

C1QBP Modulates DNA Damage Response and Radiosensitivity in Hepatocellular Carcinoma by Regulating NF-κB Activity


Abstract
:1. Introduction
2. Results
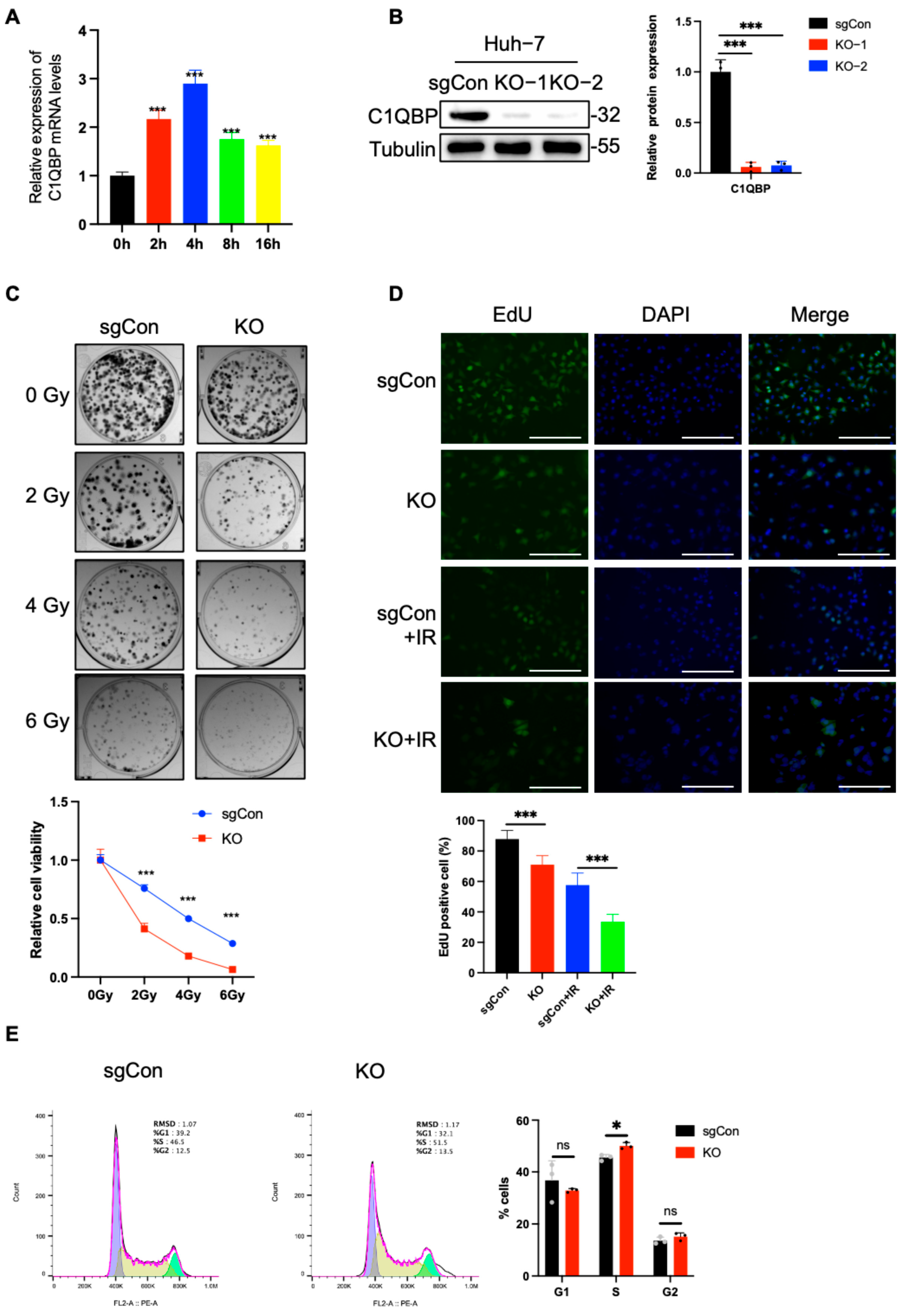
2.1. C1QBP KO Increases the Radiosensitivity of HCC Cells
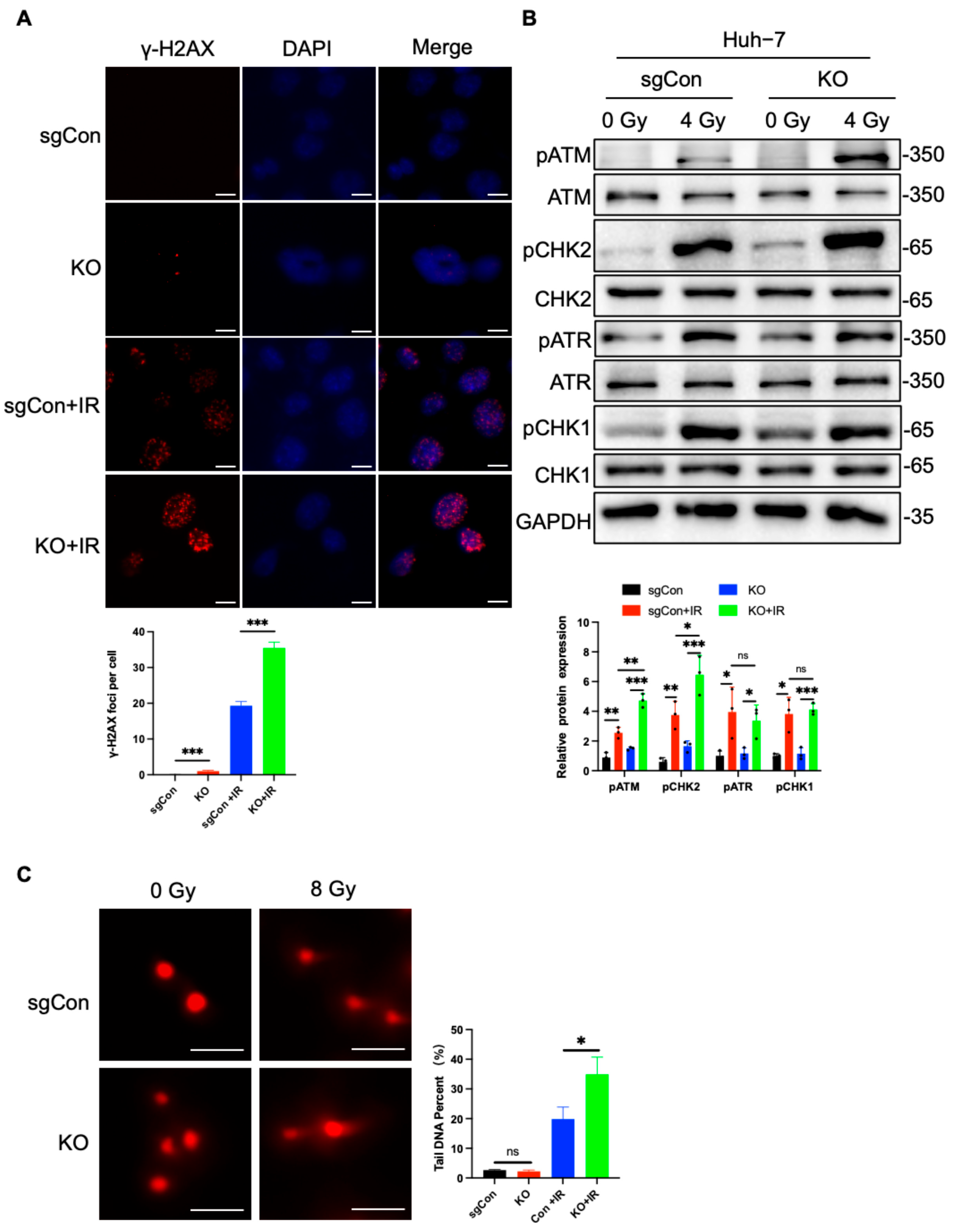
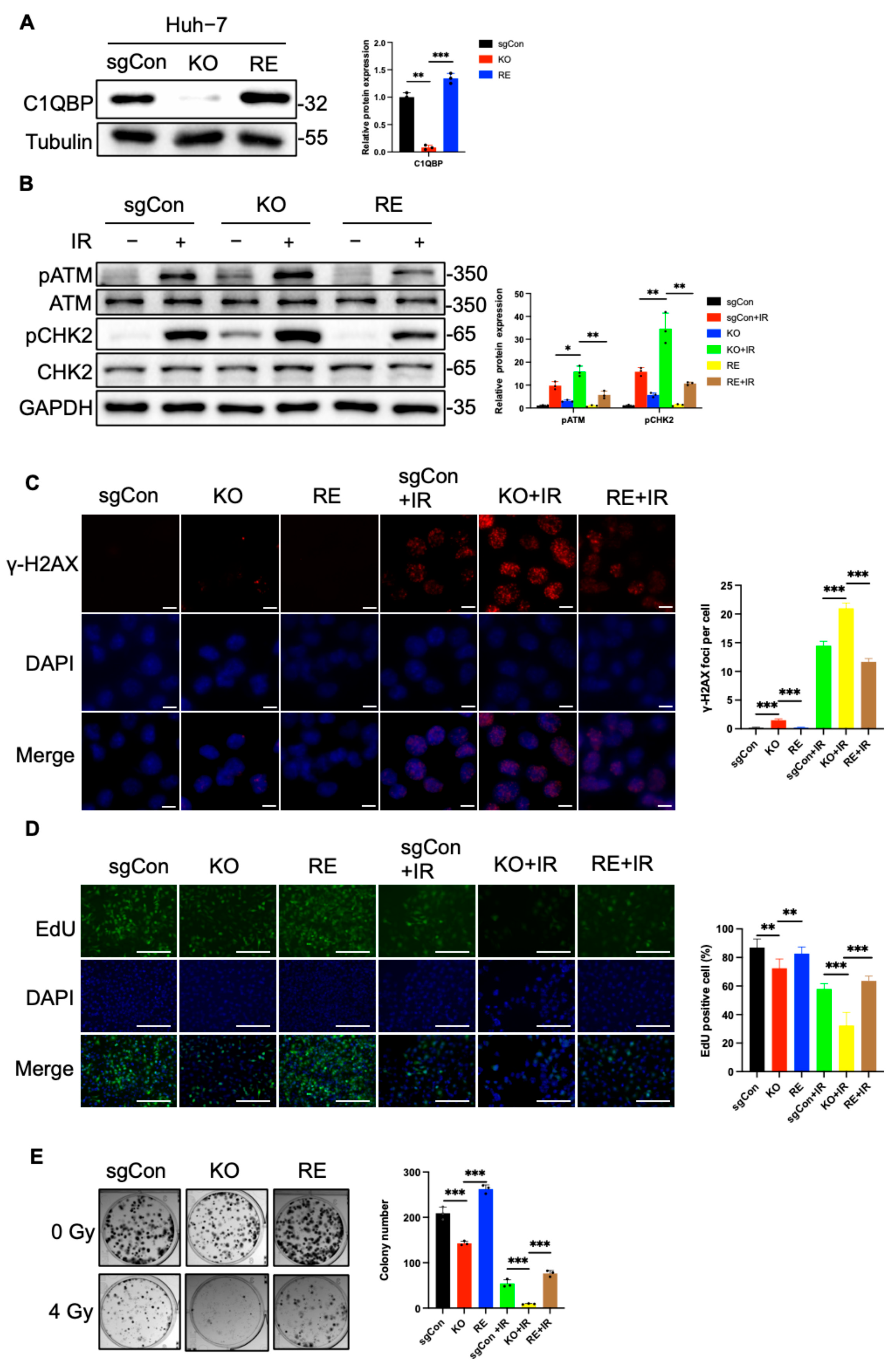
2.2. C1QBP KO Aggravates Radiation-Induced DNA Damage
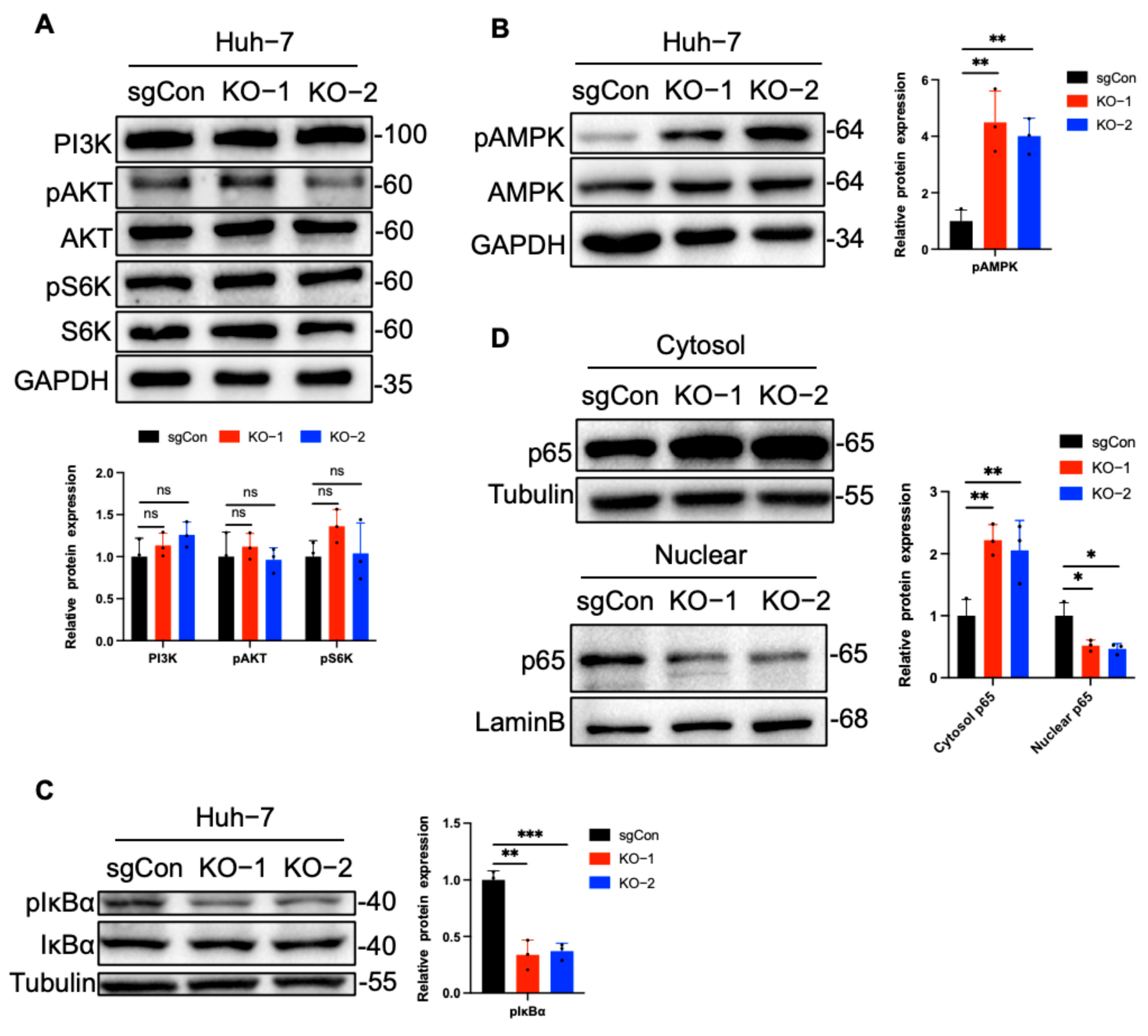
2.3. C1QBP Regulates NF-κB Activity Via AMPK
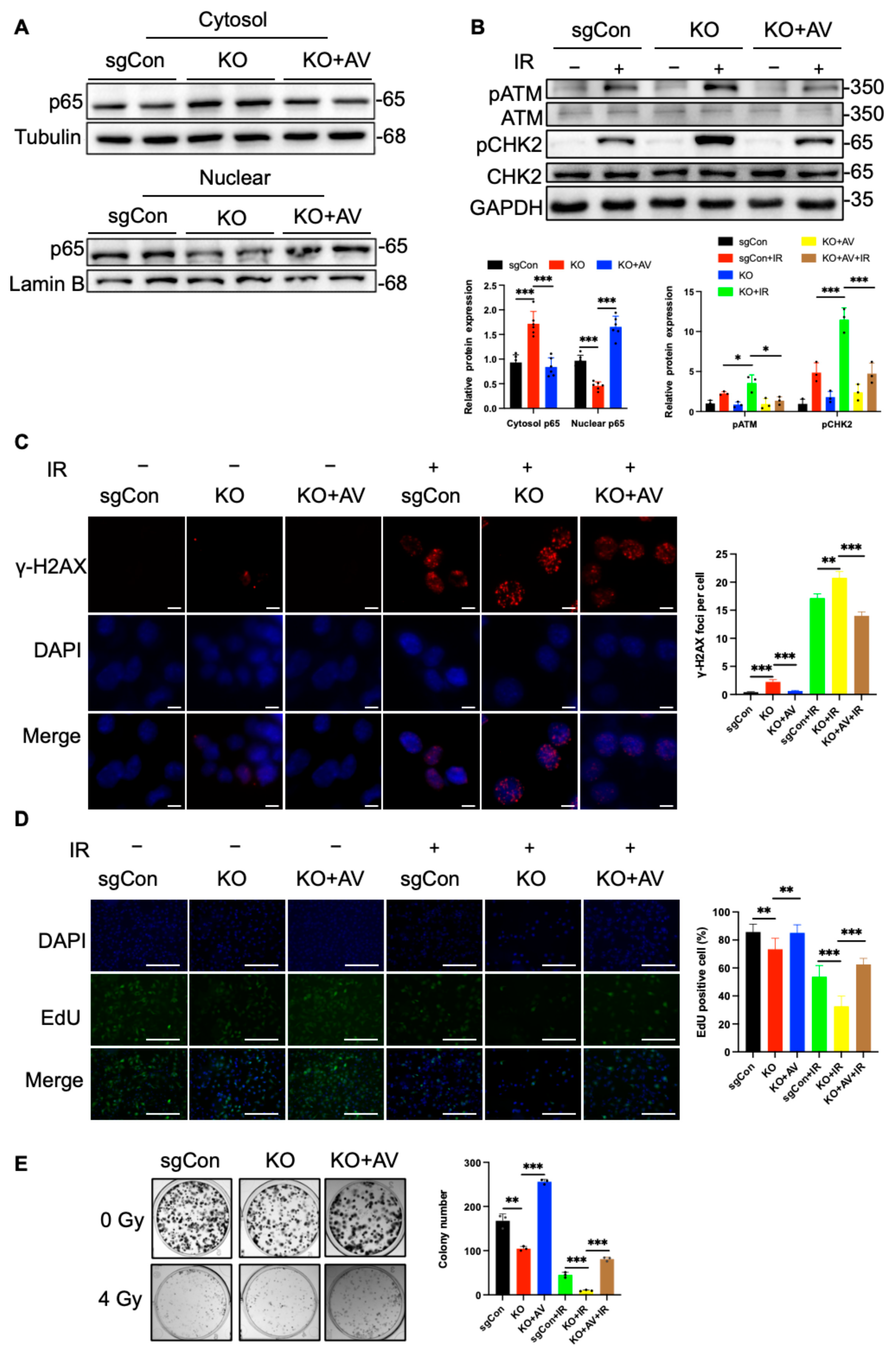
2.4. C1QBP Affects Radiation-Induced DNA Damage by Regulating NF-κΒ Activity
2.5. Reintroduction of C1QBP Ameliorates Radiation-Induced DNA Damage
3. Materials and Methods
3.1. Cell Culture
3.2. Colony Formation
3.3. Western Blot
3.4. q-PCR
3.5. Cell Cycle Arrest Analysis
3.6. EdU Staining
3.7. Immunofluorescence Staining
3.8. Comet Assay
3.9. Plasmid Transfection
3.10. CRISPR/Cas9
3.11. Statistical Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Yang, X.; Yang, C.; Zhang, S.; Geng, H.; Zhu, A.X.; Bernards, R.; Qin, W.; Fan, J.; Wang, C.; Gao, Q. Precision treatment in advanced hepatocellular carcinoma. Cancer Cell 2024, 42, 180–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiong, L.; Li, M.; Jiang, P.; Wang, J.; Li, C. Advances in radiotherapy and immunity in hepatocellular carcinoma. J. Transl. Med. 2023, 21, 526. [Google Scholar] [CrossRef]
- Ghebrehiwet, B.; Geisbrecht, B.V.; Xu, X.; Savitt, A.G.; Peerschke, E.I.B. The C1q Receptors: Focus on gC1qR/p33 (C1qBP, p32, HABP-1)(1). Semin. Immunol. 2019, 45, 101338. [Google Scholar] [CrossRef] [PubMed]
- Raschdorf, A.; Sünderhauf, A.; Skibbe, K.; Ghebrehiwet, B.; Peerschke, E.I.; Sina, C.; Derer, S. Heterozygous P32/C1QBP/HABP1 Polymorphism rs56014026 Reduces Mitochondrial Oxidative Phosphorylation and Is Expressed in Low-grade Colorectal Carcinomas. Front. Oncol. 2020, 10, 631592. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Liu, K.; Fang, H.; Zhang, Q.; Gao, X.; Liu, F.; Zhou, S.; Wang, X.; Niu, Y.; Hong, Y.; et al. Mitochondrial C1qbp promotes differentiation of effector CD8+ T cells via metabolic-epigenetic reprogramming. Sci. Adv. 2021, 7, eabk0490. [Google Scholar] [CrossRef]
- Noh, S.; Phorl, S.; Naskar, R.; Oeum, K.; Seo, Y.; Kim, E.; Kweon, H.S.; Lee, J.Y. p32/C1QBP regulates OMA1-dependent proteolytic processing of OPA1 to maintain mitochondrial connectivity related to mitochondrial dysfunction and apoptosis. Sci. Rep. 2020, 10, 10618. [Google Scholar] [CrossRef]
- Lei, Y.; Li, X.; Qin, D.; Zhang, Y.; Wang, Y. gC1qR: A New Target for Cancer Immunotherapy. Front. Immunol. 2023, 14, 1095943. [Google Scholar] [CrossRef]
- Wang, J.; Huang, C.L.; Zhang, Y. Complement C1q Binding Protein (C1QBP): Physiological Functions, Mutation-Associated Mitochondrial Cardiomyopathy and Current Disease Models. Front. Cardiovasc. Med. 2022, 9, 843853. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, W.; Li, S.; Zhan, J.; Li, H.; Zhao, M.; Zhou, X.A.; Li, S.; Li, X.; Huo, Y.; et al. C1QBP Promotes Homologous Recombination by Stabilizing MRE11 and Controlling the Assembly and Activation of MRE11/RAD50/NBS1 Complex. Mol. Cell 2019, 75, 1299–1314.e1296. [Google Scholar] [CrossRef]
- Tian, H.; Wang, G.; Wang, Q.; Zhang, B.; Jiang, G.; Li, H.; Chai, D.; Fang, L.; Wang, M.; Zheng, J. Complement C1q binding protein regulates T cells’ mitochondrial fitness to affect their survival, proliferation, and anti-tumor immune function. Cancer Sci. 2022, 113, 875–890. [Google Scholar] [CrossRef]
- Ghebrehiwet, B.; Zaniewski, M.; Fernandez, A.; DiGiovanni, M.; Reyes, T.N.; Ji, P.; Savitt, A.G.; Williams, J.L.; Seeliger, M.A.; Peerschke, E.I.B. The C1q and gC1qR axis as a novel checkpoint inhibitor in cancer. Front. Immunol. 2024, 15, 1351656. [Google Scholar] [CrossRef]
- Saha, S.K.; Kim, K.E.; Islam, S.M.R.; Cho, S.G.; Gil, M. Systematic Multiomics Analysis of Alterations in C1QBP mRNA Expression and Relevance for Clinical Outcomes in Cancers. J. Clin. Med. 2019, 8, 513. [Google Scholar] [CrossRef] [PubMed]
- Scully, O.J.; Shyamasundar, S.; Matsumoto, K.; Dheen, S.T.; Yip, G.W.; Bay, B.H. C1QBP Mediates Breast Cancer Cell Proliferation and Growth via Multiple Potential Signalling Pathways. Int. J. Mol. Sci. 2023, 24, 1343. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Lu, Z.; Wang, Z.; Yang, X. The Mitochondrial Protein C1QBP Promotes Hepatocellular Carcinoma Progression by Enhancing Cell Survival, Migration and Invasion. J. Cancer 2022, 13, 2477–2489. [Google Scholar] [CrossRef]
- Wu, H.; Chu, Y.; Sun, S.; Li, G.; Xu, S.; Zhang, X.; Jiang, Y.; Gao, S.; Wang, Q.; Zhang, J.; et al. Hypoxia-Mediated Complement 1q Binding Protein Regulates Metastasis and Chemoresistance in Triple-Negative Breast Cancer and Modulates the PKC-NF-κB-VCAM-1 Signaling Pathway. Front. Cell Dev. Biol. 2021, 9, 607142. [Google Scholar] [CrossRef]
- Panday, A.; Inda, M.E.; Bagam, P.; Sahoo, M.K.; Osorio, D.; Batra, S. Transcription Factor NF-κB: An Update on Intervention Strategies. Arch. Immunol. Ther. Exp. 2016, 64, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Simon, M.; Seluanov, A.; Gorbunova, V. DNA damage and repair in age-related inflammation. Nat. Rev. Immunol. 2023, 23, 75–89. [Google Scholar] [CrossRef]
- Yao, F.; Deng, Y.; Zhao, Y.; Mei, Y.; Zhang, Y.; Liu, X.; Martinez, C.; Su, X.; Rosato, R.R.; Teng, H.; et al. A targetable LIFR-NF-κB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat. Commun. 2021, 12, 7333. [Google Scholar] [CrossRef]
- Sun, E.J.; Wankell, M.; Palamuthusingam, P.; McFarlane, C.; Hebbard, L. Targeting the PI3K/Akt/mTOR Pathway in Hepatocellular Carcinoma. Biomedicines 2021, 9, 1639. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Sun, X.; Cao, S.; Mao, C.; Sun, F.; Zhang, X.; Song, Y. Post-translational modifications of p65: State of the art. Front. Cell Dev. Biol. 2024, 12, 1417502. [Google Scholar] [CrossRef] [PubMed]
- Egusquiza-Alvarez, C.A.; Robles-Flores, M. An approach to p32/gC1qR/HABP1: A multifunctional protein with an essential role in cancer. J. Cancer Res. Clin. Oncol. 2022, 148, 1831–1854. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Guo, Z.; Wang, S.; Gao, S.; Cao, Q. Histone Phosphorylation in DNA Damage Response. Int. J. Mol. Sci. 2025, 26, 2045. [Google Scholar] [CrossRef]
- Prabhu, K.S.; Kuttikrishnan, S.; Ahmad, N.; Habeeba, U.; Mariyam, Z.; Suleman, M.; Bhat, A.A.; Uddin, S. H2AX: A key player in DNA damage response and a promising target for cancer therapy. Biomed. Pharmacother. 2024, 175, 116663. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Huang, T.T.; Horibata, S.; Lee, J.M. Cell cycle checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor-resistant cancer. Pharmacol. Res. 2022, 178, 106162. [Google Scholar] [CrossRef]
- Simoneau, A.; Zou, L. An extending ATR-CHK1 circuitry: The replication stress response and beyond. Curr. Opin. Genet. Dev. 2021, 71, 92–98. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Dos Santos Fernandes, M.C.; Romeiro, F.; Arpini, A.P.; Dias, G.M. DNA damage and repair in patients undergoing myocardial perfusion single-photon emission computed tomography. Sci. Rep. 2024, 14, 13079. [Google Scholar] [CrossRef]
- Li, Y.J.; Liu, A.X.; Zeng, J.Y.; Miao, Y.; Zhang, M.; Liu, X.Y.; Yang, W.; Li, R.C.; Zhu, J.Q.; Liu, C.J.; et al. Repeated measurements of urinary bisphenol A and its analogues in relation to sperm DNA damage. J. Hazard. Mater. 2025, 487, 137157. [Google Scholar] [CrossRef]
- Kong, P.; Yang, M.; Wang, Y.; Yu, K.N.; Wu, L.; Han, W. Ferroptosis triggered by STAT1- IRF1-ACSL4 pathway was involved in radiation-induced intestinal injury. Redox Biol. 2023, 66, 102857. [Google Scholar] [CrossRef]
- Shimada, T.; Yabuki, Y.; Noguchi, T.; Tsuchida, M.; Komatsu, R.; Hamano, S.; Yamada, M.; Ezaki, Y.; Hirata, Y.; Matsuzawa, A. The Distinct Roles of LKB1 and AMPK in p53-Dependent Apoptosis Induced by Cisplatin. Int. J. Mol. Sci. 2022, 23, 10064. [Google Scholar] [CrossRef]
- Rae, C.; Mairs, R.J. AMPK activation by AICAR sensitizes prostate cancer cells to radiotherapy. Oncotarget 2019, 10, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Fasih, A.; Elbaz, H.A.; Hüttemann, M.; Konski, A.A.; Zielske, S.P. Radiosensitization of pancreatic cancer cells by metformin through the AMPK pathway. Radiat. Res. 2014, 182, 50–59. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Wang, Y.; Su, P.; Zhuo, Z.; Jin, Y.; Zeng, R.; Wu, H.; Huang, H.; Chen, H.; Li, Z.; Sha, W. Ginsenoside Rk1 attenuates radiation-induced intestinal injury through the PI3K/AKT/mTOR pathway. Biochem. Biophys. Res. Commun. 2023, 643, 111–120. [Google Scholar] [CrossRef]
- Kma, L.; Baruah, T.J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 2022, 69, 248–264. [Google Scholar] [CrossRef]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e2046. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chai, D.; Sobhani, N.; Sun, N.; Neeli, P.; Zheng, J.; Tian, H. C1QBP regulates mitochondrial plasticity to impact tumor progression and antitumor immune response. Front. Physiol. 2022, 13, 1012112. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Derler, M.; Basílio, J.; Schmid, J.A. NF-κB in monocytes and macrophages—An inflammatory master regulator in multitalented immune cells. Front. Immunol. 2023, 14, 1134661. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Source | Identifer |
|---|---|---|
| BAY117082 | MCE | Cat# HY-13453 |
| NF-κB activator 1 | MCE | Cat# HY-134476 |
| BSA-V | Solarbio | Cat#A8020 |
| Tween 20 | Solomen | Cat#T8220 |
| Triton X-100 | Solarbio | Cat#T8200 |
| Antibody | Source | Identifer |
|---|---|---|
| Anti-C1QBP | Cell Signaling | Cat# 6502 |
| Anti-Tubulin | Proteintech | Cat# 11224 |
| Anti-p-ATM | Cell Signaling | Cat# 5883 |
| Anti-ATM | Cell Signaling | Cat# 2873 |
| Anti-p-CHK2 | Cell Signaling | Cat# 2197 |
| Anti-CHK2 | Cell Signaling | Cat# 2662 |
| Anti-p-ATR | Cell Signaling | Cat# 30632 |
| Anti-ATR | Cell Signaling | Cat# 2790 |
| Anti-p-CHK1 | Cell Signaling | Cat# 12302 |
| Anti-CHK1 | Cell Signaling | Cat# 2360 |
| Anti-p-AMPK | Cell Signaling | Cat# 50081 |
| Anti-AMPK | Cell Signaling | Cat# 5831 |
| Anti-p65 | Cell Signaling | Cat# 8242 |
| Anti-GAPDH | Proteintech | Cat# 60004 |
| Anti-γH2AX | Sigma | Cat# 05636 |
| Anti-FLAG | Proteintech | Cat# 20543 |
| Gene | Forward | Reverse |
|---|---|---|
| C1QBP KO1 | GCGTGCGCGCAGGTTCCGAG | CTCGGAACCTGCGCGCACGC |
| C1QBP KO2 | ACGGAGGAGCCCAGCACACG | CGTGTGCTGGGCTCCTCCGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Wu, Y.; Meng, J.; Zhao, X.; Hou, Y.; Wang, Q.; Liu, Y. C1QBP Modulates DNA Damage Response and Radiosensitivity in Hepatocellular Carcinoma by Regulating NF-κB Activity. Int. J. Mol. Sci. 2025, 26, 4513. https://doi.org/10.3390/ijms26104513
Zhou H, Wu Y, Meng J, Zhao X, Hou Y, Wang Q, Liu Y. C1QBP Modulates DNA Damage Response and Radiosensitivity in Hepatocellular Carcinoma by Regulating NF-κB Activity. International Journal of Molecular Sciences. 2025; 26(10):4513. https://doi.org/10.3390/ijms26104513
Chicago/Turabian StyleZhou, Haitao, Yanjin Wu, Jiahui Meng, Xiaotong Zhao, Yujia Hou, Qin Wang, and Yang Liu. 2025. "C1QBP Modulates DNA Damage Response and Radiosensitivity in Hepatocellular Carcinoma by Regulating NF-κB Activity" International Journal of Molecular Sciences 26, no. 10: 4513. https://doi.org/10.3390/ijms26104513
APA StyleZhou, H., Wu, Y., Meng, J., Zhao, X., Hou, Y., Wang, Q., & Liu, Y. (2025). C1QBP Modulates DNA Damage Response and Radiosensitivity in Hepatocellular Carcinoma by Regulating NF-κB Activity. International Journal of Molecular Sciences, 26(10), 4513. https://doi.org/10.3390/ijms26104513