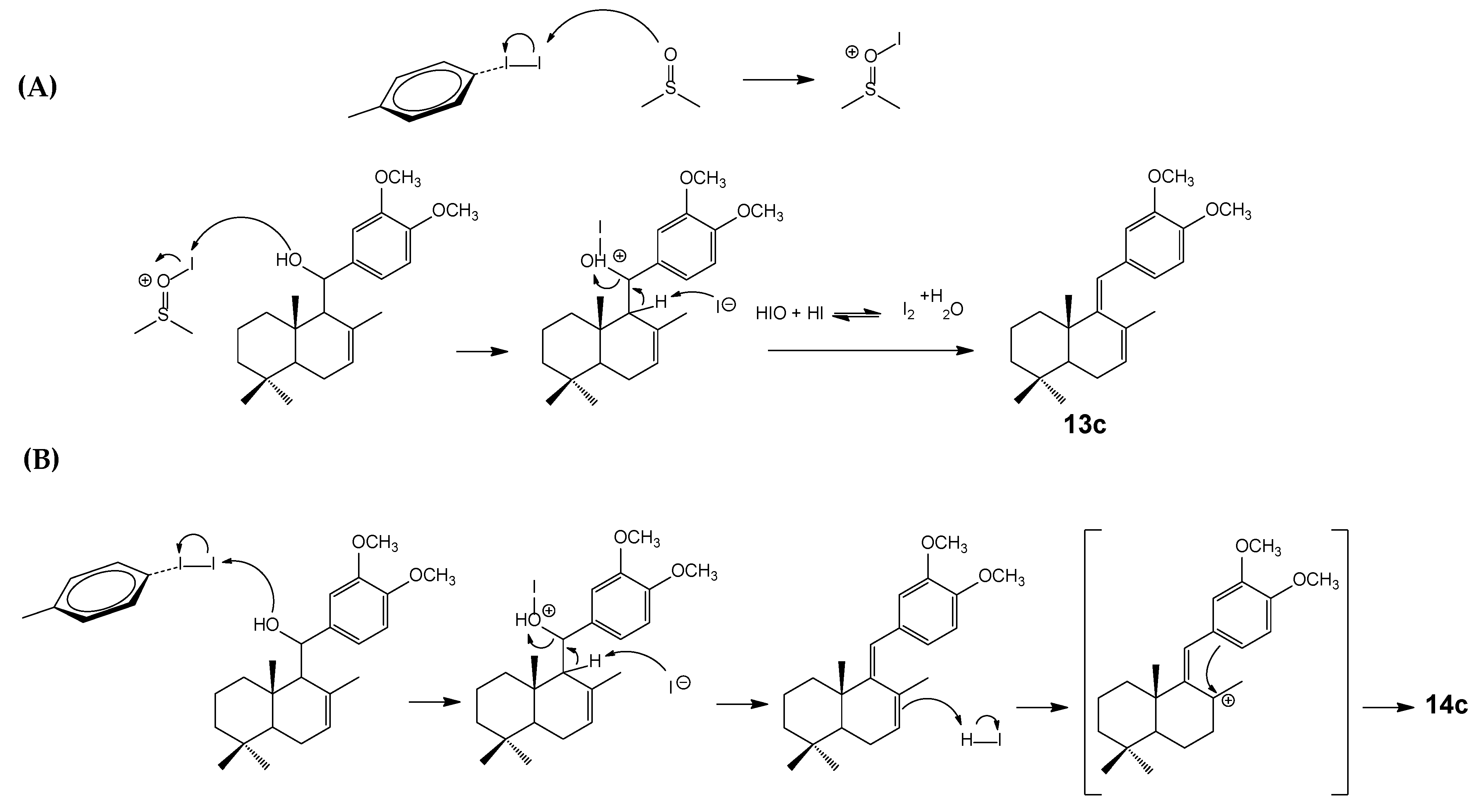
2.1. Chemistry
2.2. Biological Evaluation
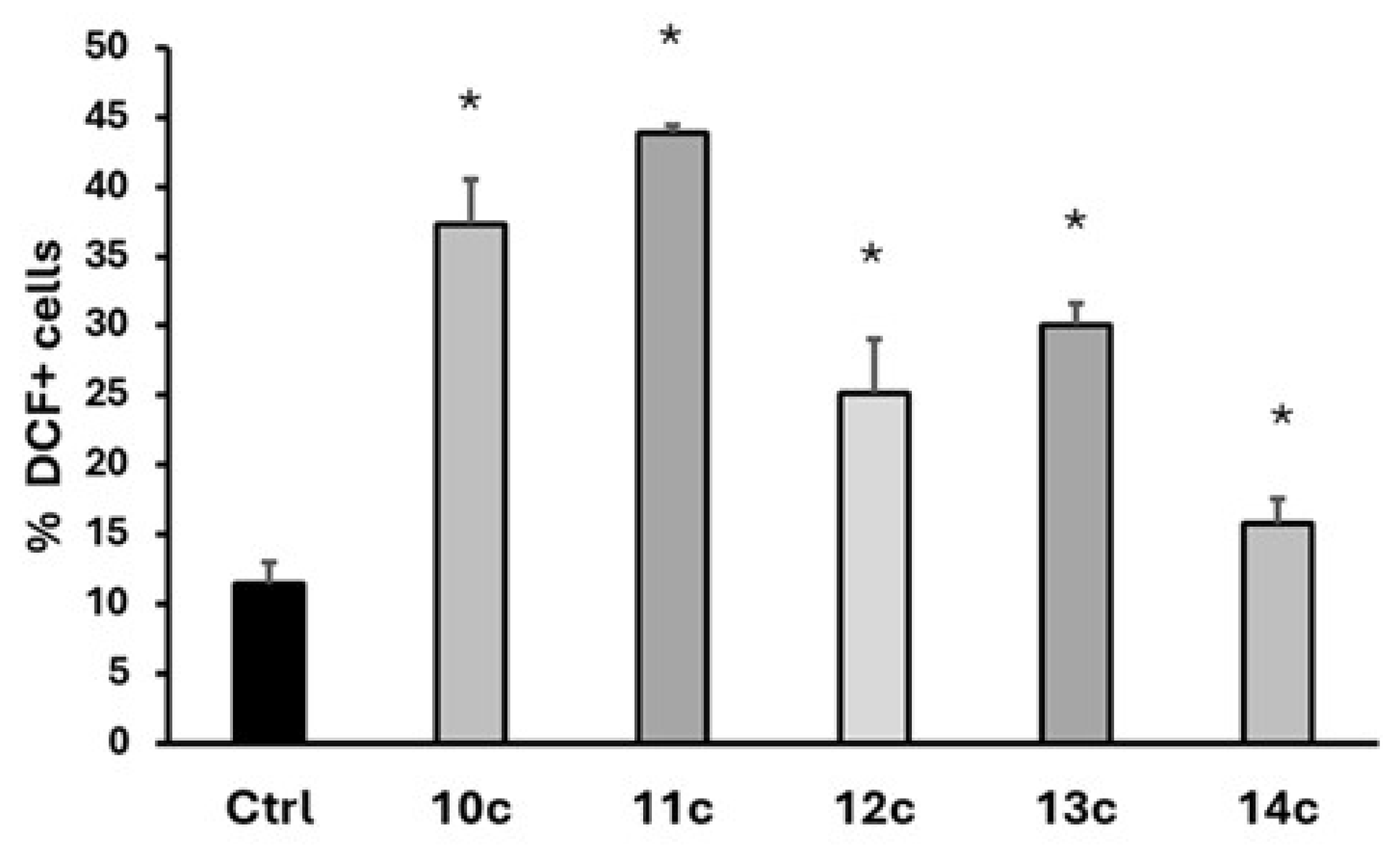
2.3. Intracellular ROS Generation
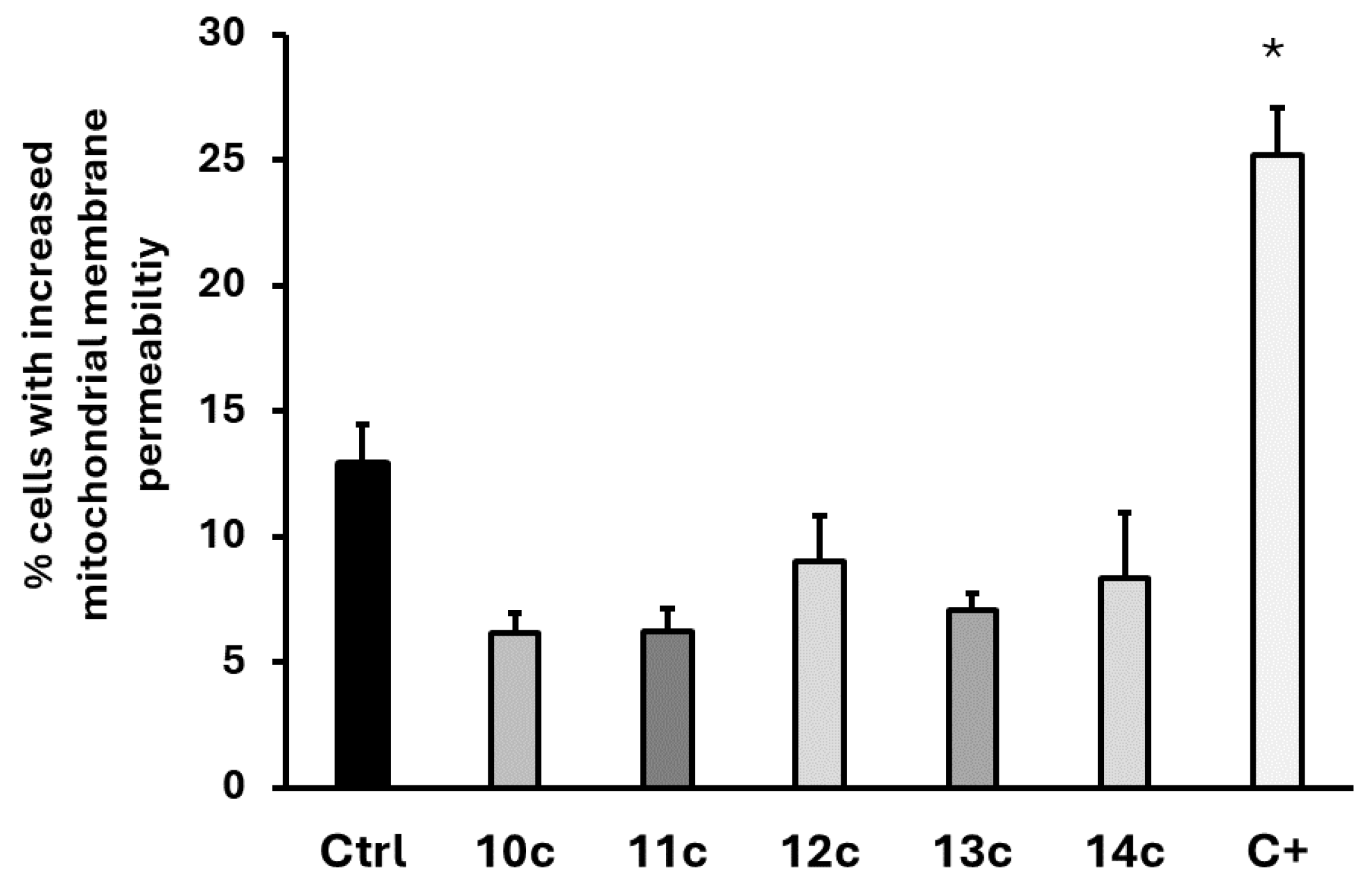
2.4. Mitochondrial Membrane Potential Analysis
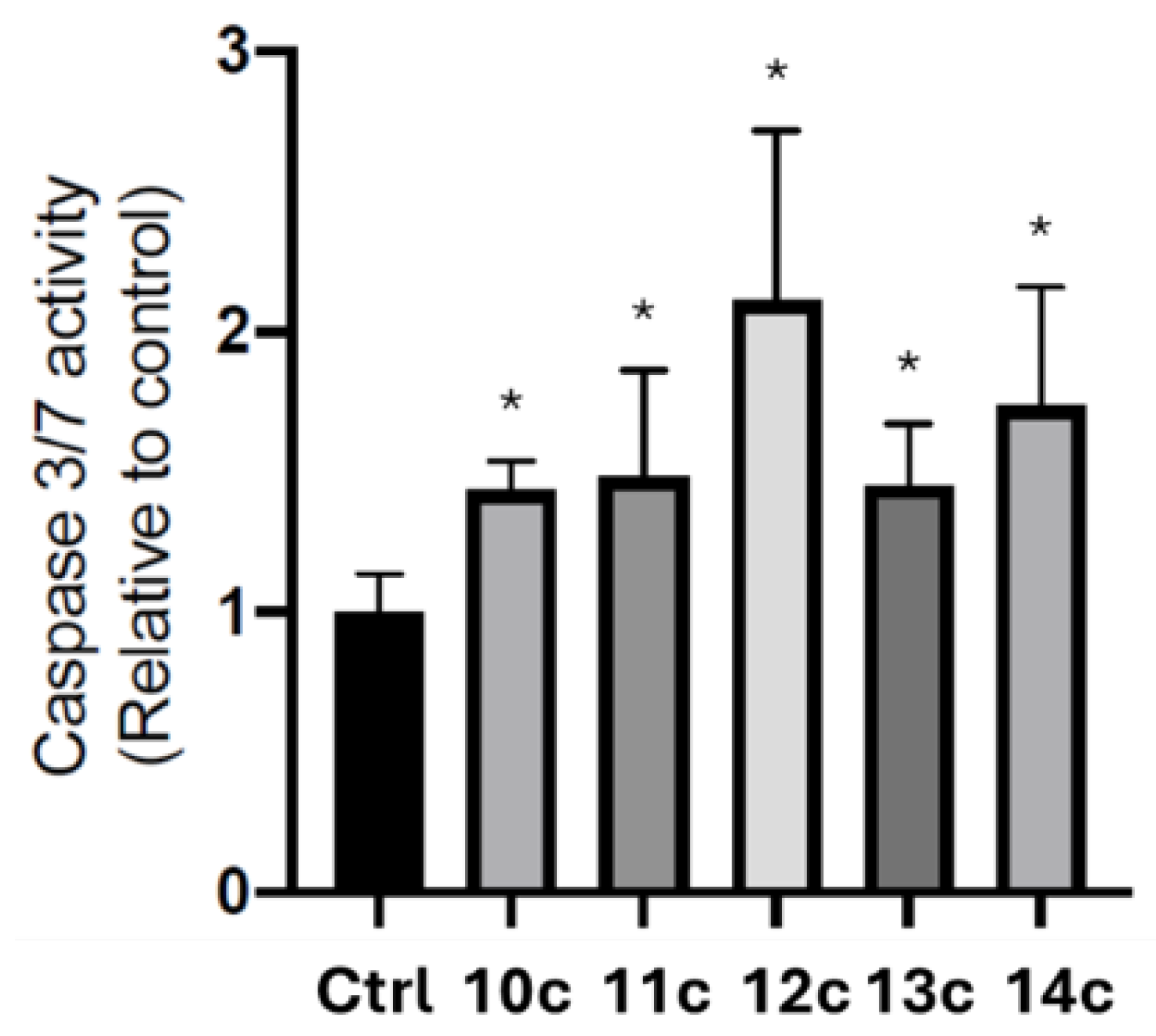
2.5. Determination of Caspases 3/7 Activation (Caspase-Glo 3/7 Assay)
2.6. Assays of TOP 1/2 Activity
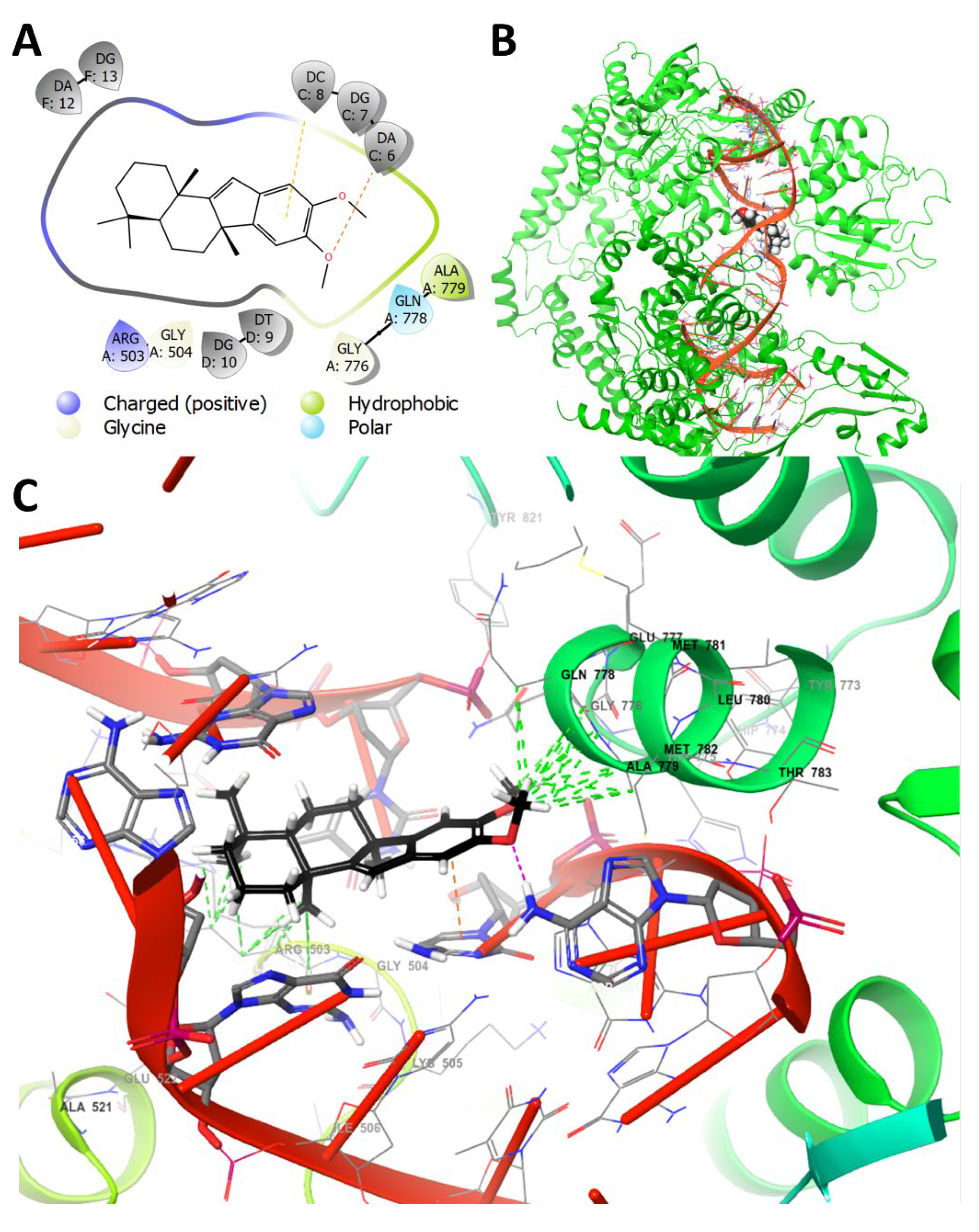
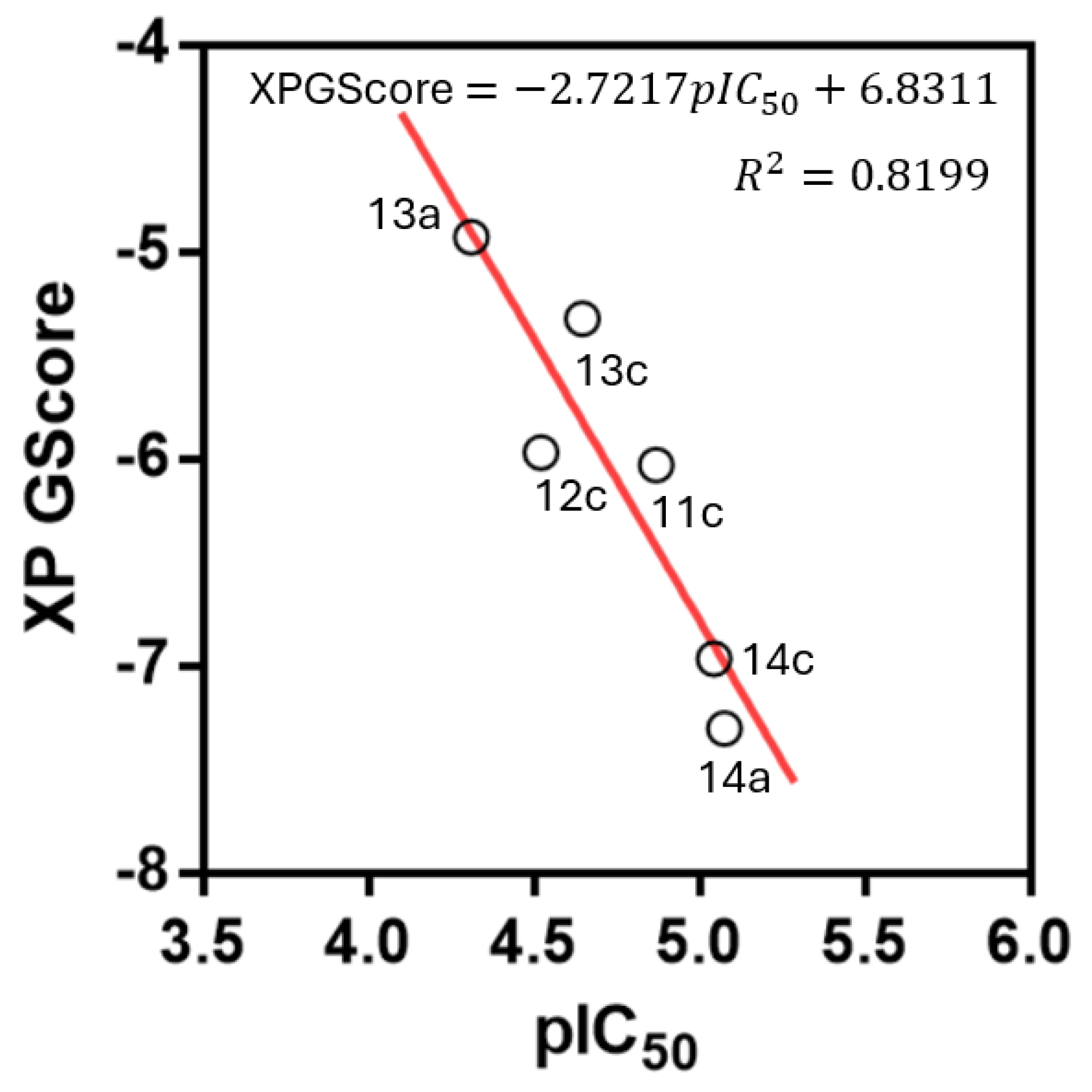
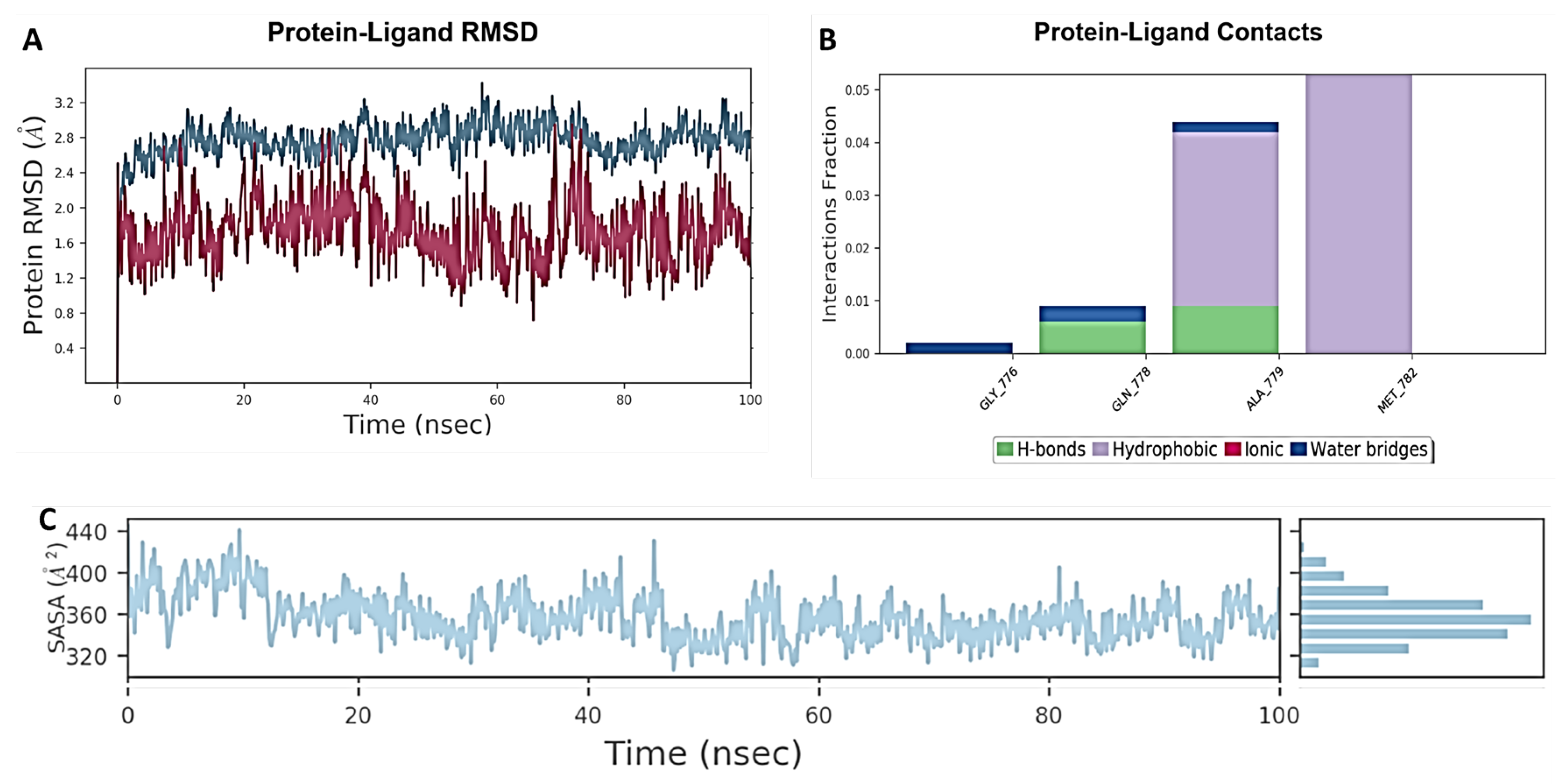
2.7. Molecular Docking and Molecular Dynamics Studies
2.8. Calculated Physicochemical Properties and ADME Parameters
3. Materials and Methods
3.1. General Information
3.2. Synthetic Procedures
3.2.1. Synthesis of Drimenal (8)
3.2.2. The General Procedure for the Synthesis of Alcohols 10a–d
3.2.3. The General Procedure for the Synthesis of Reduced Compounds 11a–d
3.2.4. General Procedure for the Synthesis of Ketone Compounds 12a–d
3.2.5. The General Procedure for the Synthesis of Diene Compounds 13a–d
3.2.6. The General Procedure for the Synthesis of Cyclic Compounds 14a–d
3.3. Biological Assays
3.3.1. Cultured Cell Lines
3.3.2. In Vitro Cytotoxicity Screening by Using MTT Assay
3.3.3. ROS in Cell Lines Exposed to Compounds
3.3.4. Determination of Mitochondrial Membrane Potential (ΔΨmt) by Flow Cytometry
3.3.5. Determination of Caspases-3/7 Activation
3.3.6. Topoisomerase I/II Activity Assay
3.4. In Silico Studies
3.4.1. Docking
3.4.2. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Burstein, H.J.; Gnant, M.; Loibl, S.; Cameron, D.; Regan, M.M.; Denkert, C.; Poortmans, P.; Weber, W.P.; Thurlimann, B.; et al. Understanding breast cancer complexity to improve patient outcomes: The St Gallen International Consensus Conference for the Primary Therapy of Individuals with Early Breast Cancer 2023. Ann. Oncol. 2023, 34, 970–986. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ouyang, Z.; Du, H.; Wang, M.; Wang, J.; Sun, H.; Kong, L.; Xu, Q.; Ma, H.; Sun, Y. New opportunities and challenges of natural products research: When target identification meets single-cell multiomics. Acta Pharm. Sin. B 2022, 12, 4011–4039. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Marcos, I.S.; Conde, A.; Moro, R.F.; Basabe, P.; Díez, D.; Urones, J.G. Synthesis of quinone/hydroquinone sesquiterpenes. Tetrahedron 2010, 66, 8280–8290. [Google Scholar] [CrossRef]
- Gordaliza, M. Cytotoxic terpene quinones from marine sponges. Mar. Drugs 2010, 8, 2849–2870. [Google Scholar] [CrossRef]
- Ding, Z.-H.; Dong, Z.-J.; Liu, J.-K. Albaconol, A Novel Prenylated Resorcinol (=Benzene-1,3-diol) from Basidiomycetes Albatrellus confluens. Helv. Chim. Acta 2001, 84, 259–262. [Google Scholar] [CrossRef]
- Liu, J.K. Secondary metabolites from higher fungi in China and their biological activity. Drug Discov. Ther. 2007, 1, 94–103. [Google Scholar]
- Qing, C.; Liu, M.H.; Yangl, W.M.; Zhang, Y.L.; Wang, L.; Liu, J.K. Effects of albaconol from the basidiomycete Albatrellus confluens on DNA topoisomerase II-mediated DNA cleavage and relaxation. Planta Med. 2004, 70, 792–796. [Google Scholar] [CrossRef]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Herrador, M.M.; Chahboun, R.; Galera, P. Synthesis and antitumoral activities of marine ent-chromazonarol and related compounds. Bioorg Med. Chem. Lett. 1999, 9, 2325–2328. [Google Scholar] [CrossRef]
- Pérez-García, E.; Zubía, E.; Ortega, M.J.; Carballo, J.L. Merosesquiterpenes from Two Sponges of the Genus Dysidea. J. Nat. Prod. 2005, 68, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, T.; Yang, X.W.; Huang, R.; Yang, B.; Tang, L.; Liu, Y. Chemical and biological aspects of marine sponges of the genus Xestospongia. Chem. Biodivers. 2010, 7, 2201–2227. [Google Scholar] [CrossRef] [PubMed]
- Khodzori, F.A.; Mazlan, N.B.; Chong, W.S.; Ong, K.H.; Palaniveloo, K.; Shah, M.D. Metabolites and Bioactivity of the Marine Xestospongia Sponges (Porifera, Demospongiae, Haplosclerida) of Southeast Asian Waters. Biomolecules 2023, 13, 484. [Google Scholar] [CrossRef]
- Cardiovascular & Renal Novel CETP inhibitors isolated from marine sponge. Expert Opin. Ther. Pat. 1995, 5, 829–830. [CrossRef]
- Jiso, A.; Demuth, P.; Bachowsky, M.; Haas, M.; Seiwert, N.; Heylmann, D.; Rasenberger, B.; Christmann, M.; Dietrich, L.; Brunner, T.; et al. Natural Merosesquiterpenes Activate the DNA Damage Response via DNA Strand Break Formation and Trigger Apoptotic Cell Death in p53-Wild-Type and Mutant Colorectal Cancer. Cancers 2021, 13, 3282. [Google Scholar] [CrossRef]
- Rodríguez, J.; Quiñoá, E.; Riguera, R.; Peters, B.M.; Abrell, L.M.; Crews, P. The structures and stereochemistry of cytotoxic sesquiterpene quinones from dactylospongia elegans. Tetrahedron 1992, 48, 6667–6680. [Google Scholar] [CrossRef]
- Arjona, O.; Garranzo, M.; Mahugo, J.; Maroto, E.; Plumet, J.; Sáez, B. Total synthesis of both enantiomers of 15-oxopuupehenol methylendioxy derivatives. Tetrahedron Lett. 1997, 38, 7249–7252. [Google Scholar] [CrossRef]
- Nasu, S.S.; Yeung, B.K.S.; Hamann, M.T.; Scheuer, P.J.; Kelly-Borges, M.; Goins, K. Puupehenone-related metabolites from two Hawaiian sponges, Hyrtios spp. J. Org. Chem. 1995, 60, 7290–7292. [Google Scholar] [CrossRef]
- Carpi, S.; Scoditti, E.; Polini, B.; Brogi, S.; Calderone, V.; Proksch, P.; Ebada, S.S.; Nieri, P. Pro-Apoptotic Activity of the Marine Sponge Dactylospongia elegans Metabolites Pelorol and 5-epi-Ilimaquinone on Human 501Mel Melanoma Cells. Mar. Drugs 2022, 20, 427. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Ho, V.W.; Kuroda, E.; Ruschmann, J.; Antignano, F.; Lam, V.; Krystal, G. Role of SHIP in cancer. Exp. Hematol. 2011, 39, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Araque, I.; Ramírez, J.; Vergara, R.; Mella, J.; Aránguiz, P.; Espinoza, L.; Vera, W.; Montenegro, I.; Salas, C.O.; Villena, J.; et al. Cytotoxic Activity, Topoisomerase I Inhibition and In Silico Studies of New Sesquiterpene-aryl Ester Derivatives of (-) Drimenol. Molecules 2023, 28, 3959. [Google Scholar] [CrossRef]
- Sierra, J.R.; López, J.T.; Cortés, M.J. (−)-3β-Acetoxydrimenin from the leaves of Drimys winteri. Phytochemistry 1985, 25, 253–254. [Google Scholar] [CrossRef]
- Zárraga, M.; Zárraga, A.M.; Rodríguez, B.; Pérez, C.; Paz, C.; Paz, P.; Sanhueza, C. Synthesis of a new nitrogenated drimane derivative with antifungal activity. Tetrahedron Lett. 2008, 49, 4775–4776. [Google Scholar] [CrossRef]
- Alvarez-Manzaneda, E.J.; Chahboun, R.; Barranco Pérez, I.; Cabrera, E.; Alvarez, E.; Alvarez-Manzaneda, R. First Enantiospecific Synthesis of the Antitumor Marine Sponge Metabolite (−)-15-Oxopuupehenol from (−)-Sclareol. Org. Lett. 2005, 7, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R. Synthesis of wiedendiol-A and wiedendiol-B from labdane diterpenes. Tetrahedron 1998, 54, 5635–5650. [Google Scholar] [CrossRef]
- Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R.; Cortés, M.; Armstrong, V. Synthesis and antitumor activity of puupehedione and related compounds. Tetrahedron 1999, 55, 15181–15208. [Google Scholar] [CrossRef]
- Laube, T.; Schröder, J.; Stehle, R.; Seifert, K. Total synthesis of yahazunol, zonarone and isozonarone. Tetrahedron 2002, 58, 4299–4309. [Google Scholar] [CrossRef]
- Barclay, L.R.C.; Sonawane, H.R.; MacDonald, M.C. Sterically Hindered Aromatic Compounds. III. Acid-catalyzed Reactions of 2,4,6-Tri-t-butyl- and 2-Methyl-4,6-di-t-butylbenzyl Alcohols and Chlorides. Can. J. Chem. 1972, 50, 281–290. [Google Scholar] [CrossRef]
- Domingo, V.; Prieto, C.; Silva, L.; Rodilla, J.M.L.; Quílez del Moral, J.F.; Barrero, A.F. Iodine, a Mild Reagent for the Aromatization of Terpenoids. J. Nat. Prod. 2016, 79, 831–837. [Google Scholar] [CrossRef]
- Domingo, V.; Prieto, C.; Castillo, A.; Silva, L.; Quílez del Moral, J.F.; Barrero, A.F. Iodine-Promoted Metal-Free Aromatization: Synthesis of Biaryls, Oligo p-Phenylenes and A-Ring Modified Steroids. Adv. Synth. Catal. 2015, 357, 3359–3364. [Google Scholar] [CrossRef]
- Meimetis, L.G.; Nodwell, M.; Yang, L.; Wang, X.; Wu, J.; Harwig, C.; Stenton, G.R.; Mackenzie, L.F.; MacRury, T.; Patrick, B.O.; et al. Synthesis of SHIP1-Activating Analogs of the Sponge Meroterpenoid Pelorol. Eur. J. Org. Chem. 2012, 2012, 5195–5207. [Google Scholar] [CrossRef]
- Montenegro, I.; Tomasoni, G.; Bosio, C.; Quiñones, N.; Madrid, A.; Carrasco, H.; Olea, A.; Martinez, R.; Cuellar, M.; Villena, J. Study on the Cytotoxic Activity of Drimane Sesquiterpenes and Nordrimane Compounds against Cancer Cell Lines. Molecules 2014, 19, 18993–19006. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Angulo-Elizari, E.; Raza, A.; Encío, I.; Sharma, A.K.; Sanmartín, C.; Plano, D. Seleno-Warfare against Cancer: Decoding Antitumor Activity of Novel Acylselenoureas and Se-Acylisoselenoureas. Pharmaceutics 2024, 16, 272. [Google Scholar] [CrossRef]
- Bartolini, D.; Sancineto, L.; Fabro de Bem, A.; Tew, K.D.; Santi, C.; Radi, R.; Toquato, P.; Galli, F. Selenocompounds in Cancer Therapy: An Overview. Adv. Cancer Res. 2017, 136, 259–302. [Google Scholar] [CrossRef]
- Krasowska, D.; Iraci, N.; Santi, C.; Drabowicz, J.; Cieslak, M.; Kaźmierczak-Barańska, J.; Palomba, M.; Królewska-Golińska, K.; Magiera, J.; Sancineto, L. Diselenides and Benzisoselenazolones as Antiproliferative Agents and Glutathione-S-Transferase Inhibitors. Molecules 2019, 24, 2914. [Google Scholar] [CrossRef] [PubMed]
- Astrain-Redin, N.; Paoletti, N.; Plano, D.; Bonardi, A.; Gratteri, P.; Angeli, A.; Sanmartin, C.; Supuran, C.T. Selenium-analogs based on natural sources as cancer-associated carbonic anhydrase isoforms IX and XII inhibitors. J. Enzym. Inhib. Med. Chem. 2023, 38, 2191165. [Google Scholar] [CrossRef]
- Lorenzoni, S.; Cerra, S.; Angulo-Elizari, E.; Salamone, T.A.; Battocchio, C.; Marsotto, M.; Scaramuzzo, F.A.; Sanmartín, C.; Plano, D.; Fratoddi, I. Organoselenium compounds as functionalizing agents for gold nanoparticles in cancer therapy. Colloids Surf. B Biointerfaces 2022, 219, 112828. [Google Scholar] [CrossRef]
- Ferlini, C.; Scambia, G. Assay for apoptosis using the mitochondrial probes, Rhodamine123 and 10-N-nonyl acridine orange. Nat. Protoc. 2007, 2, 3111–3114. [Google Scholar] [CrossRef]
- Foglesong, P.D.; Reckord, C.; Swink, S. Doxorubicin inhibits human DNA topoisomerase I. Cancer Chemother. Pharmacol. 1992, 30, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Improving Drug Candidates by Design: A Focus on Physicochemical Properties As a Means of Improving Compound Disposition and Safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Aricu, A.N.; Kuchkova, K.I.; Secara, E.S.; Barba, A.N.; Dragalin, I.P.; Ungur, N.D.; Mel′nik, E.; Kravtsov, V.K. Synthesis and Structure of Drimane Sesquiterpenoids Containing Pyrimidine, Pyrazine, 1,2,4-triazole, and Carbazole Rings. Chem Nat Compd+ 2018, 54, 455–460. [Google Scholar] [CrossRef]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. In vitro assays and techniques utilized in anticancer drug discovery. J. Appl. Toxicol. 2019, 39, 38–71. [Google Scholar] [CrossRef] [PubMed]
- Rothe, G.; Valet, G. Flow Cytometric Analysis of Respiratory Burst Activity in Phagocytes With Hydroethidine and 2′,7′-Dichlorofluorescin. J. Leukoc. Biol. 1990, 47, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Burden, D.A.; Froelich-Ammon, S.J.; Osheroff, N. Topoisomerase II-mediated cleavage of plasmid DNA. Methods Mol. Biol. 2001, 95, 283–289. [Google Scholar] [CrossRef]
- Anderson, V.E.; Zaniewski, R.P.; Kaczmarek, F.S.; Gootz, T.D.; Osheroff, N. Quinolones inhibit DNA religation mediated by Staphylococcus aureus topoisomerase IV. Changes in drug mechanism across evolutionary boundaries. J. Biol. Chem. 1999, 274, 35927–35932. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) [a] | Selectivity Index (SI) [b] | |
|---|---|---|---|
| MCF-7 | MCF-10 | ||
| 10a | 74.2 ± 7.0 | 53.2 ± 10.0 | 0.7 |
| 10b | 130.7 ± 26.6 | 10.1 ± 3.0 | 0.1 |
| 10c | 25.1 ± 2.2 | 247.9 ± 42.3 | 9.9 |
| 10d | 125.6 ± 11.5 | 12.3 ± 3.1 | 0.1 |
| 11a | 75.2 ± 11.3 | 76.7 ± 3.0 | 1.0 |
| 11b | 132.7 ± 14.9 | 54.4 ± 3.5 | 0.4 |
| 11c | 13.5 ± 1.8 | 87.7 ± 17.8 | 6.5 |
| 11d | 187.2 ± 9.0 | 164.6 ± 12.5 | 0.9 |
| 12a | 56.2 ± 7.3 | 45.4 ± 4.8 | 0.8 |
| 12b | 88.1 ± 19.9 | 72.7 ± 4.3 | 0.8 |
| 12c | 30.1 ± 6.6 | 130.8 ± 29.3 | 4.3 |
| 12d | 111.8 ± 25.4 | 107.4 ± 15.4 | 1.0 |
| 13a | 48.9 ± 8.0 | 70.9 ± 15.1 | 1.4 |
| 13c | 22.5 ± 2.6 | 134.9 ± 26.6 | 6.0 |
| 14a | 8.4 ± 1.8 | 52.4 ± 3.5 | 6.3 |
| 14c | 9.0 ± 2.8 | 393.0 ± 47.1 | 44 |
| 14d | 119.7 ± 29.8 | 31.4 ± 4.6 | 0.3 |
| Daunorubicin | 0.33 ± 0.02 [c] | 9.32 ± 1.01 [c] | 28 |
| Compound | MW (Da) | HBA | HBD | cLogP | TPSA (Å2) | NRB | hERG | CYP Substrate | Pgp Inhibition |
|---|---|---|---|---|---|---|---|---|---|
| Desirable value | ≤500 | ≤10 | ≤5 | ≤5 | ≤140 | ≤10 | M or L | - | Yes |
| 14c | 340.5 | 2 | 0 | 5.49 | 18.46 | 2 | M | 3A4 | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araque, I.; Vergara, R.; Mella, J.; Aránguiz, P.; Espinoza-Catalán, L.; Salas, C.O.; Barrero, A.F.; Quílez del Moral, J.; Villena, J.; Cuellar, M.A. New Dimethoxyaryl-Sesquiterpene Derivatives with Cytotoxic Activity Against MCF-7 Breast Cancer Cells: From Synthesis to Topoisomerase I/II Inhibition and Cell Death Mechanism Studies. Int. J. Mol. Sci. 2025, 26, 4539. https://doi.org/10.3390/ijms26104539
Araque I, Vergara R, Mella J, Aránguiz P, Espinoza-Catalán L, Salas CO, Barrero AF, Quílez del Moral J, Villena J, Cuellar MA. New Dimethoxyaryl-Sesquiterpene Derivatives with Cytotoxic Activity Against MCF-7 Breast Cancer Cells: From Synthesis to Topoisomerase I/II Inhibition and Cell Death Mechanism Studies. International Journal of Molecular Sciences. 2025; 26(10):4539. https://doi.org/10.3390/ijms26104539
Chicago/Turabian StyleAraque, Ileana, Rut Vergara, Jaime Mella, Pablo Aránguiz, Luis Espinoza-Catalán, Cristian O. Salas, Alejandro F. Barrero, José Quílez del Moral, Joan Villena, and Mauricio A. Cuellar. 2025. "New Dimethoxyaryl-Sesquiterpene Derivatives with Cytotoxic Activity Against MCF-7 Breast Cancer Cells: From Synthesis to Topoisomerase I/II Inhibition and Cell Death Mechanism Studies" International Journal of Molecular Sciences 26, no. 10: 4539. https://doi.org/10.3390/ijms26104539
APA StyleAraque, I., Vergara, R., Mella, J., Aránguiz, P., Espinoza-Catalán, L., Salas, C. O., Barrero, A. F., Quílez del Moral, J., Villena, J., & Cuellar, M. A. (2025). New Dimethoxyaryl-Sesquiterpene Derivatives with Cytotoxic Activity Against MCF-7 Breast Cancer Cells: From Synthesis to Topoisomerase I/II Inhibition and Cell Death Mechanism Studies. International Journal of Molecular Sciences, 26(10), 4539. https://doi.org/10.3390/ijms26104539