
Identification and Characterization of a Rare Exon 22 Duplication in CFTR in Two Families

 and
and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Clinical Description
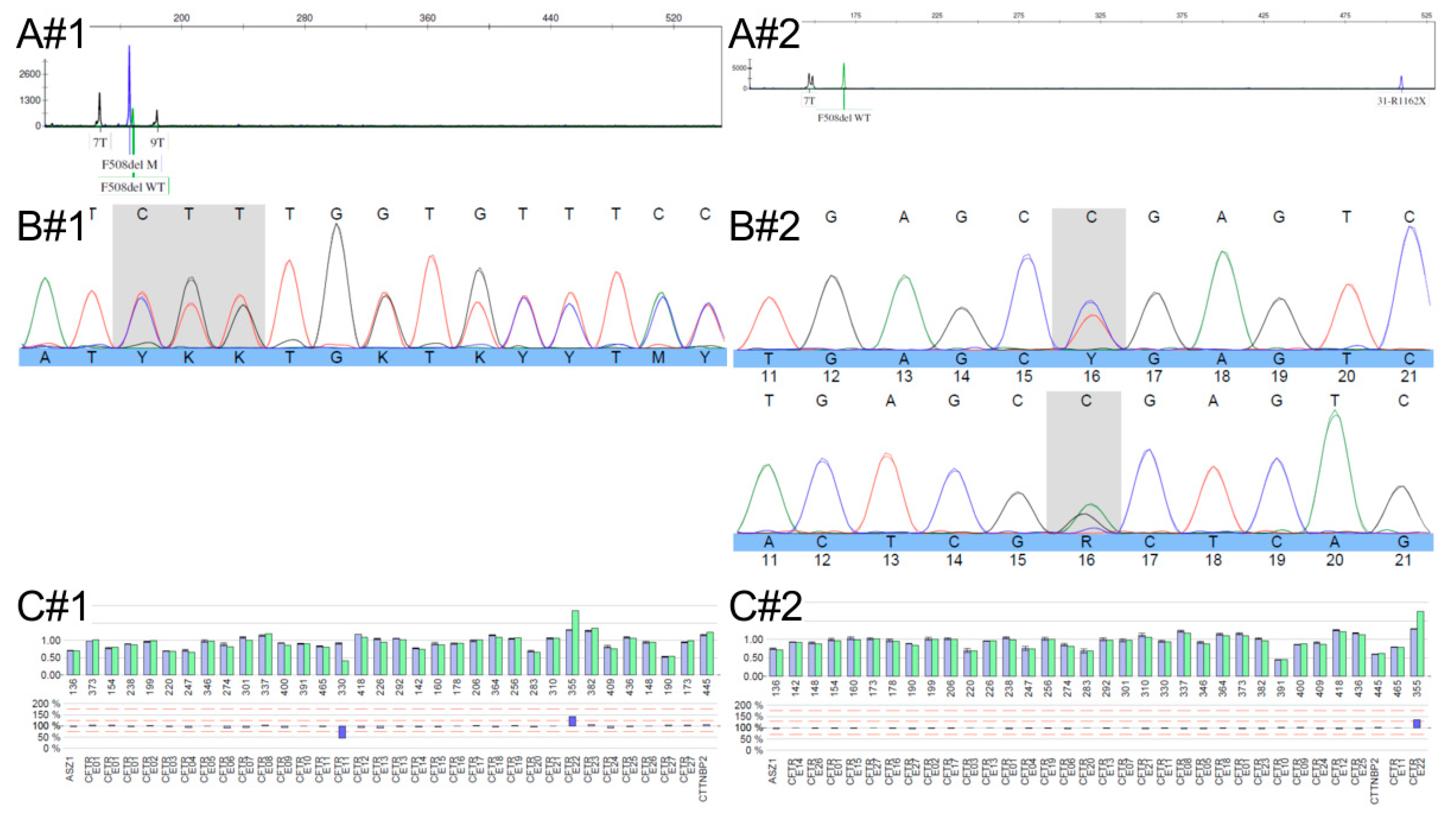
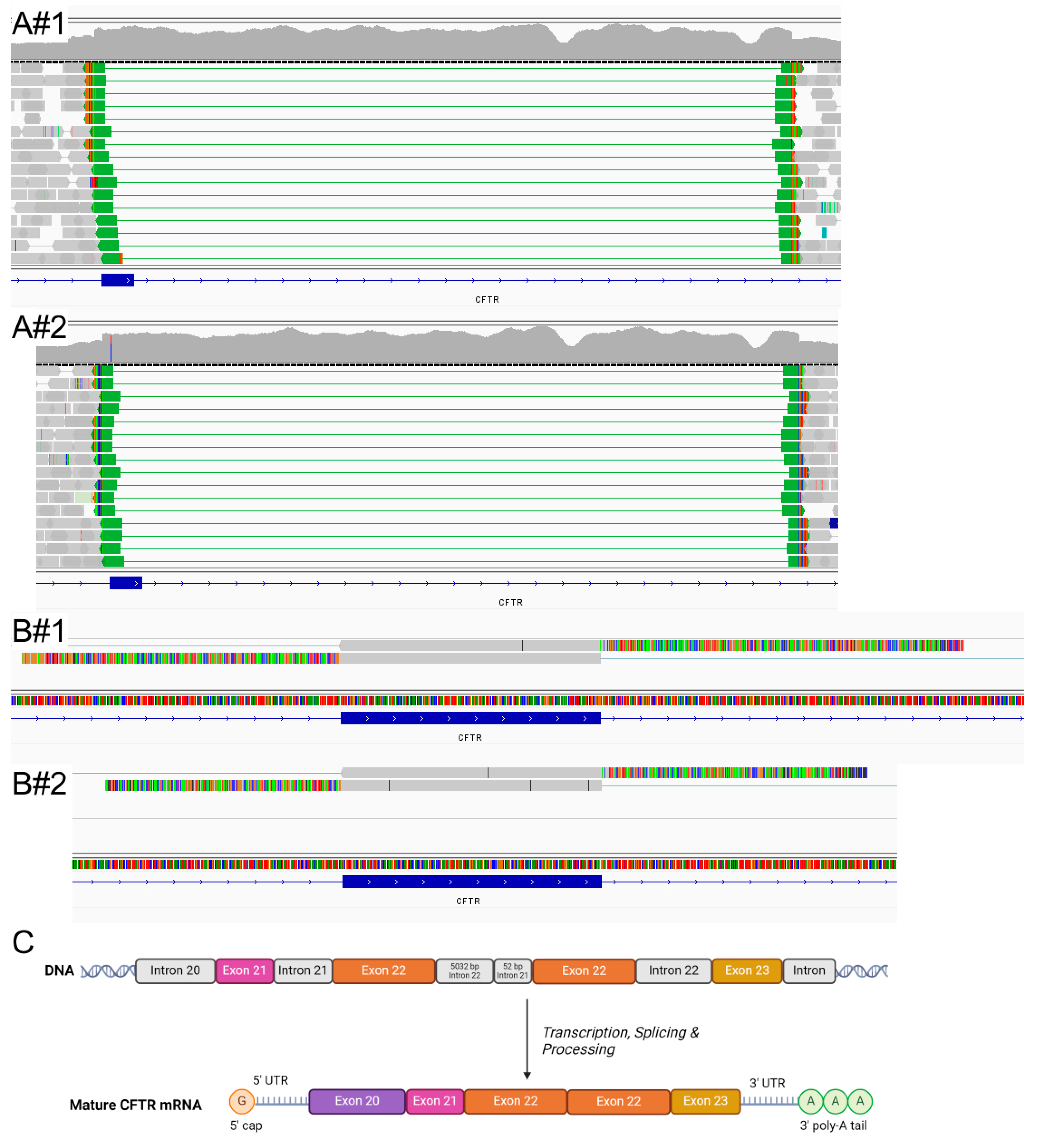
2.2. Molecular Analysis
3. Discussion
4. Materials and Methods
4.1. Ethics and Consent
4.2. CF-EU50
4.3. Sanger/MLPA
4.4. NGS
4.5. Variant Interpretation
4.6. RNA Analyses
4.7. Intestinal Current Measurement (ICM)
4.8. Multiple Breath Washout (MBW)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, S.; Hermetz, K.E.; Weckselblatt, B.; Rudd, M.K. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am. J. Hum. Genet. 2015, 96, 208–220. [Google Scholar] [CrossRef]
- Richardson, M.E.; Chong, H.; Mu, W.; Conner, B.R.; Hsuan, V.; Willett, S.; Lam, S.; Tsai, P.; Pesaran, T.; Chamberlin, A.C.; et al. DNA breakpoint assay reveals a majority of gross duplications occur in tandem reducing VUS classifications in breast cancer predisposition genes. Genet. Med. 2019, 21, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Da Martins, R.S.; Campos Junior, M.; Dos Santos Moreira, A.; Marques Zembrzuski, V.; Da Fonseca, A.C.P.; Abreu, G.d.M.; Cabello, P.H.; Cabello, G.M.K.d. Identification of a novel large deletion and other copy number variations in the CFTR gene in patients with Cystic Fibrosis from a multiethnic population. Mol. Genet. Genomic Med. 2019, 7, e00645. [Google Scholar] [CrossRef]
- Ahting, S.; Nährlich, L.; Held, I.; Henn, C.; Krill, A.; Landwehr, K.; Meister, J.; Nährig, S.; Nolde, A.; Remke, K.; et al. Every CFTR variant counts—Target-capture based next-generation-sequencing for molecular diagnosis in the German CF Registry. J. Cyst. Fibros. 2024, 23, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Sommerburg, O.; Yu, Y.; Berges, J.; Hirtz, S.; Scheuermann, H.; Berger, J.; Duerr, J.; Mall, M.A. Intestinal current measurement detects age-dependent differences in CFTR function in rectal epithelium. Front. Pharmacol. 2025, 16, 1537095. [Google Scholar] [CrossRef] [PubMed]
- Kopp, B.T.; Pastore, M.T.; Sturm, A.C.; Holtzlander, M.J.; Westman, J.A. A novel exon duplication of the cystic fibrosis transmembrane conductance regulator in a patient presenting with adult-onset recurrent pancreatitis. Pancreas 2011, 40, 773–777. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, gating, and regulation of the cftr anion channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef]
- Vernon, R.M.; Chong, P.A.; Lin, H.; Yang, Z.; Zhou, Q.; Aleksandrov, A.A.; Dawson, J.E.; Riordan, J.R.; Brouillette, C.G.; Thibodeau, P.H.; et al. Stabilization of a nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator yields insight into disease-causing mutations. J. Biol. Chem. 2017, 292, 14147–14164. [Google Scholar] [CrossRef]
- Rapino, D.; Sabirzhanova, I.; Lopes-Pacheco, M.; Grover, R.; Guggino, W.B.; Cebotaru, L. Rescue of NBD2 mutants N1303K and S1235R of CFTR by small-molecule correctors and transcomplementation. PLoS ONE 2015, 10, e0119796. [Google Scholar] [CrossRef]
- Erdoğan, M.; Köse, M.; Pekcan, S.; Hangül, M.; Balta, B.; Kiraz, A.; Akıncı Gönen, G.; Zamani, A.G.; Yıldırım, M.S.; Ramaslı Gürsoy, T.; et al. The Genetic Analysis of Cystic Fibrosis Patients With Seven Novel Mutations in the CFTR Gene in the Central Anatolian Region of Turkey. Balkan Med. J. 2021, 38, 357–364. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Vitzthum, C.; Mall, M.A. Potential of Intestinal Current Measurement for Personalized Treatment of Patients with Cystic Fibrosis. J. Pers. Med. 2021, 11, 384. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Johns Hopkins University; The Hospital for Sick Childre. The Clinical and Functional TRanslation of CFTR (CFTR2). Available online: http://cftr2.org (accessed on 22 April 2025).
- Claustres, M.; Thèze, C.; Des Georges, M.; Baux, D.; Girodon, E.; Bienvenu, T.; Audrezet, M.-P.; Dugueperoux, I.; Férec, C.; Lalau, G.; et al. CFTR-France, a national relational patient database for sharing genetic and phenotypic data associated with rare CFTR variants. Hum. Mutat. 2017, 38, 1297–1315. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Perez, G.; Barber, G.P.; Benet-Pages, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, C.M.; et al. The UCSC Genome Browser database: 2025 update. Nucleic Acids Res. 2024, 53, D1243–D1249. [Google Scholar] [CrossRef]
- Hirtz, S.; Gonska, T.; Seydewitz, H.H.; Thomas, J.; Greiner, P.; Kuehr, J.; Brandis, M.; Eichler, I.; Rocha, H.; Lopes, A.-I.; et al. CFTR Cl- channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology 2004, 127, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Graeber, S.Y.; Vitzthum, C.; Pallenberg, S.T.; Naehrlich, L.; Stahl, M.; Rohrbach, A.; Drescher, M.; Minso, R.; Ringshausen, F.C.; Rueckes-Nilges, C.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on CFTR Function in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2022, 205, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Wielpütz, M.O.; Graeber, S.Y.; Joachim, C.; Sommerburg, O.; Kauczor, H.-U.; Puderbach, M.; Eichinger, M.; Mall, M.A. Comparison of Lung Clearance Index and Magnetic Resonance Imaging for Assessment of Lung Disease in Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 349–359. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Renz, D.M.; Stahl, M.; Pallenberg, S.T.; Sommerburg, O.; Naehrlich, L.; Berges, J.; Dohna, M.; Ringshausen, F.C.; Doellinger, F.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on Lung Clearance Index and Magnetic Resonance Imaging in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2022, 206, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Dohna, M.; Graeber, S.Y.; Sommerburg, O.; Renz, D.M.; Pallenberg, S.T.; Voskrebenzev, A.; Schütz, K.; Hansen, G.; Doellinger, F.; et al. Impact of elexacaftor/tezacaftor/ivacaftor therapy on lung clearance index and magnetic resonance imaging in children with cystic fibrosis and one or two F508del alleles. Eur. Respir. J. 2024, 64, 2400004. [Google Scholar] [CrossRef]
- Wyler, F.; Oestreich, M.-A.; Frauchiger, B.S.; Ramsey, K.A.; Latzin, P. Correction of sensor crosstalk error in Exhalyzer D multiple-breath washout device significantly impacts outcomes in children with cystic fibrosis. J. Appl. Physiol. 2021, 131, 1148–1156. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahting, S.; Henn, C.; vom Hove, M.; Strehlow, V.; Duffek, P.; Behrendt, S.; Drukewitz, S.; Berger, J.; Graeber, S.Y.; Hentschel, J. Identification and Characterization of a Rare Exon 22 Duplication in CFTR in Two Families. Int. J. Mol. Sci. 2025, 26, 4487. https://doi.org/10.3390/ijms26104487
Ahting S, Henn C, vom Hove M, Strehlow V, Duffek P, Behrendt S, Drukewitz S, Berger J, Graeber SY, Hentschel J. Identification and Characterization of a Rare Exon 22 Duplication in CFTR in Two Families. International Journal of Molecular Sciences. 2025; 26(10):4487. https://doi.org/10.3390/ijms26104487
Chicago/Turabian StyleAhting, Simone, Constance Henn, Maike vom Hove, Vincent Strehlow, Patricia Duffek, Sophie Behrendt, Stephan Drukewitz, Jasmin Berger, Simon Y. Graeber, and Julia Hentschel. 2025. "Identification and Characterization of a Rare Exon 22 Duplication in CFTR in Two Families" International Journal of Molecular Sciences 26, no. 10: 4487. https://doi.org/10.3390/ijms26104487
APA StyleAhting, S., Henn, C., vom Hove, M., Strehlow, V., Duffek, P., Behrendt, S., Drukewitz, S., Berger, J., Graeber, S. Y., & Hentschel, J. (2025). Identification and Characterization of a Rare Exon 22 Duplication in CFTR in Two Families. International Journal of Molecular Sciences, 26(10), 4487. https://doi.org/10.3390/ijms26104487