3.2. Syntheses
3.2.1. Synthesis of Functionalized Amides 2–8
Method A. A solution of the corresponding acid chloride (1 equiv.) in dichloromethane (5–15 mL) was slowly added dropwise to a stirred solution of [amino(phenyl)methyl]diphenylphosphine sulfide 1a (1 equiv.) or (aminomethyl)diphenylphosphine sulfide hydrochloride 1b (1 equiv.) and triethylamine (1–2 (in the case of 1a) or 2–3 (in the case of 1b) equiv.) in CH2Cl2 (15–20 mL) at 5–10 °C. The reaction mixture was stirred at room temperature for 6 h (for ligands 3a and 6b) or 12 h (for the other cases) and was then sequentially washed with water, an aqueous solution of NaHCO3, and again with water. The organic layer was separated, dried over anhydrous Na2SO4, and evaporated to dryness. In the case of ligands 2, 3a, and 8, the residue obtained was purified by column chromatography on silica gel (gradient elution with petroleum ether–CH2Cl2 from 7:1 to 1:3 (2), elution with neat CH2Cl2 (3a), or gradient elution with a petroleum ether–CH2Cl2 mixture from 2:1 to 1:3 and then with neat CH2Cl2 (8)) to yield the target products as light crystalline solids. In the other cases, the resulting residue was recrystallized from EtOAc (3b, 6a) or EtOAc–hexane (1:1 (6b), 1:4 (7a), 1:2 (7b)) to provide the target ligands as light crystals.
Method B. A solution of (aminomethyl)diphenylphosphine sulfide hydrochloride 1b (1 equiv.) and triethylamine (~2 equiv.) in CH2Cl2 (20 mL) was slowly added dropwise to a solution of the corresponding acid chloride (1 equiv.) in CH2Cl2 (20 mL) at 5–10 °C. The reaction mixture was stirred at room temperature for 10 h and then sequentially washed with water, an aqueous solution of NaHCO3, and again with water. The organic layer was separated, dried over anhydrous Na2SO4, and evaporated to dryness. The residue obtained was recrystallized from EtOAc to give the target ligand as a white crystalline solid.
Method C. A solution of [amino(phenyl)methyl]diphenylphosphine sulfide 1a (1 equiv.) in CH2Cl2 (10 mL) was slowly added dropwise to a stirred suspension of benzo[d]thiazole-2-carboxylic acid (1 equiv.) and 4-dimethylaminopyridine (DMAP) (~0.1 equiv.) in CH2Cl2 (20 mL) at –5 to 0 °C under an argon atmosphere. The reaction mixture was stirred upon cooling for 20 min, and then a solution of N,N′-diisopropylcarbodiimide (1.3 equiv.) in CH2Cl2 (5 mL) was slowly added dropwise at −5 to 0 °C. The resulting mixture was stirred at room temperature for 30 min and left overnight. The precipitate obtained was filtered off, and the filtrate was evaporated to dryness. The resulting residue was purified by column chromatography on silica gel (eluent: CH2Cl2) to give the target ligand as a light yellow crystalline solid.
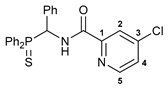
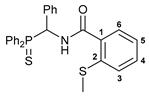
4-Chloro-N-[(diphenylthiophosphoryl)(phenyl)methyl]picolinamide 2
![Ijms 26 04536 i001]()
The compound was obtained by method A from amine 1a (0.31 g, 0.96 mmol), 4-chloropicolinoyl chloride (0.17 g, 0.97 mmol), and Et3N (0.15 g, 1.48 mmol). Yield: 0.24 g (54%). Mp: 120–122 °C. 31P{1H} NMR (161.98 MHz): δ 51.81 ppm. 1H NMR (400.13 MHz, CDCl3): δ 6.45 (dd, 1H, CH, 2JHP = 8.9 Hz, 3JHH = 10.2 Hz), 7.12–7.23 (m, 5H, HAr), 7.27–7.32 (m, 2H, HAr), 7.40–7.44 (m, 2H, HAr), 7.47–7.54 (m, 5H, HAr), 8.05–8.10 (m, 3H, HAr), 8.52 (d, 1H, H(C5), 3JHH = 5.2 Hz), 9.47–9.50 (m, 1H, NH) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 51.72 (d, CH2, 1JCP = 60.0 Hz), 122.93 and 126.64 (both s, C2 and C4), 127.79 (d, m-C in Ph, 4JCP = 2.0 Hz), 128.18 (d, p-C in Ph, 5JCP = 2.7 Hz), 128.19 (d, m-C in P(S)Ph, 3JCP = 12.4 Hz), 128.63 (d, o-C in Ph, 3JCP = 4.6 Hz), 128.89 (d, m-C in P(S)Ph, 3JCP = 12.1 Hz), 129.88 (d, ipso-C in P(S)Ph, 1JCP = 81.2 Hz), 130.60 (d, ipso-C in P(S)Ph, 1JCP = 78.8 Hz), 131.62 (d, o-C in P(S)Ph, 2JCP = 9.7 Hz), 131.87 (d, p-C in P(S)Ph, 4JCP = 2.7 Hz), 132.03 (d, o-C in P(S)Ph, 2JCP = 9.8 Hz), 132.06 (d, p-C in P(S)Ph, 4JCP = 3.2 Hz), 134.08 (s, ipso-C in Ph), 145.68 (s, C3), 149.50 (s, C5), 150.47 (s, C1), 162.42 (d, C=O, 3JCP = 7.2 Hz) ppm. IR (KBr, ν/cm−1): 476(w), 497(w), 534(m), 605(w) and 624(w) (both νP=S), 691(m), 698(m), 722(m), 756(w), 779(w), 801(w), 840(w), 907(vw), 998(vw), 1031(vw), 1103(m), 1179(w), 1234(w), 1292(w), 1308(w), 1350(w), 1395(w), 1437(m), 1462(w), 1495(m), 1510(br, s) (C(O)NH), 1555(w), 1579(w), 1670(s) (νC=O), 2849(w), 2919(w), 3059(w), 3341(br, w) (νNH). Anal. Calcd for C25H20ClN2OPS: C, 64.86; H, 4.35; N, 6.05. Found: C, 64.37; H, 4.70; N, 5.82%.
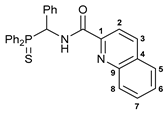
N-[(Diphenylthiophosphoryl)(phenyl)methyl]quinoline-2-carboxamide 3a
![Ijms 26 04536 i002]()
The compound was obtained by method A from amine 1a (0.34 g, 1.05 mmol), quinoline-2-carbonyl chloride derived from quinaldic acid (0.18 g, 1.04 mmol), and an excess of SOCl2, and Et3N (0.21 g, 2.08 mmol). Yield: 0.31 g (62%). Mp: 238–240 °C. 31P{1H} NMR (161.98 MHz): δ 51.55 ppm. 1H NMR (400.13 MHz, CDCl3): δ 6.50–6.55 (m, 1H, CH), 7.14–7.22 (m, 3H, HAr), 7.27–7.34 (m, 4H, HAr), 7.42–7.51 (m, 4H, HAr), 7.55–7.65 (m, 3H, HAr), 7.78–7.82 (m, 1H, HAr), 7.86 (d, 1H, H(C5) or H(C8), 3JHH = 8.2 Hz), 8.07–8.13 (m, 2H, o-H in P(S)Ph), 8.19 (d, 1H, H(C2) or H(C3), 3JHH = 8.5 Hz), 8.26 (d, 1H, HC(8) or H(C5), 3JHH = 7.9 Hz), 8.27 (d, 1H, H(C3) or H(C2), 3JHH = 8.5 Hz), 9.79 (dd, 1H, NH, 3JHP = 5.4 Hz, 3JHH = 10.1 Hz) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 51.98 (d, CH, 1JCP = 59.8 Hz), 118.72 (s, C2), 127.55 (s, C5 or C6), 127.77 (d, m-C in Ph, 4JCP = 2.0 Hz), 128.11 (d, p-C in Ph, 5JCP = 3.0 Hz), 128.17 (s, C6 or C5), 128.21 (d, m-C in P(S)Ph, 3JCP = 12.5 Hz), 128.71 (d, o-C in Ph, 3JCP = 4.6 Hz), 128.85 (d, m-C in P(S)Ph, 3JCP = 11.9 Hz), 129.44 (s, C4), 130.10 (s, C7 or C8), 130.17 (d, ipso-C in P(S)Ph, 1JCP = 81.1 Hz), 130.43 (s, C8 and C7), 131.62 (d, ipso-C in P(S)Ph, 1JCP = 83.7 Hz), 131.71 (d, o-C in P(S)Ph, 2JCP = 9.5 Hz), 131.82 (d, p-C in P(S)Ph, 4JCP = 2.8 Hz), 131.97 (d, p-C in P(S)Ph, 4JCP = 2.6 Hz), 132.09 (d, o-C in P(S)Ph, 2JCP = 9.5 Hz), 134.39 (s, ipso-C in Ph), 137.36 (s, C3), 146.58 and 148.79 (both s, C1 and C9), 163.80 (d, C=O, 3JCP = 7.1 Hz) ppm. IR (KBr, ν/cm−1): 481(w), 492(w), 522(m), 527(m), 562(w), 608(m), and 632(w) (both νP=S), 692(m), 699(m), 714(m), 725(m), 753(m), 773(m), 839(w), 917(vw), 998(vw), 1029(w), 1103(m), 1210(vw), 1343(w), 1380(w), 1428(m), 1438(m), 1495(br, s), 1525(br, m) (C(O)NH), 1566(w), 1673(s) (νC=O), 2853(vw), 2925(vw), 2953(vw), 3057(w). Anal. Calcd for C29H23N2OPS: C, 72.79; H, 4.84; N, 5.85. Found: C, 73.37; H, 5.02; N, 5.83%.
N-[(Diphenylthiophosphoryl)methyl]quinoline-2-carboxamide 3b
![Ijms 26 04536 i003]()
The compound was obtained by method A from amine hydrochloride 1b (0.85 g, 3.00 mmol), quinoline-2-carbonyl chloride derived from quinaldic acid (0.52 g, 3.00 mmol), and an excess of SOCl2, and Et3N (1.3 mL, 9.33 mmol). Yield: 1.00 g (83%). Mp: 154–156 °C. 31P{1H} NMR (161.98 MHz, CDCl3): δ 41.80 ppm. 1H NMR (400.13 MHz, CDCl3): δ 4.62 (t, 2H, CH2, 2JHP = 6.2 Hz), 7.47–7.56 (m, 6H, m-H and p-H in P(S)Ph2), 7.61–7.65 and 7.75–7.80 (both m, 1H + 1H, H(C6) and H(C7)), 7.87 (d, 1H, H(5) or H(C8), 3JHH = 8.1 Hz), 7.94 (ddd, 4H, o-H in P(S)Ph2, 3JHP = 13.0 Hz, 3JHH = 7.5 Hz, 4JHH = 1.6 Hz), 8.14 (d, 1H, H(C8) or H(C5), 3JHH = 8.5 Hz), 8.20 and 8.29 (both d, 1H + 1H, H(C2) and H(C3), 3JHH = 8.4 Hz), 9.02 (br. s, 1H, NH) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 41.84 (d, CH2, 1JCP = 63.2 Hz), 118.74 (s, C2), 127.62 and 128.19 (both s, C5 and C6), 128.89 (d, m-C in P(S)Ph2, 3JCP = 12.5 Hz), 129.41 (s, C4), 130.12 and 130.21 (both s, C7 and C8), 130.64 (d, ipso-C in P(S)Ph2, 1JCP = 80.0 Hz), 131.49 (d, o-C in P(S)Ph2, 2JCP = 10.3 Hz), 132.13 (d, p-C in P(S)Ph2, 2JCP = 2.8 Hz), 137.49 (s, C3), 146.49 and 148.73 (both s, C1 and C9), 164.29 (d, C=O, 3JCP = 4.7 Hz) ppm. IR (KBr, ν/cm−1): 481(w), 504(w), 521(w), 611(m) and 620(w) (both νP=S), 692(m), 709(m), 740(m), 774(m), 795(vw), 845(w), 862(w), 924(w), 999(vw), 1106(m), 1171(w), 1212(w), 1309(vw), 1378(w), 1428(m), 1437(m), 1500(s), 1522(br, s) (C(O)NH), 1566(w), 1592(w), 1618(w), 1678(br, s) (νC=O), 2853(vw), 2923(w), 2962(vw), 3055(w). Anal. Calcd for C23H19N2OPS: C, 68.64; H, 4.76; N, 6.96. Found: C, 68.49; H, 4.74; N, 6.91%.
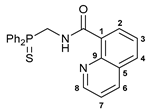
N-[(Diphenylthiophosphoryl)methyl]quinoline-8-carboxamide 4
![Ijms 26 04536 i004]()
The compound was obtained by method B from amine hydrochloride 1b (0.57 g, 2.01 mmol), quinoline-8-carbonyl chloride derived from quinoline-8-carboxylic acid (0.35 g, 2.02 mmol), and an excess of SOCl2, and Et3N (0.6 mL, 4.30 mmol). Yield: 0.60 g (71%). Mp: 199–201 °C. 31P{1H} NMR (161.98 MHz, CDCl3): δ 41.65 ppm. 1H NMR (400.13 MHz, CDCl3): δ 4.80 (dd, 2H, CH2, 2JHP = 3JHH = 5.9 Hz), 7.45–7.54 (m, 7H, HAr), 7.64–7.68 (m, 1H, HAr), 7.94–7.99 (m, 5H, HAr), 8.26 (d, 1H, HAr, 3JHH = 8.2 Hz), 8.79 (d, 1H, HAr, 3JHH = 7.3 Hz), 8.86 (dd, 1H, H(C8), 3JHH = 4.0 Hz, 4JHH = 1.6 Hz), 12.34 (br. s, 1H, NH) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 43.08 (d, CH2, 1JCP = 64.1 Hz), 120.89 (s, C7), 126.22 (s, C3), 127.69 (d, C1, 4JCP = 4.3 Hz), 128.21 (s, C5), 128.59 (d, m-C in P(S)Ph2, 3JCP = 11.7 Hz), 131.03 (d, ipso-C in P(S)Ph2, 1JCP = 88.0 Hz), 131.42 (d, o-C in P(S)Ph2, 2JCP = 10.3 Hz), 131.72 (d, p-C in P(S)Ph2, 4JCP = 2.6 Hz), 132.10 and 133.71 (both s, C2 and C4), 137.45 (s, C6), 145.23 (s, C9), 149.33 (s, C8), 165.73 (d, C=O, 3JCP = 5.9 Hz) ppm. IR (KBr, ν/cm−1): 484(w), 509(w), 519(m), 610(m), 620(w), and 648(m) (three νP=S), 690(m), 709(m), 733(m), 757(m), 794(m), 839(vw), 919(w), 998(vw), 1014(vw), 1028(vw), 1050(vw), 1103(m), 1158(w), 1194(w), 1205(w), 1255(w), 1287(w), 1388(w), 1420(w), 1437(m), 1463(w), 1490(w), 1499(m), 1535(br, m) (C(O)NH), 1575(m), 1592(m), 1609(w), 1652(s) (νC=O), 2889(w), 2965(w), 3058(w), 3164(br, w) (νNH). Anal. Calcd for C23H19N2OPS·H2O: C, 65.70; H, 5.03; N, 6.66. Found: C, 65.63; H, 4.67; N, 6.66%.
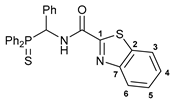
N-[(Diphenylthiophosphoryl)(phenyl)methyl]benzo[d]thiazole-2-carboxamide 5
![Ijms 26 04536 i005]()
The compound was obtained by method C from amine 1a (0.52 g, 1.61 mmol), benzo[d]thiazole-2-carboxylic acid (0.29 g, 1.62 mmol), DMAP (16 mg, 0.13 mmol), and N,N′-diisopropylcarbodiimide 0.27 g (2.14 mmol). Yield: 0.62 g (80%). Mp: 220–225 °C. 31P{1H} NMR (161.98 MHz, CDCl3): δ 51.65 ppm. 1H NMR (400.13 MHz, CDCl3): δ 6.43–6.48 (m, 1H, CH), 7.15–7.23 (m, 3H, HAr), 7.26–7.33 (m, 4H, HAr), 7.42–7.59 (m, 8H, HAr), 7.94 (d, 1H, H(C3) or H(C6), 3JHH = 8.0 Hz), 8.08–8.13 (m, 2H, o-H in P(S)Ph), 8.17 (d, 1H, H(C6) or H(C3), 3JHH = 8.3 Hz), 8.95 (dd, 1H, NH, 2JHP = 5.4 Hz, 3JHH = 9.8 Hz) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 51.84 (d, CH, 1JCP = 59.7 Hz), 122.24 and 124.92 (both s, C3 and C6), 126.91 and 126.96 (both s, C4 and C5), 127.91 (d, m-C in Ph, 4JCP = 1.9 Hz), 128.27 (d, m-C in P(S)Ph, 3JCP = 12.5 Hz), 128.36 (d, p-C in Ph, 5JCP = 3.3 Hz), 128.69 (d, o-C in Ph, 3JCP = 4.4 Hz), 129.03 (d, m-C in P(S)Ph, 3JCP = 11.7 Hz), 129.62 (d, ipso-C in P(S)Ph, 1JCP = 81.7 Hz), 130.35 (d, ipso-C in P(S)Ph, 1JCP = 79.3 Hz), 131.64 (d, o-C in P(S)Ph, 2JCP = 9.5 Hz), 132.01 (d, p-C in P(S)Ph, 4JCP = 3.1 Hz), 132.07 (d, o-C in P(S)Ph, 2JCP = 9.5 Hz), 132.23 (d, p-C in P(S)Ph, 4JCP = 2.8 Hz), 133.79 (d, ipso-C in Ph), 137.11 (s, C2), 152.89 (s, C7), 159.24 (d, C=O, 3JCP = 7.7 Hz), 162.11 (s, C1) ppm. IR (KBr, ν/cm−1): 482(w), 522(m), 538(w), 582(vw), 607(m) and 626(w) (both νP=S), 691(m), 698(m), 717(m), 728(m), 751(m), 767(w), 831(vw), 892(w), 918(vw), 998(vw), 1029(vw), 1103(m), 1142(w), 1249(vw), 1290(w), 1318(w), 1351(w), 1437(m), 1455(w), 1513(br, s) (C(O)NH), 1556(w), 1585(vw), 1600(vw), 1675(m) (νC=O), 2946(vw), 3058(w), 3295(br, w) (νNH). Anal. Calcd for C27H21N2OPS2: C, 66.92; H, 4.37; N, 5.78. Found: C, 67.29; H, 4.61; N, 5.33%.
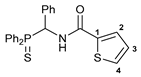
N-[(Diphenylthiophosphoryl)(phenyl)methyl]thiophene-2-carboxamide 6a
![Ijms 26 04536 i006]()
The compound was obtained by method A from amine 1a (0.31 g, 0.96 mmol), thiophene-2-carbonyl chloride (0.14 g, 0.96 mmol), and Et3N (0.11 g, 1.09 mmol). Yield: 0.33 g (80%). Mp: 206–208 °C. 31P{1H} NMR (161.98 MHz, CDCl3): δ 52.64 ppm. 1H NMR (400.13 MHz, CDCl3): δ 6.35–6.40 (m, 1H, CH), 7.08 (dd, 1H, H(C3), 3JHH = 4.8 Hz, 3JHH = 3.8 Hz), 7.11–7.22 (m, 5H, HAr), 7.25–7.30 (m, 2H, HAr), 7.39–7.58 (m, 8H, HAr), 7.69–7.73 (br. m, 1H, NH), 8.03–8.08 (m, 2H, o-H in P(S)Ph) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 51.15 (d, CH2, 1JCP = 58.8 Hz), 127.74 (s, C2 or C3), 127.86 (d, m-C in Ph, 4JCP = 1.9 Hz), 128.18 (d, p-C in Ph, 5JCP = 2.8 Hz), 128.19 (d, m-C in P(S)Ph, 3JCP = 12.5 Hz), 128.46 (d, o-C in Ph, 3JCP = 4.6 Hz), 128.58 (s, C3 or C2), 129.06 (d, m-C in P(S)Ph, 3JCP = 11.7 Hz), 129.38 (d, ipso-C in P(S)Ph, 1JCP = 82.4 Hz), 130.64 (d, ipso-C in P(S)Ph, 1JCP = 79.2 Hz), 130.84 (s, C4), 131.49 (d, o-C in P(S)Ph, 2JCP = 9.5 Hz), 131.97 (d, p-C in P(S)Ph, 4JCP = 3.1 Hz), 132.06 (d, o-C in P(S)Ph, 2JCP = 10.3 Hz), 132.20 (d, p-C in P(S)Ph, 4JCP = 2.9 Hz), 134.27 (s, ipso-C in Ph), 137.97 (s, C1), 160.77 (d, C=O, 3JCP = 8.0 Hz) ppm. IR (KBr, ν/cm−1): 482(w), 525(m), 551(vw), 604(w) and 625(w) (both νP=S), 696(m), 722(s), 739(w), 744(w), 752(w), 856(w), 1099(m), 1261(vw), 1291(w), 1307(w), 1355(w), 1418(w), 1437(m), 1500(s), 1504(s), 1527(br, s) (C(O)NH), 1654(s) (νC=O), 2951(vw), 3056(w), 3112(vw), 3334(br, w) (νNH). Anal. Calcd for C24H20NOPS2: C, 66.49; H, 4.65; N, 3.23. Found: C, 66.41; H, 4.57; N, 3.31%.
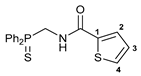
N-[(Diphenylthiophosphoryl)methyl]thiophene-2-carboxamide 6b
![Ijms 26 04536 i007]()
The compound was obtained by method A from amine hydrochloride 1b (0.29 g, 1.02 mmol), thiophene-2-carbonyl chloride (0.15 g, 1.02 mmol), and Et3N (0.22 g, 2.17 mmol). Yield: 0.31 g (85%). Mp: 147–149 °C. 31P{1H} NMR (161.98 MHz, CDCl3): δ 42.41 ppm. 1H NMR (400.13 MHz, CDCl3): δ 4.47 (dd, 2H, CH2, 2JHP = 3JHH = 5.8 Hz), 6.93 (br. s, 1H, NH), 7.07–7.09 (m, 1H, H(C3)), 7.48–7.58 (m, 8H, HAr), 7.86–7.91 (m, 4H, o-H in P(S)Ph2) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 40.87 (d, CH2, 1JCP = 62.8 Hz), 127.76 and 128.53 (both s, C2 and C3), 128.98 (d, m-C in P(S)Ph2, 3JCP = 11.7 Hz), 130.33 (d, ipso-C in P(S)Ph2, 1JCP = 80.9 Hz), 130.73 (s, C4), 131.30 (d, o-C in P(S)Ph2, 2JCP = 10.3 Hz), 132.29 (d, p-C in P(S)Ph2, 4JCP = 3.0 Hz), 137.88 (s, C1), 161.49 (d, C=O, 3JCP = 5.7 Hz) ppm. IR (KBr, ν/cm−1): 494(w), 519(w), 574(w), 606(m) and 618(vw) (both νP=S), 692(m), 704(m), 712(m), 725(m), 738(m), 750(w), 767(w), 858(w), 901(m), 998(vw), 1107(m), 1237(w), 1267(m), 1300(m), 1352(w), 1395(w), 1416(w), 1435(m), 1505(s), 1528(br, s) (C(O)NH), 1650 (νC=O), 2921(vw), 2976(w), 3059(vw), 3104(vw), 3351(br, w) (νNH). Anal. Calcd for C18H16NOPS2: C, 60.49; H, 4.51; N, 3.92. Found: C, 61.04; H, 4.41; N, 3.99%.
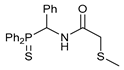
N-[(Diphenylthiophosphoryl)(phenyl)methyl]-2-(methylthio)acetamide 7a
The compound was obtained by method A from amine 1a (0.34 g, 1.05 mmol), 2-(methylthio)acetyl chloride (0.13 g, 1.04 mmol), and Et3N (0.11 g, 1.09 mmol). Yield: 0.37 g (86%). Mp: 195–197 °C. 31P{1H} NMR (161.98 MHz, CDCl3): δ 52.30 ppm. 1H NMR (400.13 MHz, CDCl3): δ 1.79 (s, 3H, Me), 3.09 and 3.17 (ABq, 2H, CH2S, JAB = 16.5 Hz), 6.28–6.33 (m, 1H, CH), 7.15–7.29 (m, 7H, HAr), 7.38–7.49 (m, 3H, HAr), 7.54–7.62 (m, 3H, HAr), 8.09–8.14 (m, 2H, o-H in P(S)Ph), 8.41–8.44 (m, 1H, NH) ppm. IR (KBr, ν/cm−1): 481(w), 518(m), 534(m), 548(w), 592(w), 625(w) (νP=S), 693(m), 719(m), 738(w), 804(w), 1030(vw), 1106(br, m), 1133(w), 1179(vw), 1291(vw), 1313(w), 1437(m), 1455(w), 1495(m), 1503(br, s) (C(O)NH), 1669(s) (νC=O), 2914(vw), 2946(vw), 3058(w), 3331(w) (νNH). Anal. Calcd for C22H22NOPS2: C, 64.21; H, 5.39; N, 3.40. Found: C, 64.18; H, 5.36; N, 3.41%.
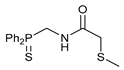
N-[(Diphenylthiophosphoryl)methyl]-2-(methylthio)acetamide 7b
The compound was obtained by method A from amine hydrochloride 1b (0.34 g, 1.20 mmol), 2-(methylthio)acetyl chloride (0.15 g, 1.20 mmol), and Et3N (0.25 g, 2.47 mmol). Yield: 0.30 g (75%). Mp: 82–84 °C. 31P{1H} NMR (161.98 MHz, CDCl3): δ 42.08 ppm. 1H NMR (400.13 MHz, CDCl3): δ 1.89 (s, 3H, Me), 3.14 (s, 2H, CH2S), 4.38 (dd, 2H, CH2P, 2JHP = 3JHH = 5.8 Hz), 7.48–7.58 (m, 6H, m-H and p-H in P(S)Ph2), 7.65 (br. s, 1H, NH), 7.89 (ddd, o-H in P(S)Ph2, 3JHP = 13.0 Hz, 3JHH = 7.6 Hz, 4JHH = 1.2 Hz) ppm. IR (KBr, ν/cm−1): 487(w), 509(w), 566(w), 597(m), 642(w) (νP=S), 691(m), 704(m), 713(m), 744(m), 776(w), 824(m), 962(w), 1000(w), 1107(m), 1170(vw), 1237(w), 1258(w), 1386(m), 1437(m), 1479(w), 1516(br, s) (C(O)NH), 1660(s) (νC=O), 2912(w), 2960(vw), 3051(vw), 3329(m) (νNH). Anal. Calcd for C16H18NOPS2: C, 57.29; H, 5.41; N, 4.18. Found: C, 57.41; H, 5.37; N, 4.23%.
N-[(Diphenylthiophosphoryl)(phenyl)methyl]-2-(methylthio)benzamide 8
![Ijms 26 04536 i010]()
The compound was obtained by method A from a hydrochloride salt of amine 1a (0.25 g, 0.69 mmol), 2-(methylthio)benzoyl chloride (0.13 g, 0.70 mmol), and Et3N (0.18 g, 1.78 mmol). Yield: 0.15 g (44%). Mp: 140–145 °C. 31P{1H} NMR (202.45 MHz, CDCl3): δ 52.32 ppm. 1H NMR (500.13 MHz, CDCl3): δ 2.33 (s, 3H, Me), 6.55 (dd, 1H, CH, 2JHP = 3JHH = 9.3 Hz), 7.14–7.17 (m, 3H, H(C5) + m-H in Ph), 7.20–7.23 (m, 3H, o-H and p-H in Ph), 7.26–7.31 (m, 3H, m-H in P(S)Ph + H(C4)), 7.36–7.43 (m, 3H, H(C3) + H(C6) + p-H in P(S)Ph), 7.46–7.50 (m, 2H, o-H in P(S)Ph), 7.54–7.60 (m, 3H, m-H and p-H in P(S)Ph), 7.95–7.98 (m, 1H, NH), 8.13–8.18 (m, 2H, o-H in P(S)Ph) ppm. 13C{1H} NMR (125.76 MHz, CDCl3): δ 16.97 (s, Me), 51.90 (d, CH, 1JCP= 59.2 Hz), 125.27 (s, C5), 127.69 (s, C4), 127.73 (d, m-C in Ph, 4JCP = 2.2 Hz), 128.11 (d, p-C in Ph, 5JCP = 3.0 Hz), 128.18 (d, m-C in P(S)Ph, 3JCP = 12.7 Hz), 128.48 (s, C6), 128.75 (d, o-C in Ph, 3JCP = 4.7 Hz), 128.98 (d, m-C in P(S)Ph, 3JCP = 12.0 Hz), 129.82 (d, ipso-C in P(S)Ph, 1JCP = 80.9 Hz), 130.51 (d, ipso-C in P(S)Ph, 1JCP = 79.3 Hz), 131.09 (s, C3), 131.82 (d, o-C in P(S)Ph, 2JCP = 9.8 Hz), 131.88 (d, p-C in P(S)Ph, 4JCP = 3.1 Hz), 131.97 (d, o-C in P(S)Ph, 2JCP = 10.2 Hz), 132.11 (d, p-C in P(S)Ph, 4JCP = 2.9 Hz), 133.96 (s, C1), 133.98 (d, ipso-C in Ph, 2JCP = 1.2 Hz), 137.83 (s, C6), 166.96 (d, C=O, 3JCP = 7.2 Hz) ppm. IR (KBr, ν/cm−1): 479(w), 527(s), 605(w) and 628(m) (both νP=S), 652(w), 695(s), 714(m), 747(m), 787(w), 894(vw), 999(vw), 1028(vw), 1102(m), 1149(vw), 1185(vw), 1247(w), 1286(w), 1309(m), 1345(w), 1386(w), 1436(s), 1461(m), 1493(br, s) (C(O)NH), 1586(m), 1656(br, s) (νC=O), 2852(vw), 2922(w), 3056(w), 3319(br, w) (νNH). Anal. Calc. for C27H24NOPS2·0.15CH2Cl2: C, 67.05; H, 5.04; N, 2.88. Found: C, 67.09; H, 5.17; N, 2.94%.
3.2.2. Synthesis of Pd(II) Pincer Complexes 9–14
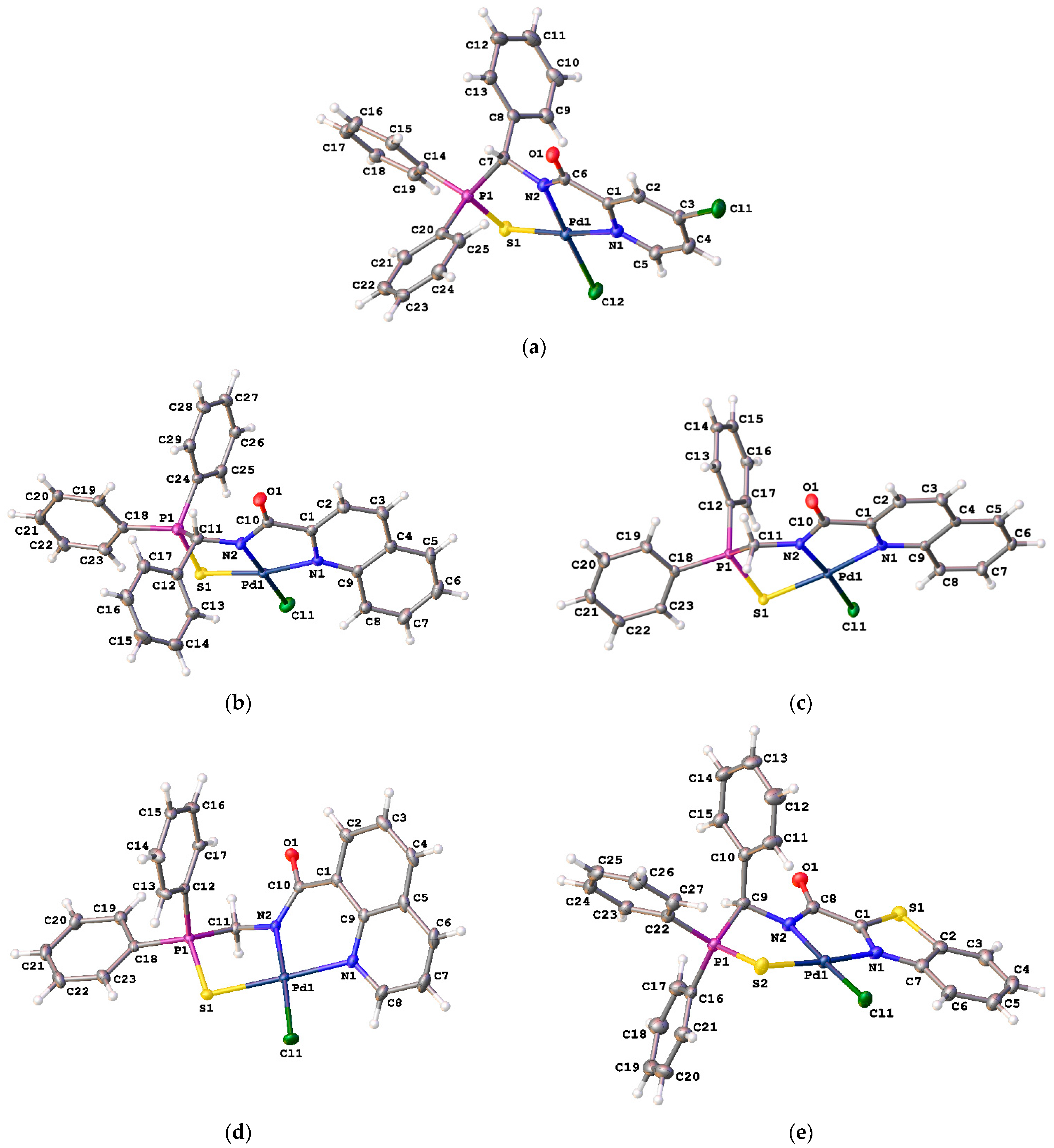
General procedure. A solution of PdCl2(NCPh)2 (61 mg, 0.159 mmol) in CH2Cl2 (4 mL) was slowly added dropwise to a solution of the corresponding ligand (0.159 mmol) and Et3N (23 μL, 0.165 mmol) in CH2Cl2 (6 mL). The reaction mixture was left under ambient conditions for 2 h (in the case of ligand 7a) or 1 day (in the other cases). After the mentioned period of time, the resulting mixture was purified by column chromatography on silica gel (eluent: CH2Cl2–MeOH (50:1 (9), 100:1 (14), 200:1 (12)), CH2Cl2–EtOH (50:1 (10a), 20:1 (10b)), CHCl3–EtOH (20:1 (11), 50:1 (13a,b)) to provide the target complexes as beige (9), yellow (11, 12, 13a, 14), or orange (10a,b, 13b) crystalline solids.
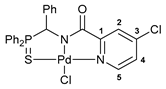
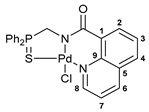
[κ3-S,N,N-(L)Pd(II)Cl] complex 9
![Ijms 26 04536 i011]()
Yield: 70 mg (73%). 31P{1H} NMR (161.98 MHz, CDCl3): δ 67.75 ppm. 1H NMR (500.13 MHz, CDCl3): δ 6.48 (d, 1H, CH, 2JHP = 2.7 Hz), 7.13 (dd, 2H, o-H in P(S)Ph, 3JHP = 12.8 Hz, 3JHH = 7.8 Hz), 7.17–7.20 (m, 2H, m-H in Ph), 7.23–7.28 (m, 3H, m-H in P(S)Ph + p-H in Ph), 7.48–7.51 (m, 2H, H(C4) + p-H in P(S)Ph), 7.58–7.60 (m, 2H, o-H in Ph), 7.66–7.70 (m, 2H, m-H in P(S)Ph), 7.72–7.76 (m, 1H, p-H in P(S)Ph), 7.81 (d, 1H, H(C2), 4JHH = 2.4 Hz), 8.14 (dd, 2H, o-H in P(S)Ph, 3JHP = 12.8 Hz, 3JHH = 7.5 Hz), 8.97 (d, 1H, H(C5), 3JHH = 5.9 Hz) ppm. 13C{1H} NMR (125.76 MHz, CDCl3–(CD3)2SO): δ 65.06 (d, CH, 1JCP = 71.6 Hz), 124.81 (d, ipso-C in P(S)Ph, 1JCP = 75.1 Hz), 126.00 (s, C2), 126.20 (d, ipso-C in P(S)Ph, 1JCP = 78.1 Hz), 127.33 (s, C4), 128.12 (d, o-C in Ph, 3JCP = 5.3 Hz), 128.15 (d, m-C in Ph, 4JCP = 3.1 Hz), 128.65 (d, m-C in P(S)Ph, 3JCP = 12.5 Hz), 128.84 (d, p-C in Ph, 5JCP = 3.6 Hz), 129.85 (d, m-C in P(S)Ph, 3JCP = 12.3 Hz), 132.50 (d, o-C in P(S)Ph, 2JCP = 9.4 Hz), 132.75 (d, o-C in P(S)Ph, 2JCP = 9.9 Hz), 133.58 (d, ipso-C in Ph, 2JCP = 1.2 Hz), 133.62 (d, p-C in P(S)Ph, 4JCP = 2.6 Hz), 133.64 (d, p-C in P(S)Ph, 4JCP = 2.2 Hz), 148.15 (s, C5), 148.28 (s, C3), 154.78 (s, C1), 169.76 (d, C=O, 3JCP = 14.6 Hz) ppm. IR (KBr, ν/cm−1): 488(w), 500(w), 525(w), 536(m), 575(m) and 583(w) (both νP=S), 618(vw), 692(m), 701(m), 710(m), 719(m), 732(w), 753(w), 778(w), 806(w), 841(w), 886(vw), 998(w), 1029(w), 1059(br, w), 1104(m), 1112(m), 1192(w), 1251(vw), 1340(m), 1355(m), 1421(w), 1437(m), 1454(w), 1492(w), 1507(w), 1553(w), 1594(s), 1618(s) (νC=O), 2892(vw), 3023(vw), 3058(vw). Anal. Calcd for C25H19Cl2N2OPPdS: C, 49.73; H, 3.17; N, 4.64. Found: C, 49.54; H, 3.21; N, 4.61%.
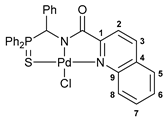
[κ3-S,N,N-(L)Pd(II)Cl] complex 10a
![Ijms 26 04536 i012]()
Yield: 81 mg (82%). 31P{1H} NMR (161.98 MHz, CDCl3): δ 66.26 ppm. 1H NMR (400.13 MHz, CDCl3): δ 6.73 (d, 1H, CH, 2JHP = 3.2 Hz), 7.13–7.31 (m, 7H, HAr), 7.49–7.53 (m, 1H, HAr), 7.65–7.75 (m, 6H, HAr), 7.84–7.90 (m, 2H, HAr), 8.02 (d, 1H, HAr, 3JHH = 8.2 Hz), 8.19–8.24 (m, 2H, o-H in P(S)Ph), 8.41 (d, 1H, HAr, 3JHH = 8.2 Hz), 9.86 (d, 1H, HAr, 3JHH = 8.9 Hz) ppm. 13C{1H} NMR (100.61 MHz, CDCl3–(CD3)2SO): δ 64.41 (d, CH, 1JCP = 68.5 Hz), 121.74 (s, C2), 123.97 (d, ipso-C in P(S)Ph, 1JCP = 73.9 Hz), 126.09 (d, ipso-C in P(S)Ph, 1JCP = 78.3 Hz), 128.01 (d, m-C in Ph, 4JCP = 2.7 Hz), 128.26, 128.31, and 128.32 (three s, C5–C8), 128.70 (d, p-C in Ph, 5JCP = 3.1 Hz), 128.85 (d, m-C in P(S)Ph, 3JCP = 11.7 Hz), 128.89 (d, o-C in Ph, 3JCP = 3.9 Hz), 129.85 (d, m-C in P(S)Ph, 3JCP = 12.4 Hz), 131.22 (s, C4), 131.40 (s, C5–C8), 132.67 (d, o-C in P(S)Ph, 2JCP = 9.8 Hz), 132.87 (d, o-C in P(S)Ph, 2JCP = 10.0 Hz), 133.84 (d, p-C in P(S)Ph, 4JCP = 2.6 Hz), 133.95 (d, p-C in P(S)Ph, 4JCP = 2.8 Hz), 134.23 (s, ipso-C in Ph), 141.30 (s, C3), 146.74 and 155.46 (both s, C1 and C9), 171.30 (d, C=O, 3JCP = 13.1 Hz) ppm. IR (KBr, ν/cm−1): 495(w), 526(m), 579(m) (νP=S), 606(w), 689(m), 696(m), 708(m), 750(w), 764(m), 848(w), 929(vw), 998(w), 1028(w), 1104(m), 1155(w), 1187(w), 1214(vw), 1342(w), 1375(m), 1385(m), 1437(m), 1461(w), 1491(w), 1514(w), 1560(w), 1618(vs) (νC=O), 2916(w), 3057(w). Anal. Calcd for C29H22ClN2OPPdS: C, 56.23; H, 3.58; N, 4.52. Found: C, 56.31; H, 3.65; N, 4.56%.
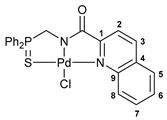
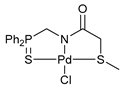
[κ3-S,N,N-(L)Pd(II)Cl] complex 10b
![Ijms 26 04536 i013]()
Yield: 56 mg (65%). 31P{1H} NMR (161.98 MHz, CDCl3): δ 59.24 ppm. 1H NMR (400.13 MHz, CDCl3): δ 4.90 (d, 2H, CH2P, 2JHP = 4.7 Hz), 7.56–7.69 (m, 7H, HAr), 7.80–7.92 (m, 6H, HAr), 8.01 (d, 1H, HAr, 3JHH = 8.3 Hz), 8.40 (d, 1H, HAr, 3JHH = 8.3 Hz), 9.72 (d, 1H, HAr, 3JHH = 9.1 Hz) ppm. 13C{1H} NMR (100.61 MHz, CDCl3–(CD3)2SO): δ 54.04 (d, CH2P, 1JCP = 74.2 Hz), 121.35 (s, C2), 124.34 (d, ipso-C in P(S)Ph2, 1JCP = 77.8 Hz), 127.76, 128.57, and 128.86 (three s, C5–C8), 129.56 (d, m-C in P(S)Ph2, 3JCP = 12.7 Hz), 130.82 (s, C4), 131.23 (s, C5–C8), 131.99 (d, o-C in P(S)Ph2, 2JCP = 10.6 Hz), 133.86 (d, p-C in P(S)Ph2, 4JCP = 2.9 Hz), 140.67 (s, C3), 146.68 and 155.31 (both s, C1 and 9) ppm (the signal of C=O carbon nucleus was not observed). IR (KBr, ν/cm−1): 478(w), 506(m), 528(w), 557(w), 584(m) (νP=S), 688(m), 702(m), 717(m), 747(w), 764(m), 848(w), 929(w), 998(w), 1028(vw), 1107(m), 1150(m), 1188(vw), 1209(vw), 1393(br, m), 1437(m), 1463(m), 1485(w), 1514(m), 1560(w), 1597(m), 1628(vs) (νC=O), 2874(vw), 2924(vw), 3053(w). Anal. Calcd for C23H18ClN2OPPdS: C, 50.85; H, 3.34; N, 5.16. Found: C, 50.81; H, 3.51; N, 5.27%.
[κ3-S,N,N-(L)Pd(II)Cl] complex 11
![Ijms 26 04536 i014]()
Yield: 74 mg (86%). 31P{1H} NMR (161.98 MHz, CDCl3): δ 53.01 ppm. 1H NMR (400.13 MHz, CDCl3): δ 5.44 (d, 2H, CH2, 2JHP = 4.4 Hz), 7.47 (dd, 1H, H(C7), 3JHH = 8.0 Hz, 3JHH = 5.4 Hz), 7.57–7.67 (m, 7H, HAr), 7.91 (d, 1H, HAr, 3JHH = 8.0 Hz), 7.97 (dd, 4H, o-H in P(S)Ph2, 3JHP = 13.3 Hz, 3JHH = 7.3 Hz), 8.37 (d, 1H, HAr, 3JHH = 8.1 Hz), 8.84 (d, 1H, HAr, 3JHH = 7.4 Hz), 9.67 (d, 1H, H(C8), 3JHH = 5.4 Hz) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 61.54 (d, CH2, 1JCP = 69.0 Hz), 121.19 (s, C7), 124.24 (d, ipso-C in P(S)Ph2, 1JCP = 77.7 Hz), 127.36 (s, C3), 128.89 (s, C5), 129.39 (d, m-C in P(S)Ph2, 3JCP = 12.5 Hz), 131.29 (s, C2, C4, or C6), 131.50 (s, C1), 132.63 (d, o-C in P(S)Ph2, 2JCP = 10.9 Hz), 133.54 (d, p-C in P(S)Ph2, 4JCP = 2.7 Hz), 136.73 (s, C2, C4, or C6), 140.41 (s, C2, C4, or C6), 143.40 (s, C9), 155.69 (s, C8), 163.72 (d, C=O, 3JCP = 6.6 Hz) ppm. IR (KBr, ν/cm−1): 492(w), 520(m), 583(m) (νP=S), 694(m), 718(m), 746(w), 756(w), 784(m), 838(w), 933(w), 998(vw), 1111(m), 1179(m), 1308(w), 1374(m), 1395(w), 1438(m), 1506(w), 1560(s), 1581(m), 1614(m) (νC=O), 2953(w), 3052(vw). Anal. Calcd for C23H18ClN2OPPdS: C, 50.85; H, 3.34; N, 5.16. Found: C, 50.97; H, 3.34; N, 5.14%.
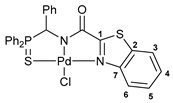
[κ3-S,N,N-(L)Pd(II)Cl] complex 12
![Ijms 26 04536 i015]()
Yield: 75 mg (75%). 31P{1H} NMR (161.98 MHz, CDCl3): δ 67.28 ppm. 1H NMR (400.13 MHz, CDCl3): δ 6.48 (d, 1H, CH, 2JHP = 2.4 Hz), 7.13 (dd, 2H, o-H in P(S)Ph, 3JHP = 12.8 Hz, 3JHH = 7.4 Hz), 7.19–7.23 (m, 2H, HAr), 7.25–7.30 (m, 3H, HAr), 7.49–7.56 (m, 2H, HAr), 7.60–7.78 (m, 6H, HAr), 7.91 (d, 1H, H(C3) or H(C6), 3JHH = 8.0 Hz), 8.16–8.21 (m, 2H, o-H in P(S)Ph), 9.36 (d, 1H, H(C6) or H(C3), 3JHH = 8.3 Hz) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 65.32 (d, CH, 1JCP = 70.5 Hz), 122.19 (s, C3 or C6), 124.17 (d, ipso-C in P(S)Ph, 1JCP = 74.8 Hz), 124.64 (s, C6 or C3), 125.70 (d, ipso-C in P(S)Ph, 1JCP = 78.3 Hz), 127.46 (s, C4 or C5), 128.17–128.22 (m, overlapping signals of o-C and m-C in Ph and C5 or C4), 128.72 (d, m-C in P(S)Ph, 3JCP = 12.4 Hz), 128.97 (d, p-C in Ph, 5JCP = 3.5 Hz), 129.97 (d, m-C in P(S)Ph, 3JCP = 12.4 Hz), 132.46 (s, ipso-C in Ph or C2), 132.58 (d, o-C in P(S)Ph, 2JCP = 9.5 Hz), 132.85 (d, o-C in P(S)Ph, 2JCP = 10.0 Hz), 133.42 (s, C2 or ipso-C in Ph), 133.79 (d, p-C in P(S)Ph2, 4JCP = 2.8 Hz), 150.26 (s, C7), 166.52 (d, C=O, 3JCP = 15.1 Hz), 170.83 (s, C1) ppm. IR (KBr, ν/cm−1): 486(w), 523(m), 581(w) (νP=S), 687(m), 708(m), 747(m), 757(m), 913(vw), 923(vw), 998(vw), 1106(m), 1157(w), 1187(w), 1251(vw), 1318(w), 1373(w), 1437(m), 1457(m), 1480(m), 1506(w), 1559(w), 1630(vs) (νC=O), 2906(vw), 3060(w). Anal. Calcd for C27H20ClN2OPPdS2: C, 51.85; H, 3.22; N, 4.48. Found: C, 51.55; H, 3.29; N, 4.42%.
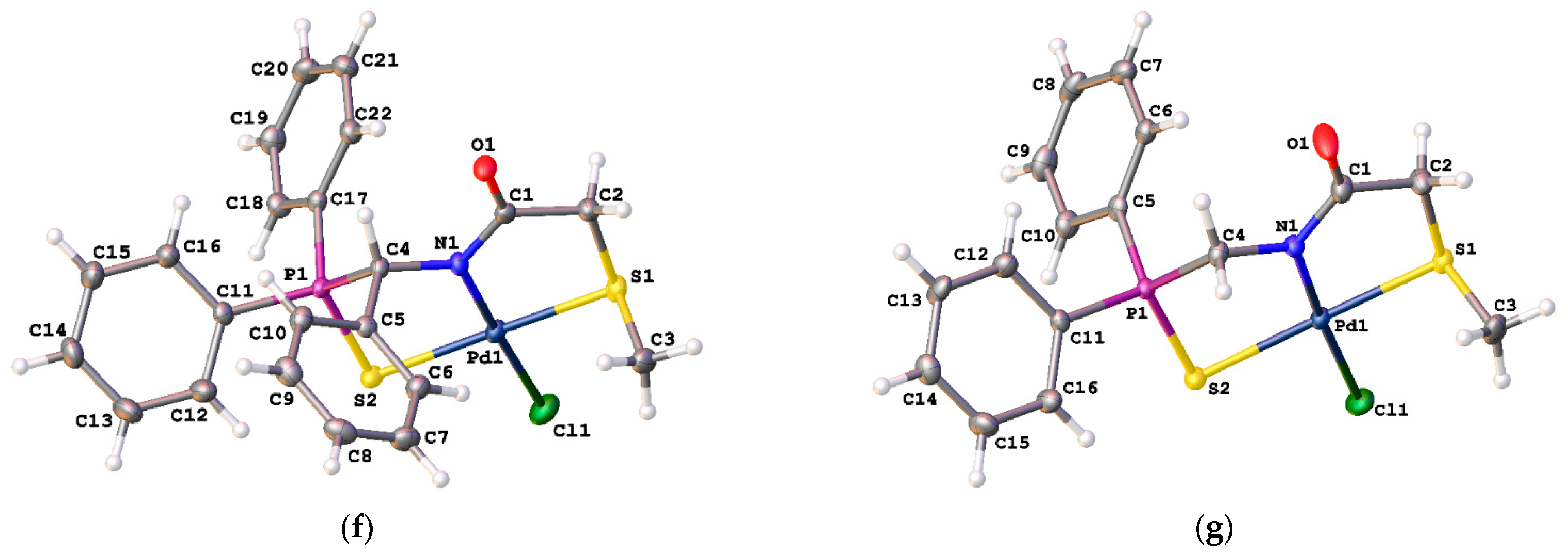
[κ3-S,N,S-(L)Pd(II)Cl] complex 13a
![Ijms 26 04536 i016]()
The compound was obtained as a mixture of diastereomers arising due to the configuration stabilization of the thioether sulfur atom as a result of coordination in addition to the existing chiral carbon center. Yield: 85 mg (97%). 31P{1H} NMR (161.98 MHz, CDCl3): δ 64.82 (minor isomers (m) 39%), 67.28 (major isomers (M) 61%) ppm. 1H NMR (400.13 MHz, CDCl3): δ 2.45 (s, 3H, Me (m)), 2.64 (s, 3H, Me (M)), 3.18 (d, 1H, CH2S (M), 2JHH = 16.8 Hz), 3.32 (d, 1H, CH2S (m), 2JHH = 16.2 Hz), 3.68 (d, 1H, CH2S (M), 2JHH = 16.8 Hz), 3.70 (d, 1H, CH2S (m), 2JHH = 16.2 Hz), 6.52 (d, 1H, CH (m), 2JHP = 3.2 Hz), 6.56 (d, 1H, CH (M), 2JHP = 3.1 Hz), 7.08–7.29 (m, 7H, HAr (M) + 7H, HAr (m)), 7.45–7.50 (m, 1H, HAr (M) + 1H, HAr (m)), 7.55–7.57 (m, 2H, HAr (m)), 7.66–7.80 (m, 5H, HAr (M) + 3H, HAr (m)), 8.07 (dd, 2H, o-H in P(S)Ph (M), 3JHP = 12.6 Hz, 3JHH = 7.7 Hz), 8.15 (dd, 2H, o-H in P(S)Ph (m), 3JHP = 12.4 Hz, 3JHH = 7.5 Hz) ppm. IR (KBr, ν/cm−1): 474(w), 520(m), 584(m) (νP=S), 687(m), 708(m), 749(m), 804(w), 851(vw), 999(w), 1027(vw), 1108(m), 1182(w), 1315(w), 1340(w), 1360(m), 1420(w), 1437(m), 1452(w), 1490(w), 1594(vs), 1602(vs) (νC=O), 2906(w), 3056(w). Anal. Calcd for C22H21ClNOPPdS2: C, 47.84; H, 3.83; N, 2.54. Found: C, 47.71; H, 3.92; N, 2.64%.
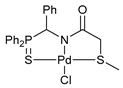
[κ3-S,N,S-(L)Pd(II)Cl] complex 13b
![Ijms 26 04536 i017]()
Yield: 61 mg (81%). 31P{1H} NMR (161.98 MHz, CDCl3): δ 59.26 ppm. 1H NMR (400.13 MHz, CDCl3): δ 2.56 (s, 3H, Me), 3.27 (d, 1H, CH2S, 2JHH = 16.2 Hz), 3.70 (d, 1H, CH2S, 2JHH = 16.2 Hz), 4.56 (dd, 1H, CH2P, 2JHP = 4.2 Hz, 2JHH = 14.8 Hz), 4.86 (dd, 1H, CH2P, 2JHP = 3.5 Hz, 2JHH = 14.8 Hz), 7.55–7.61 (m, 4H, m-H in P(S)Ph2), 7.66–7.70 (m, 2H, p-H in P(S)Ph2), 7.77–7.86 (m, 4H, o-H in P(S)Ph2) ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 23.66 (s, Me), 41.66 (s, CH2S), 54.94 (d, CH2P, 1JCP = 76.5 Hz), 125.11 (d, ipso-C in P(S)Ph, 1JCP = 79.2 Hz), 125.40 (d, ipso-C in P(S)Ph, 1JCP = 78.6 Hz), 129.58 (d, m-C in P(S)Ph, 3JCP = 12.5 Hz), 129.61 (d, m-C in P(S)Ph, 3JCP = 12.5 Hz), 132.03 (d, o-C in P(S)Ph, 2JCP = 10.6 Hz), 132.05 (d, o-C in P(S)Ph, 2JCP = 10.3 Hz), 133.72 (d, p-C in P(S)Ph, 4JCP = 2.8 Hz), 133.75 (d, p-C in P(S)Ph, 4JCP = 2.8 Hz) ppm (the signal of C=O carbon nucleus was not observed). IR (KBr, ν/cm−1): 448(w), 513(m), 584(m) (νP=S), 687(m), 703(m), 718(m), 751(m), 799(w), 845(w), 998(w), 1110(m), 1176(w), 1312(w), 1364(m), 1437(m), 1484(w), 1601(s) (νC=O), 2884(vw), 2921(w), 3055(vw). Anal. Calcd for C16H17ClNOPPdS2: C, 40.35; H, 3.60; N, 2.94. Found: C, 40.41; H, 3.65; N, 3.02%.
[κ3-S,N,S-(L)Pd(II)Cl] complex 14
The compound was obtained as a mixture of diastereomers arising due to the configuration stabilization of the thioether sulfur atom as a result of coordination in addition to the existing chiral carbon center. Yield: 75 mg (77%). 31P{1H} NMR (202.45 MHz, CDCl3, 258 K): δ 59.28 (major isomers (M) 68%), 60.46 (minor isomers (m) 32%) ppm. For the 1H and 13C{1H} NMR spectra, see the SI. IR (KBr, ν/cm−1): 485(w), 514(w), 530(m), 579(w) and 589(w) (both νP=S), 689(m), 708(m), 746(m), 786(w), 968(w), 998(w), 1027(w), 1105(m), 1145(w), 1187(w), 1338(br, m), 1414(w), 1437(m), 1492(w), 1547(s), 1580(s) (νC=O), 2908(vw), 3025(w), 3056(w). Anal. Calcd for C27H23ClNOPPdS2: C, 52.78; H, 3.77; N, 2.28. Found: C, 52.77; H, 3.91; N, 2.41%.
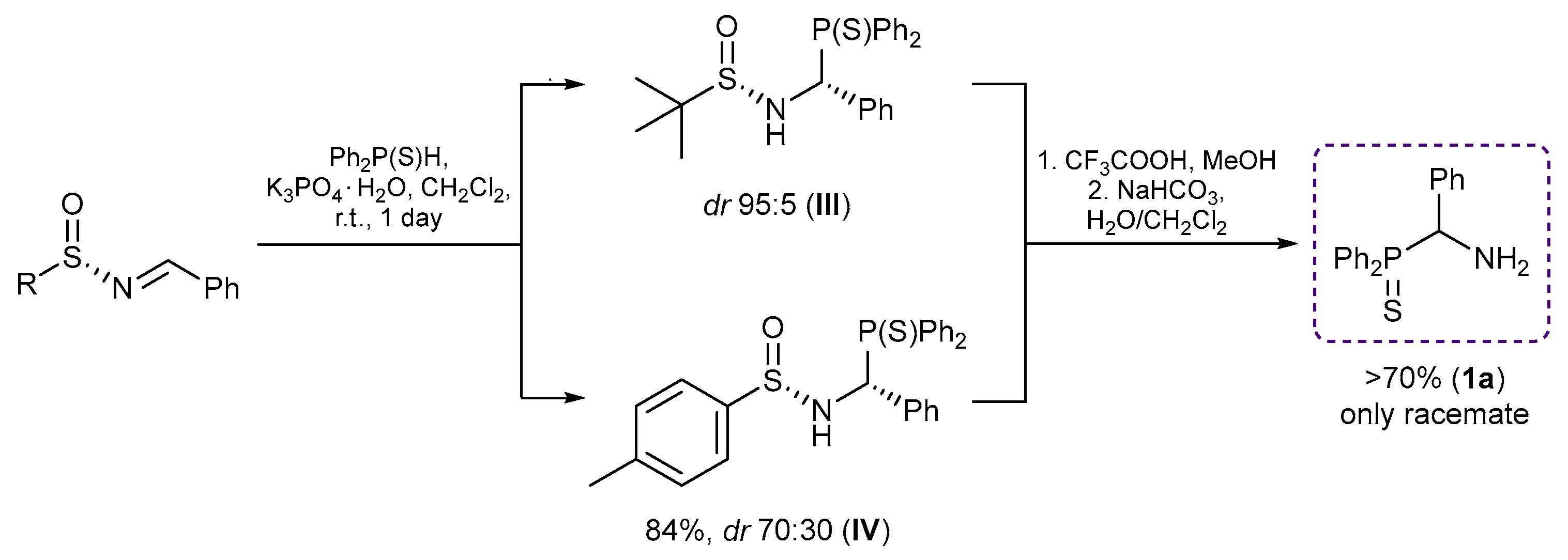
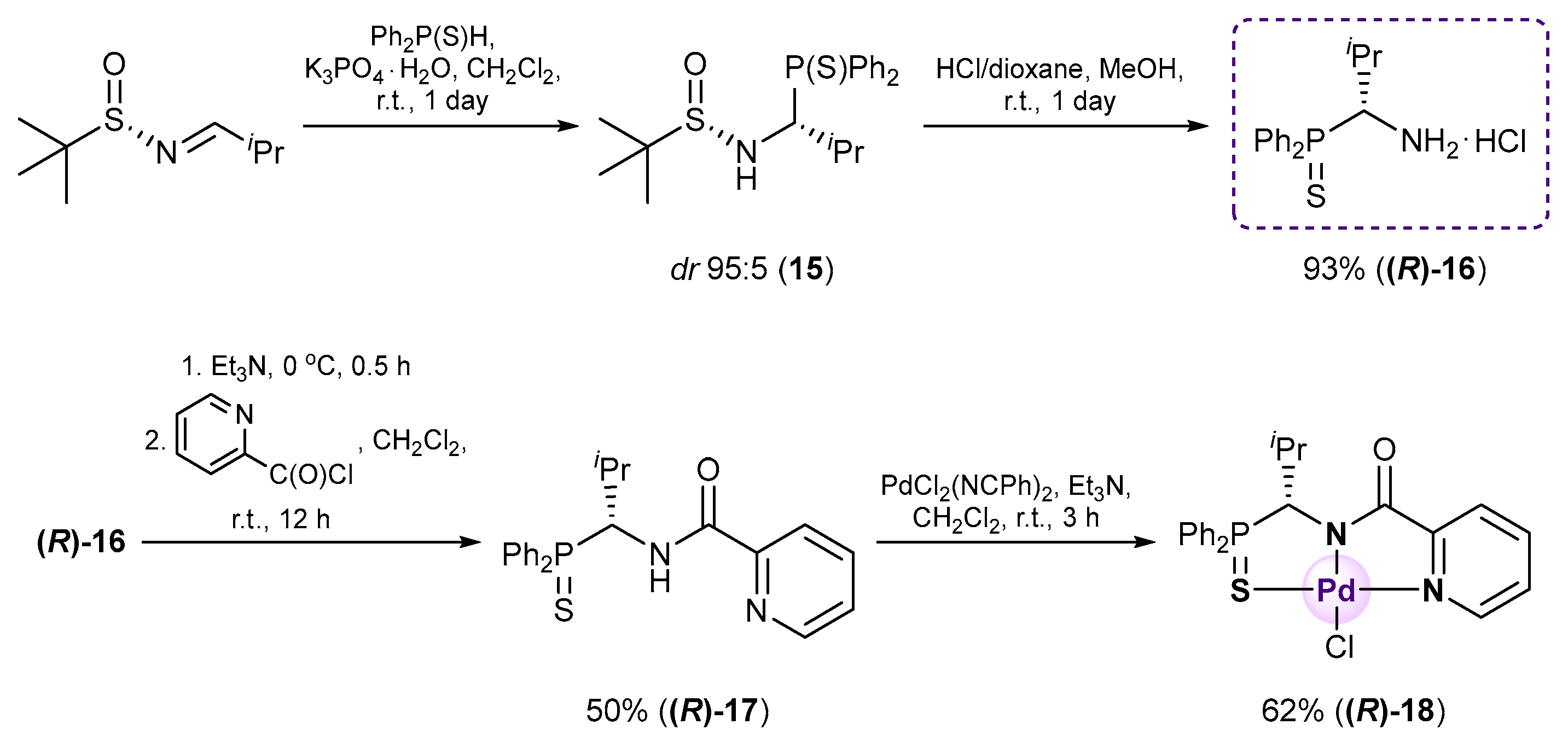
3.2.3. Synthesis of Optically Active Isopropyl-Substituted Derivatives 16–18
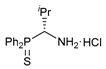
(R)-(1-Amino-2-methylpropyl)diphenylphosphine sulfide hydrochloride (R)-16
A total of 4.0 M HCl in dioxane (0.25 mL, 1.000 mmol) was slowly added dropwise to a stirred solution of compound 15 (0.200 g, 0.528 mmol) in MeOH (5 mL) at 0–5 °C under an argon atmosphere. The reaction mixture was stirred at room temperature for 3 h and left overnight. The solvent was removed under reduced pressure. The resulting residue was crystallized upon addition of Et2O. The mother liquor was decanted, and the obtained precipitate was dried under a vacuum to give 0.160 g of the target product as white crystals. Yield: 93%. Mp: 115–120 °C. 31P{1H} NMR (161.98 MHz, CD3OD): δ 44.91 ppm. 1H (400.13 MHz, CD3OD): δ 1.05 (d, 3H, Me, 3JHH = 7.0 Hz), 1.13 (d, 3H, Me, 3JHH = 7.0 Hz), 2.16–2.20 (m, 1H, CH in iPr), 3.32–3.34 (m, 3H, NH3Cl), 4.68–4.71 (m, 1H, CH), 7.57–7.66 (m, 6H, m-H and p-H in P(S)Ph2), 8.09–8.15 (m, 2H, o-H in P(S)Ph), 8.18–8.23 (m, 2H, o-H in P(S)Ph) ppm.
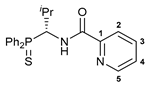
(R)-N-[1-(Diphenylthiophosphoryl)-2-methylpropyl]picolinamide (R)-17
![Ijms 26 04536 i020]()
A solution of triethylamine (0.114 g, 1.127 mmol) in CH2Cl2 (3 mL) was added dropwise to a stirred solution of amine hydrochloride (R)-16 (0.147 g, 0.451 mmol) in CH2Cl2 (5 mL) at –5 °C. The resulting mixture was stirred upon cooling for 15 min. Then, a solution of picolinoyl chloride generated in situ from picolinic acid (0.056 g, 0.455 mmol), SOCl2 (0.054 g, 0.454 mmol), and Et3N (0.070 g, 0.692 mmol) in CH2Cl2 (8 mL) was slowly added dropwise. The reaction mixture was stirred at room temperature for 12 h and then sequentially washed with water, an aqueous solution of NaHCO3, and again with water. The organic layer was separated, dried over anhydrous Na2SO4, and evaporated to dryness. The residue obtained was crystallized upon the addition of Et2O. The resulting precipitate was collected by filtration to provide 0.089 g of the target product as beige crystals. Yield: 50%. Mp: 210–212 °C. 31P{1H} NMR (161.98 MHz, CDCl3): δ 48.45 ppm. 1H NMR (500.13 MHz, CDCl3): δ 0.89 (d, 3H, Me, 3JHH = 6.7 Hz), 1.11 (d, 3H, Me, 3JHH = 6.7 Hz), 2.27–2.35 (m, 1H, CH in iPr), 5.38–5.42 (m, 1H, CH), 7.28–7.36 (m, 3H, m-H and p-H in P(S)Ph), 7.40–7.42 (m, 1H, H(C4)), 7.51–7.57 (m, 3H, m-H and p-H in P(S)Ph), 7.79 (dt, 1H, H(C3), 3JHH = 7.6 Hz, 4JHH = 1.1 Hz), 7.92–7.96 (m, 2H, o-H in P(S)Ph), 8.03–8.09 (m, 3H, H(C2) + o-H in P(S)Ph), 8.58 (d, 1H, H(C5), 3JHH = 4.4 Hz), 8.75 (br. d, 1H, NH, 3JHH = 10.5 Hz) ppm. 13C{1H} NMR (125.76 MHz, CDCl3): δ 18.28 (d, Me, 3JCP = 4.3 Hz), 22.27 (d, Me, 3JCP = 9.4 Hz), 30.03 (d, CH in iPr, 2JCP = 6.1 Hz), 52.01 (d, CH, 1JCP = 61.3 Hz), 122.21 (s, C2), 126.35 (s, C4), 128.31 (d, m-C in P(S)Ph, 3JCP = 11.8 Hz), 128.76 (d, m-C in P(S)Ph, 3JCP = 11.8 Hz), 131.21 (d, ipso-C in P(S)Ph, 1JCP = 75.4 Hz), 131.35 (d, o-C in P(S)Ph, 2JCP = 10.0 Hz), 131.40 (d, p-C in P(S)Ph, 4JCP = 2.7 Hz), 131.76 (d, o-C in P(S)Ph, 2JCP = 10.0 Hz), 131.84 (d, ipso-C in P(S)Ph, 1JCP = 80.0 Hz), 131.86 (d, p-C in P(S)Ph, 4JCP = 3.0 Hz), 137.15 (s, C3), 148.43 (s, C5), 148.96 (s, C1), 164.19 (d, C=O, 3JCP = 4.8 Hz) ppm. IR (KBr, ν/cm−1): 482(w), 512(m), 522(m), 597(w), 614(w) and 627(m) (both νP=S), 654(w), 690(m), 696(m), 713(m), 745(m), 754(m), 774(m), 817(w), 899(vw), 998(w), 1024(w), 1040(w), 1096(m), 1121(w), 1158(w), 1181(w), 1240(w), 1262(w), 1289(w), 1301(w), 1314(w), 1340(w), 1370(w), 1389(w), 1435(m), 1464(m), 1482(m), 1508(s) (C(O)NH), 1569(w), 1591(w), 1680(s) (νC=O), 2850(w), 2920(m), 2960(w), 3049(w), 3365(m) (νNH). Anal. Calcd for C22H23N2OPS: C, 66.99; H, 5.88; N, 7.10. Found: C, 67.34; H, 6.28; N, 6.81%.
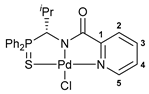
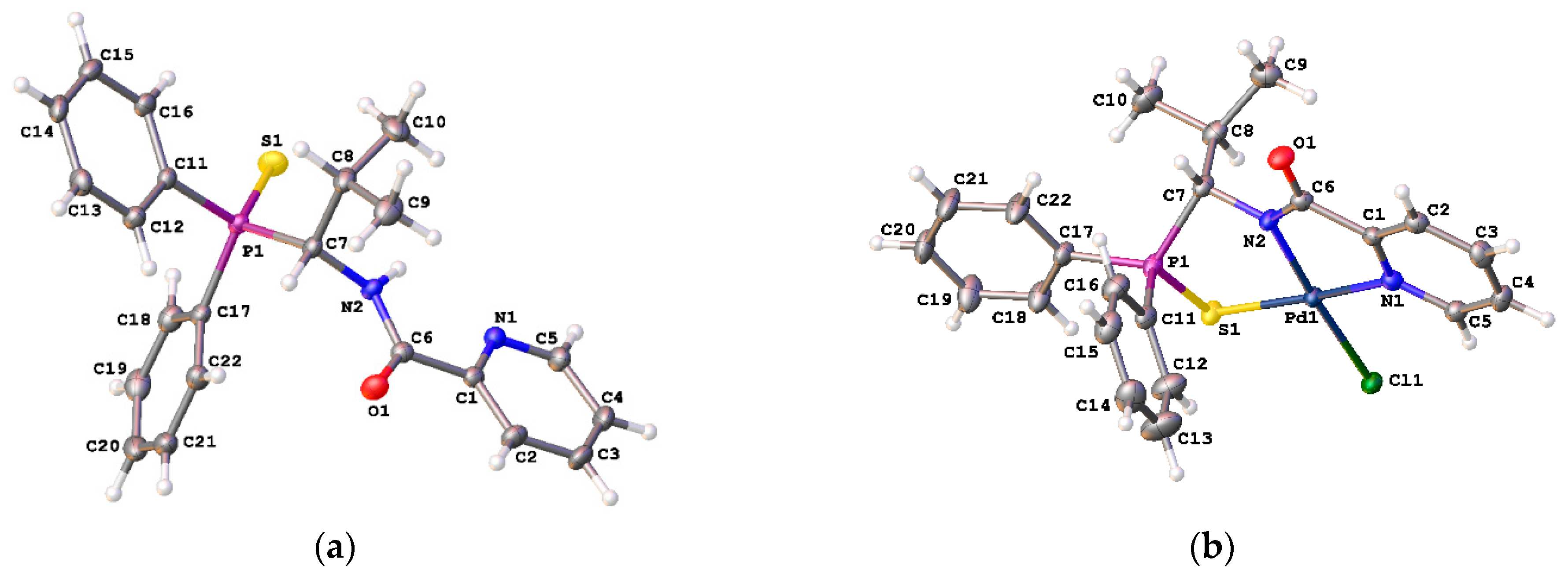
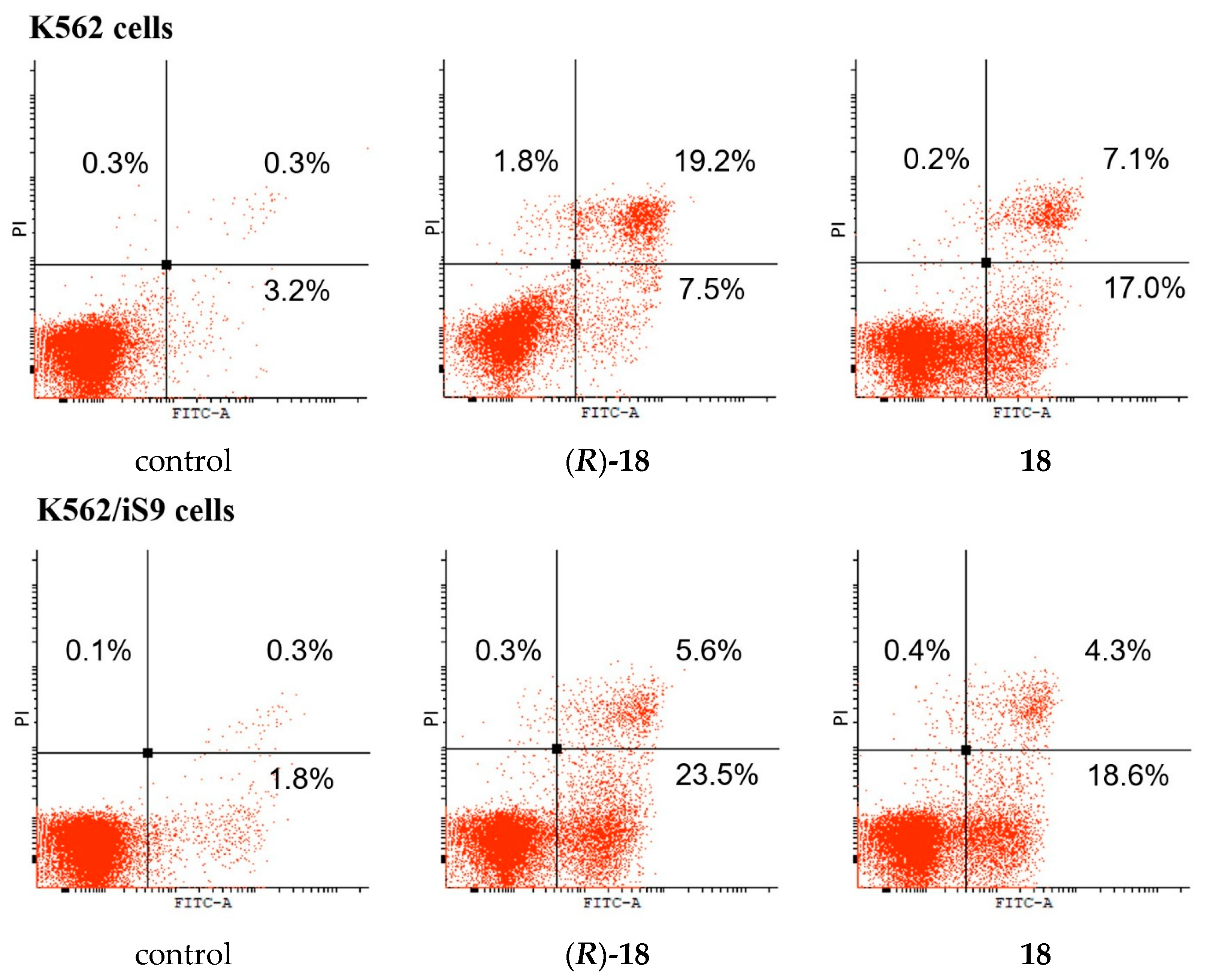
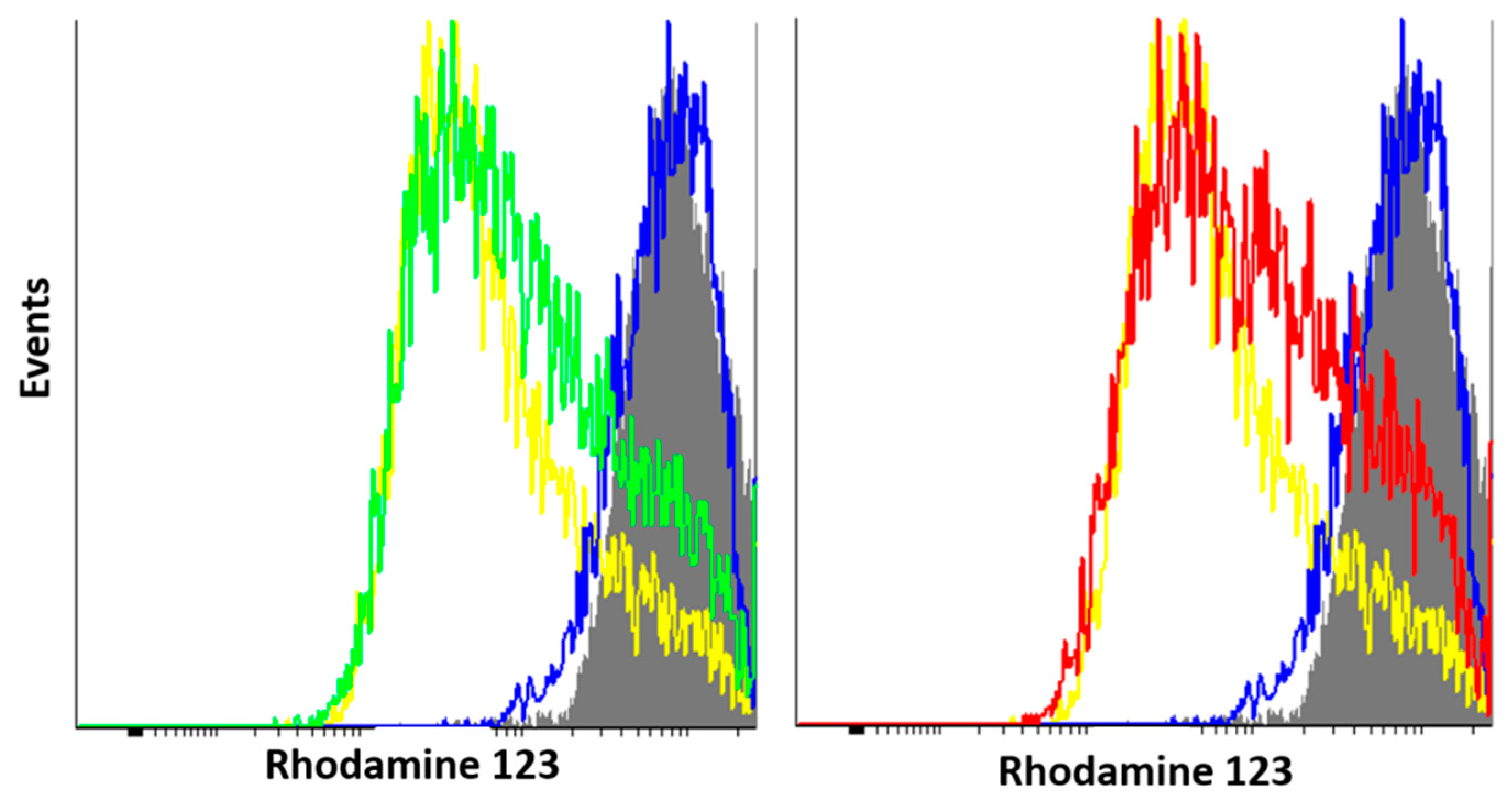
[κ3-S,N,N-(L)Pd(II)Cl] Pd(II) pincer complex (R)-18
![Ijms 26 04536 i021]()
A solution of PdCl2(NCPh)2 (38 mg, 0.099 mmol) in CH2Cl2 (3 mL) was slowly added dropwise to a solution of ligand (R)-17 (39 mg, 0.099 mmol) and Et3N (10 mg, 0.099 mmol) in CH2Cl2 (7 mL). The reaction mixture was left under ambient conditions for 3 h and then evaporated to dryness. The residue obtained was purified by column chromatography on silica gel (eluent: CH2Cl2–EtOH (100:1)) to provide the target complex as a yellow crystalline solid. Yield: 33 mg (62%). 31P{1H} NMR (121.49 MHz, CDCl3): δ 63.93 ppm. 1H NMR (500.13 MHz, CDCl3): δ 0.66 (d, 3H, Me, 3JHH = 6.8 Hz), 1.15 (d, 3H, Me, 3JHH = 6.8 Hz), 2.29–2.38 (m, 1H, CH in iPr), 5.45 (d, 1H, CH, 2JHP = 7.2 Hz), 7.48–7.51 (m, 1H, H(C4)), 7.52–7.66 (m, 6H, m-H and p-H in P(S)Ph2), 7.75 (dd, 2H, o-H in P(S)Ph, 3JHP = 13.0 Hz, 3JHH = 7.4 Hz), 7.88 (d, 1H, H(C2), 3JHH = 7.4 Hz), 7.95–7.99 (m, 3H, H(C3) + o-H in P(S)Ph), 8.99 (d, 1H, H(C5), 3JHH = 4.9 Hz) ppm. 13C{1H} NMR (125.76 MHz, CDCl3): δ 19.37 (d, Me, 3JCP = 7.7 Hz), 21.90 (d, Me, 3JCP = 4.9 Hz), 30.55 (d, CH in iPr, 2JCP = 4.1 Hz), 64.02 (d, CH, 1JCP = 70.4 Hz), 125.58 (s, C2), 126.06 (d, ipso-C in P(S)Ph, 1JCP = 69.3 Hz), 127.14 (s, C4), 128.32 (d, ipso-C in P(S)Ph, 1JCP = 82.0 Hz), 129.36 (d, m-C in P(S)Ph, 3JCP = 11.8 Hz), 129.66 (d, m-C in P(S)Ph, 3JCP = 12.7 Hz), 131.73 (d, o-C in P(S)Ph, 2JCP = 10.0 Hz), 132.69 (d, o-C in P(S)Ph, 2JCP = 10.4 Hz), 133.07 (d, p-C in P(S)Ph, 4JCP = 2.9 Hz), 133.68 (d, p-C in P(S)Ph, 4JCP = 2.9 Hz), 139.45 (s, C3), 147.54 (s, C5), 153.82 (s, C1), 171.54 (d, C=O, 3JCP = 12.4 Hz) ppm. IR (KBr, ν/cm−1): 497(m), 518(m), 579(m) (νP=S), 684(m), 708(m), 718(w), 748(m), 808(w), 998(w), 1049(w), 1105(m), 1184(vw), 1289(w), 1358(m), 1437(m), 1459(w), 1598(s), 1627(s) (νC=O), 2887(vw), 2929(w), 2963(w), 3055(vw). Anal. Calcd for C22H22N2OPPdS: C, 49.36; H, 4.14; N, 5.23. Found: C, 49.30; H, 4.23; N, 5.23%.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}