
Neurotrophic and Neurotoxic Effects of Aβ42 and Its Oligomers on Neuronal Survival: Revealed by Their Opposite Influence on the Potency of Extracellular BDNF


Abstract
1. Introduction
2. Results
2.1. Examination of the Binding Specificity of BDNF to Four Aβ42 Species
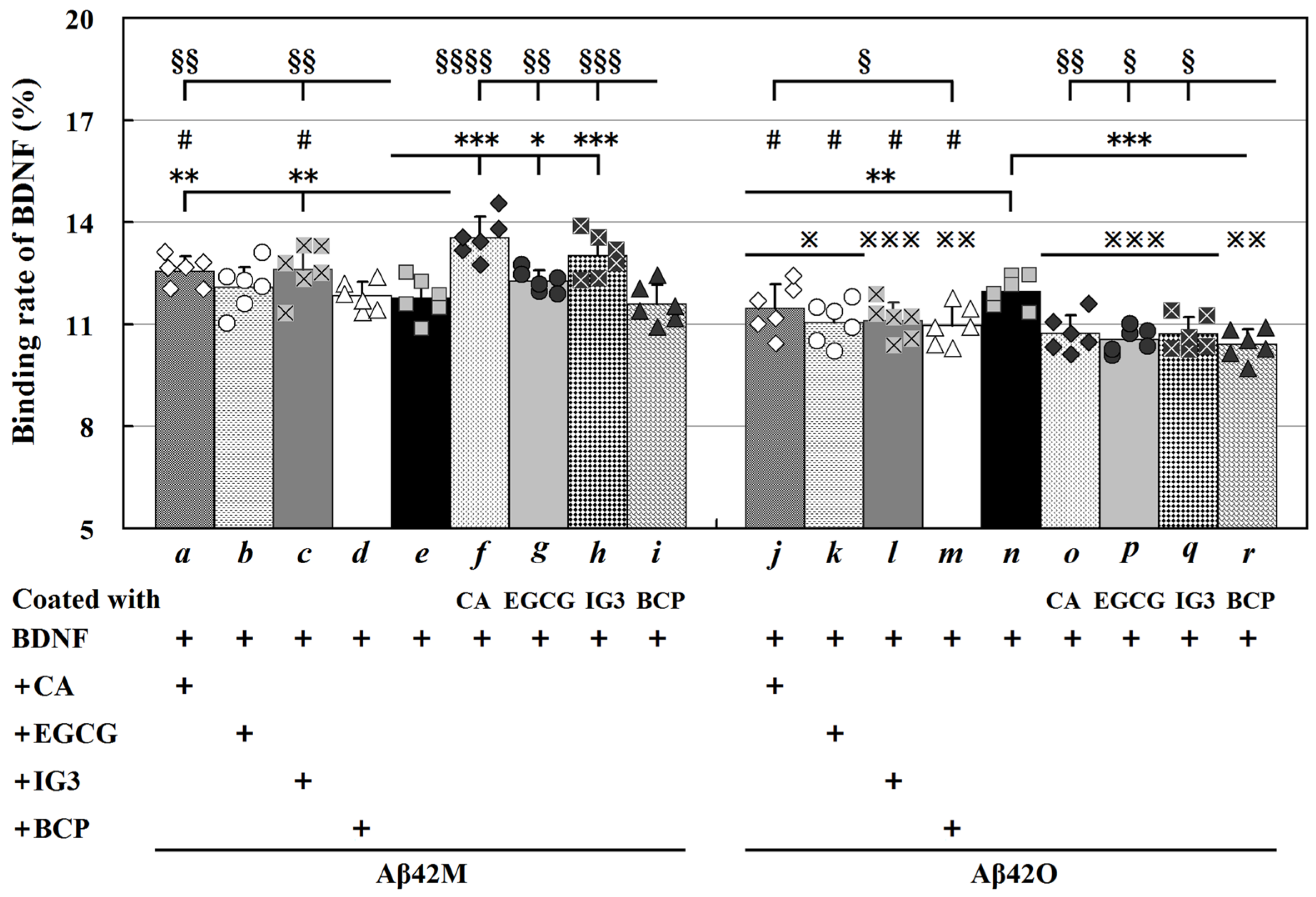
2.2. Binding Affinity of BDNF for Aβ42M and Aβ42O
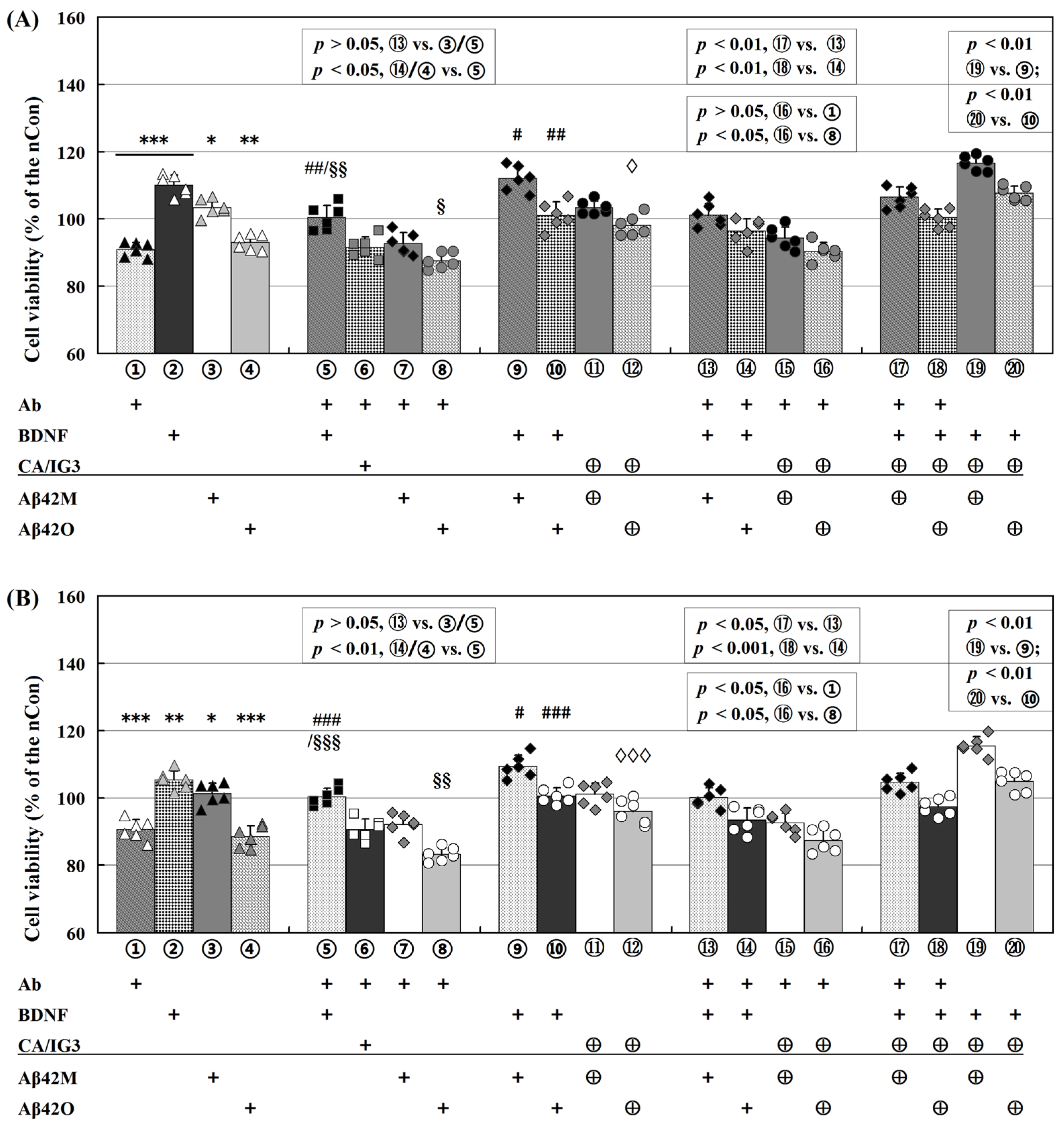
2.3. Differential Effects of Aβ42M and Aβ42O on BDNF-Supported Neural Cell Survival
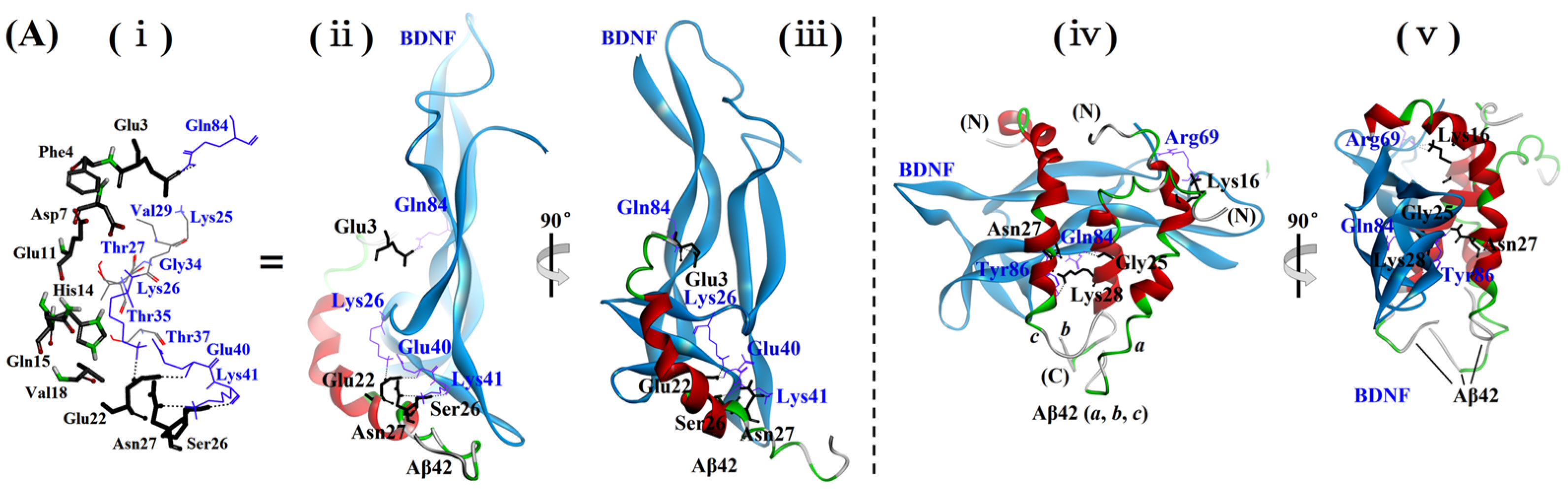
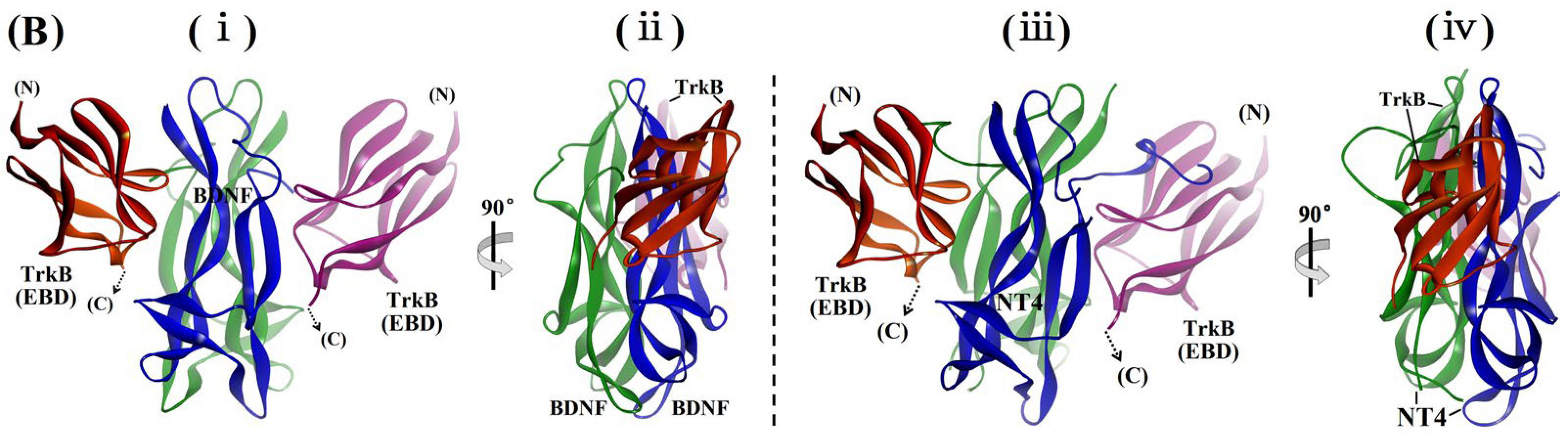
2.4. Examination of the Sites and Properties of the Interaction Between Aβ42M/Aβ42O and BDNF by Molecular Docking
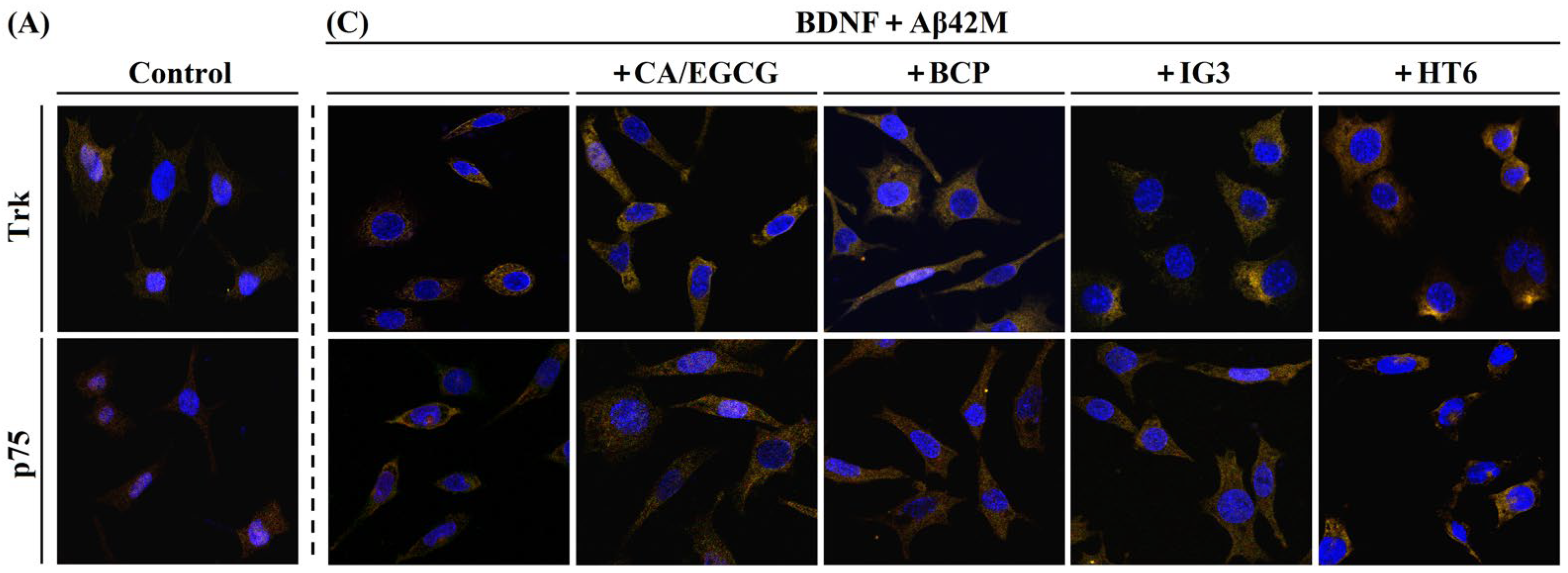
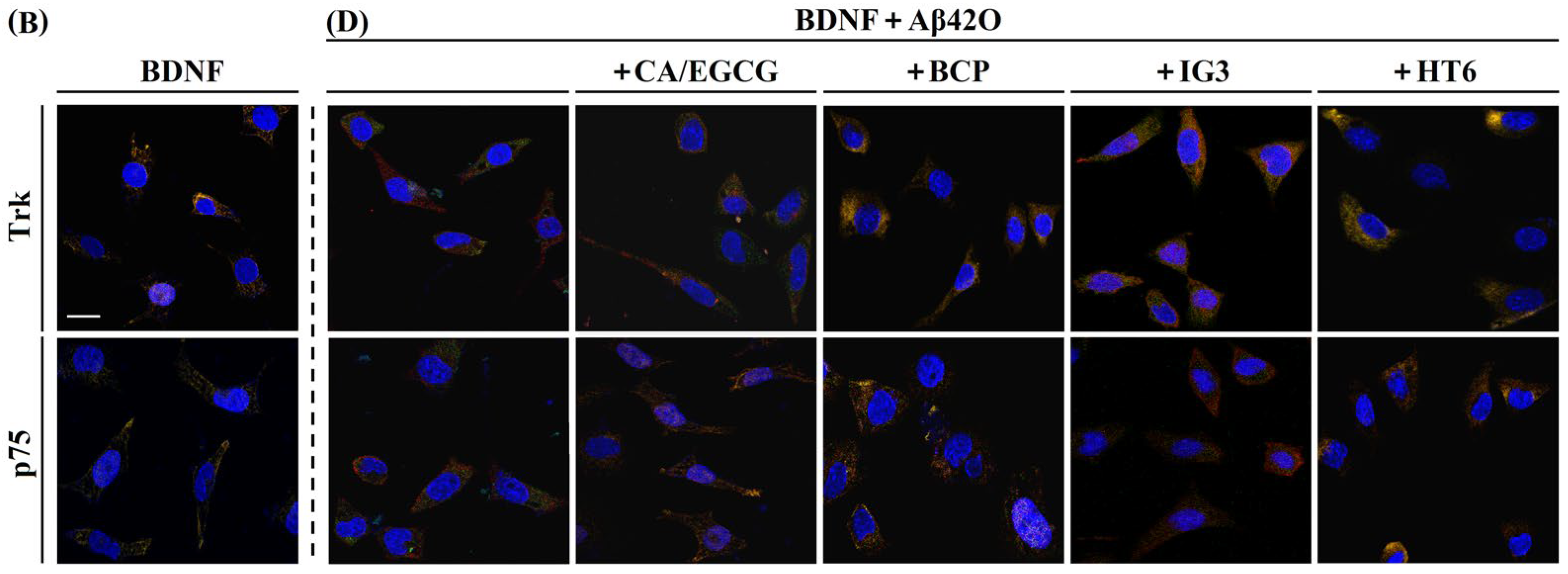
2.5. Effects of Aβ42M and Aβ42O on the Receptor-Targeting Performance of BDNF in the Presence and Absence of IG3/BCP/CA/EGCG by IF Staining
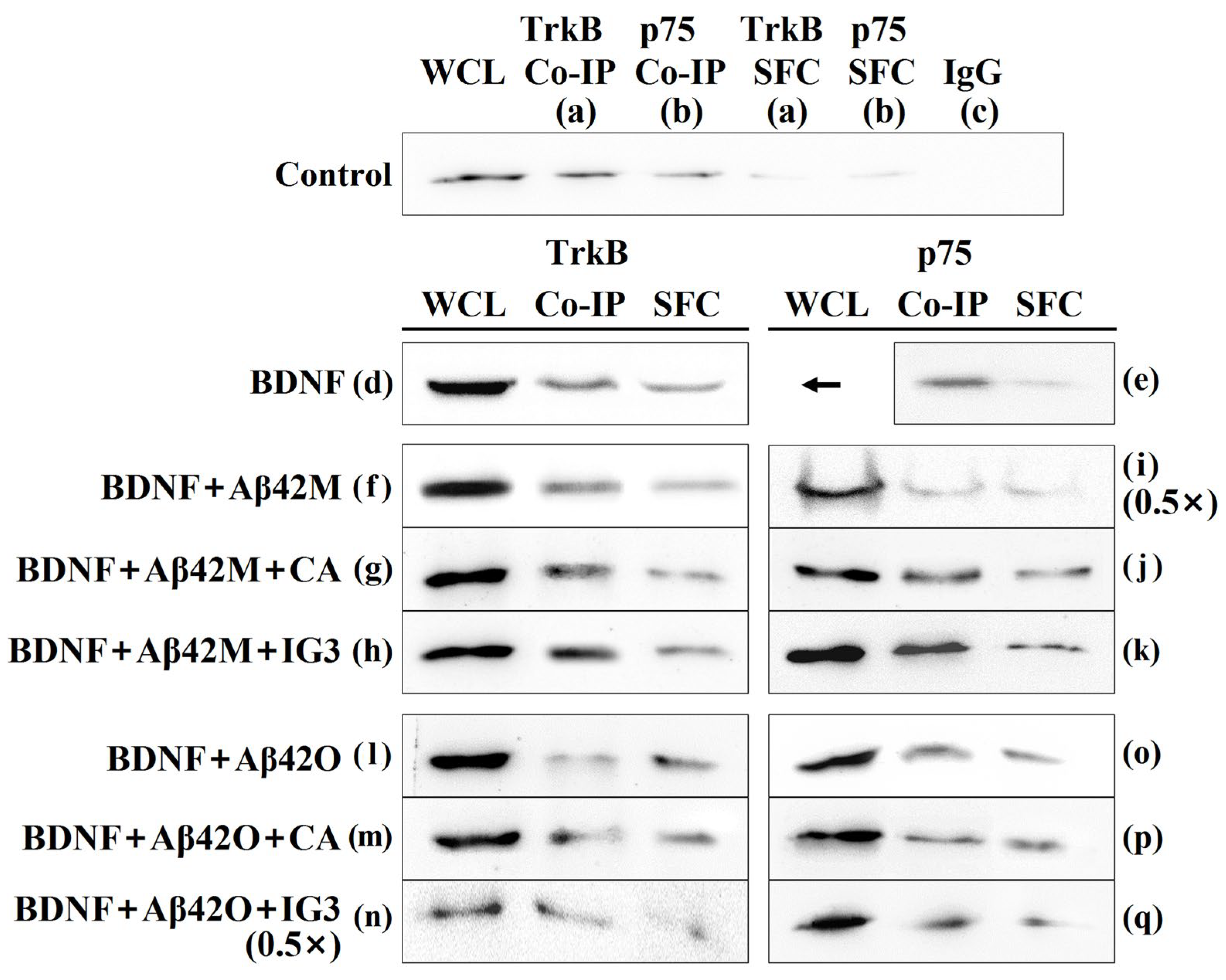
2.6. Effects of Aβ42M and Aβ42O on BDNF Binding to TrkB/p75 in the Presence and Absence of IG3/BCP/CA/EGCG by Co-IP
3. Materials and Methods
3.1. Aβ42 Species and Other Proteins
3.2. Chemicals
3.3. Cell Culture
3.4. Determination of the Binding Specificity of BDNF to Different Aβ42 Species by Indirect Enzyme-Linked Immunosorbent Assay (ELISA)
3.4.1. Construction of a Standard BDNF Curve
3.4.2. Determination of the Binding Rate of BDNF to Different Aβ42 Species
3.5. Measurement of the Equilibrium Dissociation Constant (KD) of BDNF to Aβ42M/Aβ42O by Indirect Competitive ELISA
3.6. Determination of the Effect of Aβ42M/Aβ42O with and Without BOs and CAs on the Cytological Efficacy of BDNF in Terms of Cell Survival by MTT Assay
3.7. Molecular Docking
3.8. Immunofluorescence (IF) Microscopy
3.9. Co-Immunoprecipitation (Co-IP) Assay
3.10. Statistical Analysis
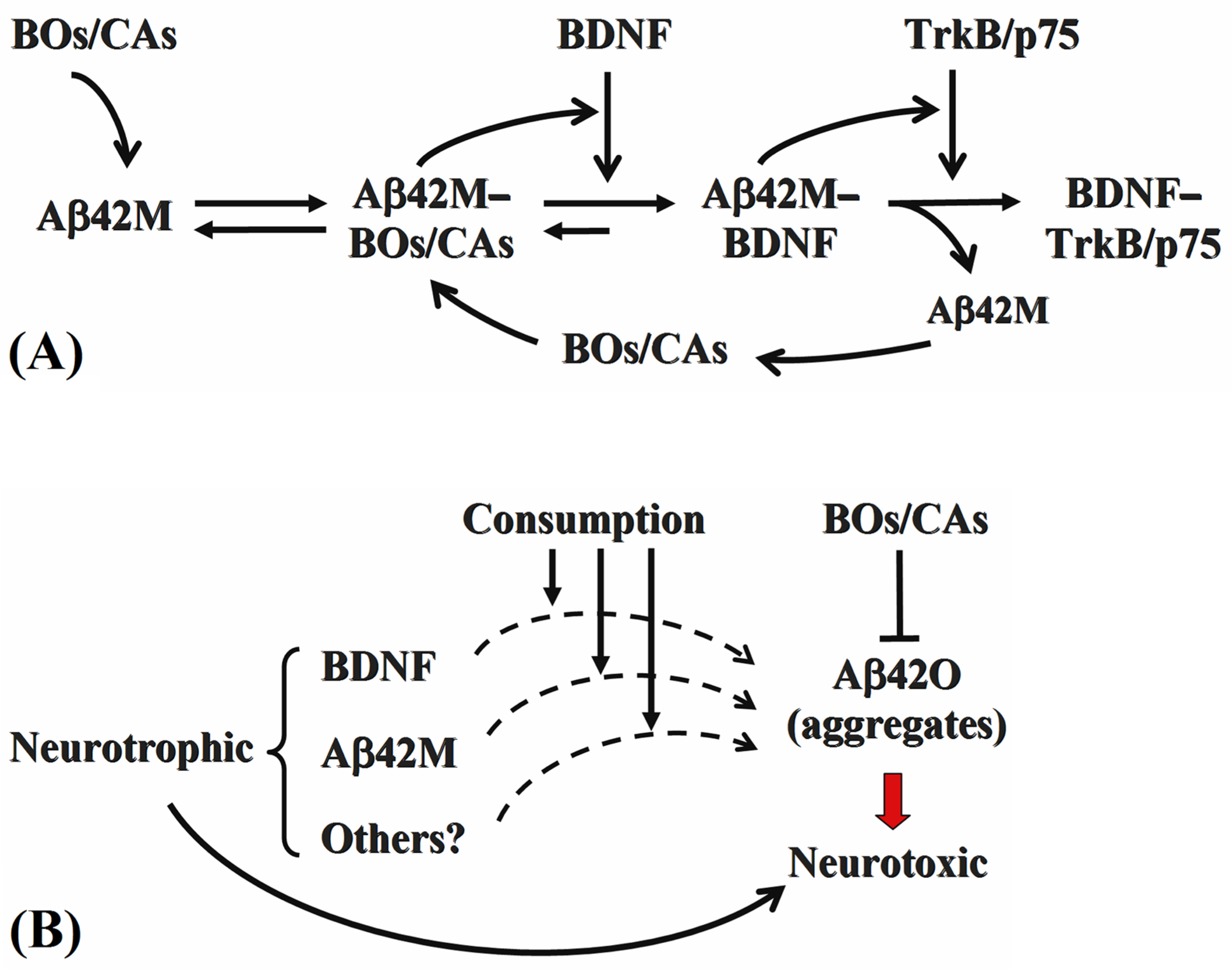
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ42 | amyloid-β protein 1–42 |
| AD | Alzheimer disease |
| CAs | catechins |
| EGCG | epigallocatechin gallate |
| BOs | branched oligosaccharides |
| IG3 | isomaltotriose |
| BCP | bianntennary N-linked core pentasaccharide |
| BDNF | brain-derived neurotrophic factor |
| NT | neurotrophin |
| NGF | nerve growth factor |
| TrkB | tyrosine kinase receptor B |
| ELISA | enzyme-linked immunosorbent assay |
| BBB | blood–brain barrier |
| DMEM | Dulbecco’s modified eagle’s medium |
| FBS | fetal bovine serum |
| HSA | human serum albumin |
References
- Song, C.; Zhang, T.; Zhang, Y. Conformational Essentials Responsible for Neurotoxicity of Aβ42 Aggregates Revealed by Antibodies against Oligomeric Aβ42. Molecules 2022, 27, 6751. [Google Scholar] [CrossRef]
- Qu, L.F.S.; Matsuzaki, K.; Hoshino, T. Computational Study on the Assembly of Amyloid β-Peptides in the Hydrophobic Environment. Chem. Pharm. Bull. 2019, 67, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Li, H.; Zhang, T.; Wang, Z.; Li, H.; Zhang, Y. Nonlinear and mixed inhibitory effect of matrine on the cytotoxicity of oligomeric amyloid-β protein. Neurochem. Int. 2020, 137, 104746. [Google Scholar] [CrossRef]
- Puzzo, D.; Privitera, L.; Fa’, M.; Staniszewski, A.; Hashimoto, G.; Aziz, F.; Sakurai, M.; Ribe, E.M.; Troy, C.M.; Mercken, M. Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 2011, 69, 819–830. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Kashif, M.; Sivaprakasam, P.; Vijendra, P.; Waseem, M.; Pandurangan, A.K. A Recent Update on Pathophysiology and Therapeutic Interventions of Alzheimer’s Disease. Curr. Pharm. Des. 2023, 29, 3428–3441. [Google Scholar] [CrossRef]
- Copani, A. The underexplored question of β-amyloid monomers. Eur. J. Pharmacol. 2017, 817, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Song, C.; Li, H.; Zheng, Y.; Zhang, Y. Different Extracellular β-Amyloid (1-42) Aggregates Differentially Impair Neural Cell Adhesion and Neurite Outgrowth through Differential Induction of Scaffold Palladin. Biomolecules 2022, 12, 1808. [Google Scholar] [CrossRef]
- Zhang, T.; Song, C.; Zheng, C.; Chen, X.; Zhang, Y. Extracellular Amyloid β-protein (1–42) Oligomers Anchor Brain Cells and Make them inert as an Unconventional Integrin-Coupled Ligand. Cell. Mol. Neurobiol. 2023, 43, 841–858. [Google Scholar] [CrossRef]
- Barbara, M.; Magdalena, G.; Ala, L.Z.; Johannes, K.; Piotr, L. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef]
- Ebadi, M.; Bashir, R.M.; Heidrick, M.L.; Hamada, F.M.; Lassi, N.K. Neurotrophins and their receptors in nerve injury and repair. Neurochem. Int. 2012, 30, 347. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fischer, I.; Tessler, A.; Houle, J.D. Transplants of fibroblasts genetically modified to express BDNF promote axonal regeneration from supraspinal neurons following chronic spinal cord injury. Exp. Neurol. 2002, 177, 265–275. [Google Scholar] [CrossRef]
- Soontornniyomkij, V.; Wang, G.; Pittman, C.A.; Hamilton, R.L.; Wiley, C.A.; Achim, C.L. Absence of brain-derived neurotrophic factor and trkB receptor immunoreactivity in glia of Alzheimer’s disease. Acta Neuropathol. 1999, 98, 345. [Google Scholar] [CrossRef] [PubMed]
- Ramu, V.; Eunhee, J.; Yeou, K.S. Phytochemicals That Regulate Neurodegenerative Disease by Targeting Neurotrophins: A Comprehensive Review. BioMed Res. Int. 2015, 2015, 814068. [Google Scholar] [CrossRef]
- Li, H.; Zheng, C.; Zheng, Y.; Wen, K.; Zhang, Y. Distinct functional diversity of branched oligosaccharides as chaperones and inhibitory-binding partners of amyloid beta-protein and its aggregates. Neuropharmacology 2024, 261, 110141. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Expression of Concern: Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef]
- Kabir, E.R.; Chowdhury, N.M.; Yasmin, H.; Kabir, M.T.; Akter, R.; Perveen, A.; Ashraf, G.M.; Akter, S.; Rahman, M.H.; Sweilam, S.H. Unveiling the Potential of Polyphenols as Anti-Amyloid Molecules in Alzheimer’s Disease. Curr. Neuropharmacol. 2023, 21, 787–807. [Google Scholar] [CrossRef]
- Gonalves, P.B.; Sodero, A.C.R.; Cordeiro, Y. Natural products targeting amyloid-β oligomer neurotoxicity in Alzheimer’s disease. Eur. J. Med. Chem. 2024, 276, 116684. [Google Scholar] [CrossRef]
- Shimazu, R.; Anada, M.; Miyaguchi, A.; Nomi, Y.; Matsumoto, H. Evaluation of Blood-Brain Barrier Permeability of Polyphenols, Anthocyanins, and Their Metabolites. J. Agric. Food Chem. 2021, 69, 11676–11686. [Google Scholar] [CrossRef]
- Acharya, A.; Stockmann, J.; Beyer, L.; Rudack, T.; Nabers, A.; Gumbart, J.C.; Gerwert, K.; Batista, V.S. The Effect of (−)-Epigallocatechin-3-Gallate on the Amyloid-β Secondary Structure. Biophys. J. 2020, 119, 349–359. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Liu, J.; Zhang, Y. The protective effects and underlying mechanism of an anti-oligomeric Aβ42 single-chain variable fragment antibody. Neuropharmacology 2015, 99, 387–395. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Huai, Y.; Zhang, Y.-J. Functional Characteristics and Molecular Mechanism of a New scFv Antibody Against Aβ42 Oligomers and Immature Protofibrils. Mol. Neurobiol. 2014, 52, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huai, Y.; Zhang, X.; Song, C.; Cai, J.; Zhang, Y. The Mode of Action of an Anti-Oligomeric Amyloid β-Protein Antibody Affects its Protective Efficacy. Neurotox. Res. 2018, 35, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huai, Y.; Cai, J.; Song, C.; Zhang, Y. Novel antibody against oligomeric amyloid-β: Insight into factors for effectively reducing the aggregation and cytotoxicity of amyloid-β aggregates. Int. Immunopharmacol. 2019, 67, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Tebar, A.; Dechant, G.; Barde, Y.A. Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron 1990, 4, 487–492. [Google Scholar] [CrossRef]
- Begum, A.; Aysegul, O.; Jens, A.; Kursad, G.; Sermin, G. Lithium protects against paraquat neurotoxicity by NRF2 activation and miR-34a inhibition in SH-SY5Y cells. Front. Cell. Neurosci. 2015, 9, 209. [Google Scholar] [CrossRef]
- Mao, X.-Y.; Zhou, H.-H.; Li, X.; Liu, Z.-Q. Huperzine A Alleviates Oxidative Glutamate Toxicity in Hippocampal HT22 Cells via Activating BDNF/TrkB-Dependent PI3K/Akt/mTOR Signaling Pathway. Cell. Mol. Neurobiol. 2016, 36, 915–925. [Google Scholar] [CrossRef]
- Banfield, M.J.; Naylor, R.L.; Robertson, A.G.; Allen, S.J.; Brady, R.L. Specificity in Trk receptor:neurotrophin interactions: The crystal structure of TrkB-d5 in complex with neurotrophin-4/5. Structure 2002, 9, 1191–1199. [Google Scholar] [CrossRef]
- Rodriguez-Tebar, A.; Barde, Y.A. Binding characteristics of brain-derived neurotrophic factor to its receptors on neurons from the chick embryo. J. Neurosci. 1988, 8, 3337–3342. [Google Scholar] [CrossRef]
- Huang, X.; Wang, J.; Cui, L.; Zou, X.; Zhang, Y. Recombinant GST-I-Aβ28-induced efficient serum antibody against Aβ42. J. Neurosci. Methods 2010, 186, 52–59. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2012, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. Neurotrophic Factors: An Overview. Methods Mol. Biol. 2018, 1727, 1–17. [Google Scholar] [CrossRef]
- Pinton, S.; Sampaio, T.B.; Savall, A.S.; Gutierrez, M.E.Z. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: Implications for pathogenesis and therapy. Neural Regen. Res. 2017, 4, 549–557. [Google Scholar] [CrossRef]
- Bahlakeh, G.; Rahbarghazi, R.; Abedelahi, A.; Sadigh-Eteghad, S.; Karimipour, M. Neurotrophic factor-secreting cells restored endogenous hippocampal neurogenesis through the Wnt/β-catenin signaling pathway in AD model mice. Stem Cell Res. Ther. 2022, 13, 343. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, 2016, 53193. [Google Scholar] [CrossRef]
- Azman, K.F.; Zakaria, R. Recent Advances on the Role of Brain-Derived Neurotrophic Factor (BDNF) in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 6827. [Google Scholar] [CrossRef]
- Josiane, B.; Tatiani, B.S.; Francielle, M.; Michelle, L.G.; Alexandra, I.Z. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331–341. [Google Scholar] [CrossRef]
- Qin, X.Y.; Cao, C.; Cawley, N.X.; Liu, T.T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N = 7277). Mol. Psychiatry 2017, 22, 312–320. [Google Scholar] [CrossRef]
- Amidfar, M.; Oliveira, J.D.; Kucharska, E.; Budni, J.; Kim, Y. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. 2020, 257, 118020. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.J.; Kim, K.J.; Choi, J.; Kang, D.H.; Lee, B.Y. Spirulina maxima extract prevents cell death through BDNF activation against amyloid beta 1-42 (Aβ 1-42) induced neurotoxicity in PC12 cells. Neurosci. Lett. 2018, 673, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Park, S.-Y.; Lee, S.-H.; Kim, Y.; Kim, Y.J.; Jun, W.; Yoon, H.G. Ameliorative Effects of Dendropanax morbifera on Cognitive Impairment via Enhancing Cholinergic Functions and Brain-Derived Neurotrophic Factor Expression in β-Amyloid-Induced Mice. J. Med. Food 2019, 22, 587–593. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aβ42 Species | In the Presence of a CA (or EGCG), b IG3, or c BCP | In the Absence of CA/EGCG/IG3/BCP |
|---|---|---|
| Aβ42M | a 4.91 ± 0.57 × 10−7 | 6.67 ± 0.26 × 10−7 |
| b 3.89 ± 0.43 × 10−7 | ||
| c 6.17 ± 0.13 × 10−7 | ||
| Aβ42O | a 3.36 ± 0.27 × 10−6 | 2.88 ± 0.33 × 10−7 |
| b 2.19 ± 0.12 × 10−6 | ||
| c 2.34 ± 0.28 × 10−5 |
| No. | Group | MOC | M1 | M2 | Receptor |
|---|---|---|---|---|---|
| 1 | Control | 0.622 ± 0.013 | 0.587 ± 0.004 | 0.611 ± 0.001 | TrkB |
| 2 | Control | 0.614 ± 0.007 | 0.556 ± 0.006 | 0.200 ± 0.003 | p75 |
| 3 | BDNF (or with BOs/CAs) | 0.778 ± 0.009 | 0.653 ± 0.002 | 0.638 ± 0.004 | TrkB |
| 4 | BDNF (or with BOs/CAs) | 0.654 ± 0.011 | 0.565 ± 0.004 | 0.208 ± 0.007 | p75 |
| 5 | AβM+BDNF+CA/EGCG | 0.714 ± 0.015 | 0.586 ± 0.006 | 0.668 ± 0.004 | TrkB |
| 6 | AβM+BDNF+BCP | 0.823 ± 0.009 | 0.592 ± 0.003 | 0.639 ± 0.002 | TrkB |
| 7 | AβM+BDNF+IG3 | 0.871 ± 0.012 | 0.597 ± 0.001 | 0.702 ± 0.005 | TrkB |
| 8 | AβM+BDNF+HT6 | 0.731 ± 0.005 | 0.593 ± 0.004 | 0.670 ± 0.003 | TrkB |
| 9 | AβM+BDNF | 0.721 ± 0.006 | 0.551 ± 0.007 | 0.650 ± 0.005 | TrkB |
| 10 | AβM+BDNF+CA/EGCG | 0.875 ± 0.010 | 0.598 ± 0.006 | 0.316 ± 0.002 | p75 |
| 11 | AβM+BDNF+BCP | 0.726 ± 0.007 | 0.583 ± 0.002 | 0.295 ± 0.003 | p75 |
| 12 | AβM+BDNF+IG3 | 0.746 ± 0.012 | 0.566 ± 0.005 | 0.309 ± 0.002 | p75 |
| 13 | AβM+BDNF+HT6 | 0.726 ± 0.012 | 0.534 ± 0.005 | 0.328 ± 0.003 | p75 |
| 14 | AβM+BDNF | 0.710 ± 0.005 | 0.536 ± 0.003 | 0.271 ± 0.002 | p75 |
| 15 | AβO+BDNF+ CA/EGCG | 0.676 ± 0.011 | 0.517 ± 0.005 | 0.523/0.581 ± 0.004 | TrkB |
| 16 | AβO+BDNF+BCP | 0.619 ± 0.012 | 0.623 ± 0.005 | 0.686 ± 0.004 | TrkB |
| 17 | AβO+BDNF+IG3 | 0.623 ± 0.011 | 0.557 ± 0.004 | 0.656 ± 0.003 | TrkB |
| 18 | AβO+BDNF+HT6 | 0.769 ± 0.015 | 0.550 ± 0.005 | 0.673 ± 0.004 | TrkB |
| 19 | AβO+BDNF | 0.617 ± 0.008 | 0.410 ± 0.003 | 0.572 ± 0.001 | TrkB |
| 20 | AβO+BDNF+CA/EGCG | 0.661 ± 0.009 | 0.524 ± 0.002 | 0.300 ± 0.002 | p75 |
| 21 | AβO+BDNF+BCP | 0.672 ± 0.006 | 0.533 ± 0.006 | 0.302 ± 0.003 | p75 |
| 22 | AβO+BDNF+IG3 | 0.712 ± 0.015 | 0.544 ± 0.004 | 0.327 ± 0.005 | p75 |
| 23 | AβO+BDNF+HT6 | 0.817 ± 0.011 | 0.561 ± 0.007 | 0.311 ± 0.003 | p75 |
| 24 | AβO+BDNF | 0.615 ± 0.007 | 0.485 ± 0.004 | 0.163 ± 0.002 | p75 |
| Group | Bait | BDNF | Aβ42M | Aβ42O | CA/ EGCG | IG3/ BCP | WCL | Co-IP | SFC |
|---|---|---|---|---|---|---|---|---|---|
| a | − | − | − | − | − | 100 | 44.07 ± 0.79 | 10.87 ± 1.09 | |
| d | TrkB | + | − | − | − | − | 243.90 ± 1.31 | 85.03 ± 0.58 | 63.22 ± 0.64 |
| f | + | + | − | − | − | 248.71 ± 1.51 | 88.33 ± 0.96 | 51.63 ± 0.74 | |
| g | + | + | − | + | − | 242.51 ± 0.89 | 89.27 ± 0.65 | 47.72 ± 0.88 | |
| h | + | + | − | − | + | 245.97 ± 1.37 | 92.01 ± 0.84 | 46.39 ± 0.73 | |
| l | + | − | + | − | − | 210.06 ± 1.08 | 58.12 ± 0.74 | 56.13 ± 0.61 | |
| m | + | − | + | + | − | 221.36 ± 0.95 | 72.18 ± 0.87 | 45.60 ± 0.72 | |
| n | + | − | + | − | + | 212.38 ± 1.35 | 73.44 ± 0.63 | 49.61 ± 0.76 | |
| b | − | − | − | − | − | 100 | 31.95 ± 1.04 | 12.57 ± 0.42 | |
| e | P75 | + | − | − | − | − | 243.90 ± 1.31 | 70.00 ± 0.69 | 22.81 ± 0.73 |
| i | + | + | − | − | − | 248.29 ± 1.31 | 71.88 ± 0.67 | 39.11 ± 0.78 | |
| j | + | + | − | + | − | 242.65 ± 1.26 | 73.40 ± 0.78 | 38.07 ± 0.91 | |
| k | + | + | − | − | + | 246.20 ± 1.53 | 83.23 ± 0.68 | 37.58 ± 0.73 | |
| o | + | − | + | − | − | 208.18 ± 1.38 | 64.28 ± 0.63 | 43.90 ± 0.54 | |
| p | + | − | + | + | − | 230.02 ± 1.32 | 62.07 ± 0.76 | 47.02 ± 0.62 | |
| q | + | − | + | − | + | 231.36 ± 0.62 | 61.68 ± 0.98 | 41.85 ± 0.81 |
| Group | Incubated Duration (h) | Anti-BDNF Antibody | BDNF | Aβ42M/ Aβ42O | CA/EGCG/ IG3/BCP |
|---|---|---|---|---|---|
| Ctrl0 | 2 | − | − | − | − |
| ① | 2 | + | − | − | − |
| ② | − | + | − | − | |
| ③/④ | − | − | + | − | |
| ⑤ | 0.5 + 1.5 | + | + | − | − |
| ⑥ | + | − | − | + | |
| ⑦/⑧ | + | − | + | − | |
| ⑨/⑩ | 2 | − | + | + | − |
| ⑪/⑫ | 0.5 + 1.5 | − | − | + premixture | |
| ⑬/⑭ | + | + | + | − | |
| ⑮/⑯ | + | − | + premixture | ||
| ⑰/⑱ | + | + | + premixture | ||
| ⑲/⑳ | − | + | + premixture | ||
| Group | Bait | Prey | BDNF | Aβ42M | Aβ42O | CA/EGCG/ IG3/BCP |
|---|---|---|---|---|---|---|
| a | TrkB | BDNF | − | − | − | − |
| d | + | − | − | − | ||
| f | + | + | − | − | ||
| g/h | + | + | − | + | ||
| l | + | − | + | − | ||
| m/n | + | − | + | + | ||
| b | p75 | − | − | − | − | |
| e | + | − | − | − | ||
| i | + | + | − | − | ||
| j/k | + | + | − | + | ||
| o | + | − | + | − | ||
| p/q | + | − | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Zheng, C.; Wen, K.; Zhang, T.; Zhang, Y. Neurotrophic and Neurotoxic Effects of Aβ42 and Its Oligomers on Neuronal Survival: Revealed by Their Opposite Influence on the Potency of Extracellular BDNF. Int. J. Mol. Sci. 2025, 26, 4501. https://doi.org/10.3390/ijms26104501
Li H, Zheng C, Wen K, Zhang T, Zhang Y. Neurotrophic and Neurotoxic Effects of Aβ42 and Its Oligomers on Neuronal Survival: Revealed by Their Opposite Influence on the Potency of Extracellular BDNF. International Journal of Molecular Sciences. 2025; 26(10):4501. https://doi.org/10.3390/ijms26104501
Chicago/Turabian StyleLi, He, Changxin Zheng, Kai Wen, Tianyu Zhang, and Yingjiu Zhang. 2025. "Neurotrophic and Neurotoxic Effects of Aβ42 and Its Oligomers on Neuronal Survival: Revealed by Their Opposite Influence on the Potency of Extracellular BDNF" International Journal of Molecular Sciences 26, no. 10: 4501. https://doi.org/10.3390/ijms26104501
APA StyleLi, H., Zheng, C., Wen, K., Zhang, T., & Zhang, Y. (2025). Neurotrophic and Neurotoxic Effects of Aβ42 and Its Oligomers on Neuronal Survival: Revealed by Their Opposite Influence on the Potency of Extracellular BDNF. International Journal of Molecular Sciences, 26(10), 4501. https://doi.org/10.3390/ijms26104501