
The Unexplored Role of Connexin Hemichannels in Promoting Facioscapulohumeral Muscular Dystrophy Progression



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
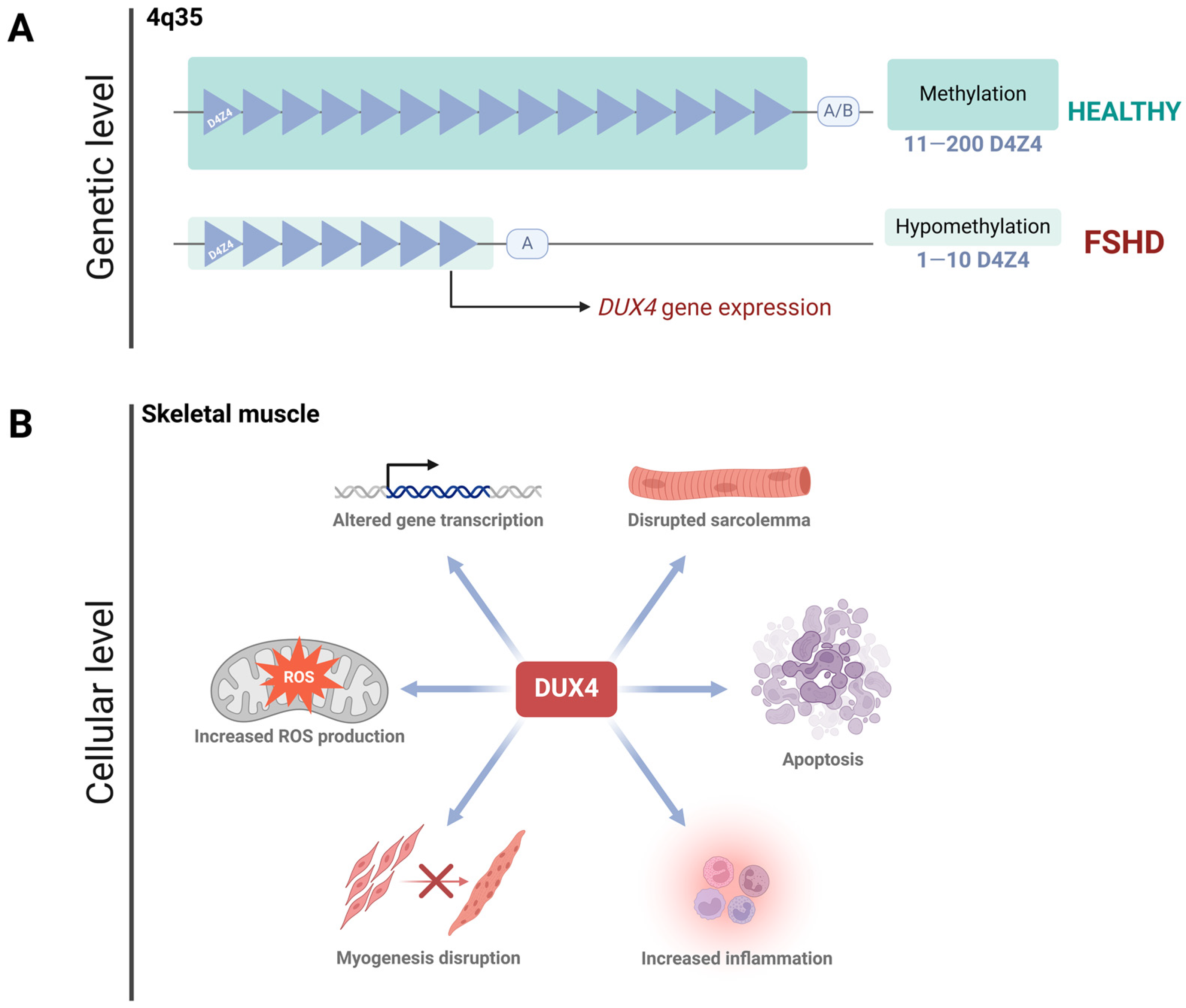
2. Facioscapulohumeral Muscular Dystrophy
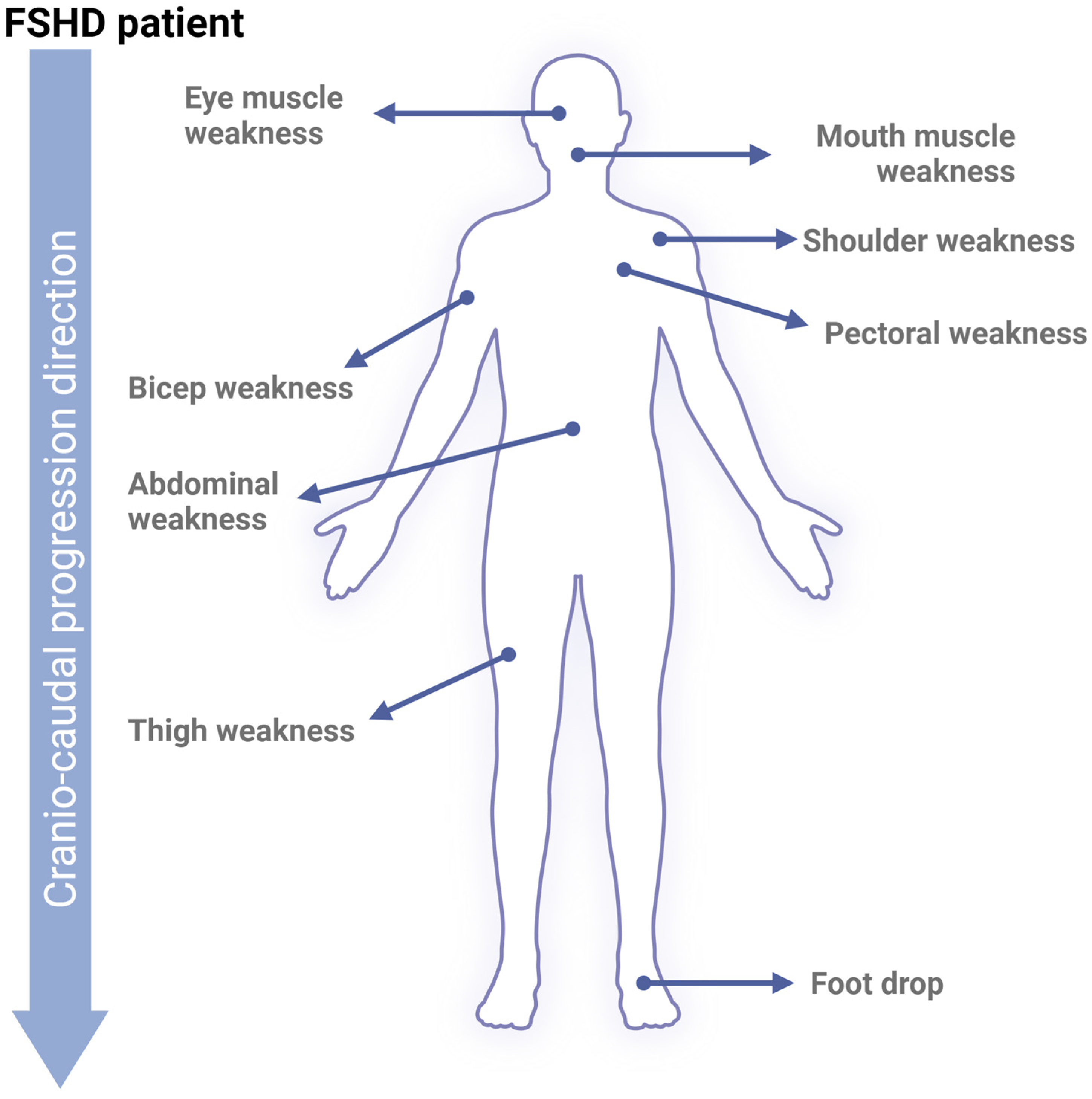
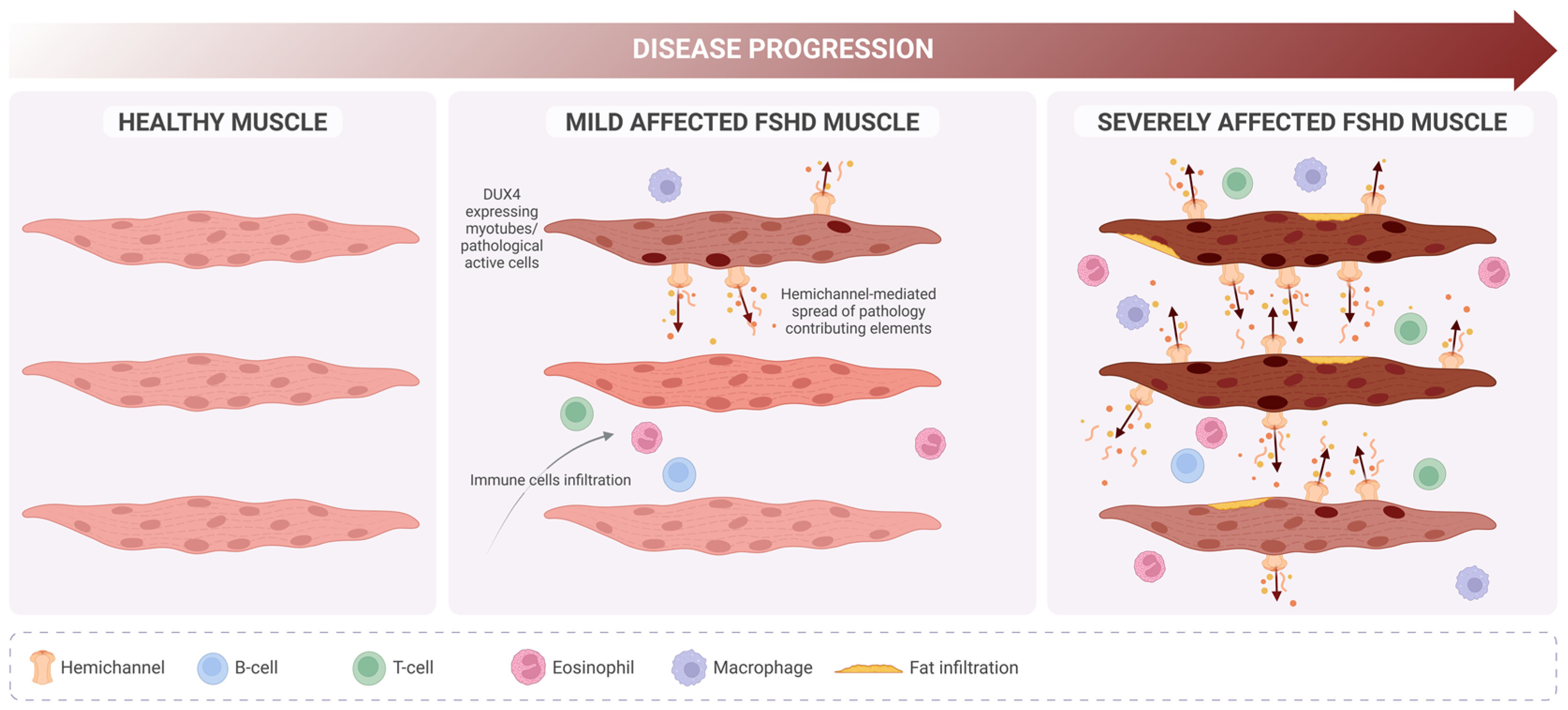
FSHD Progression
3. Connexin Hemichannels
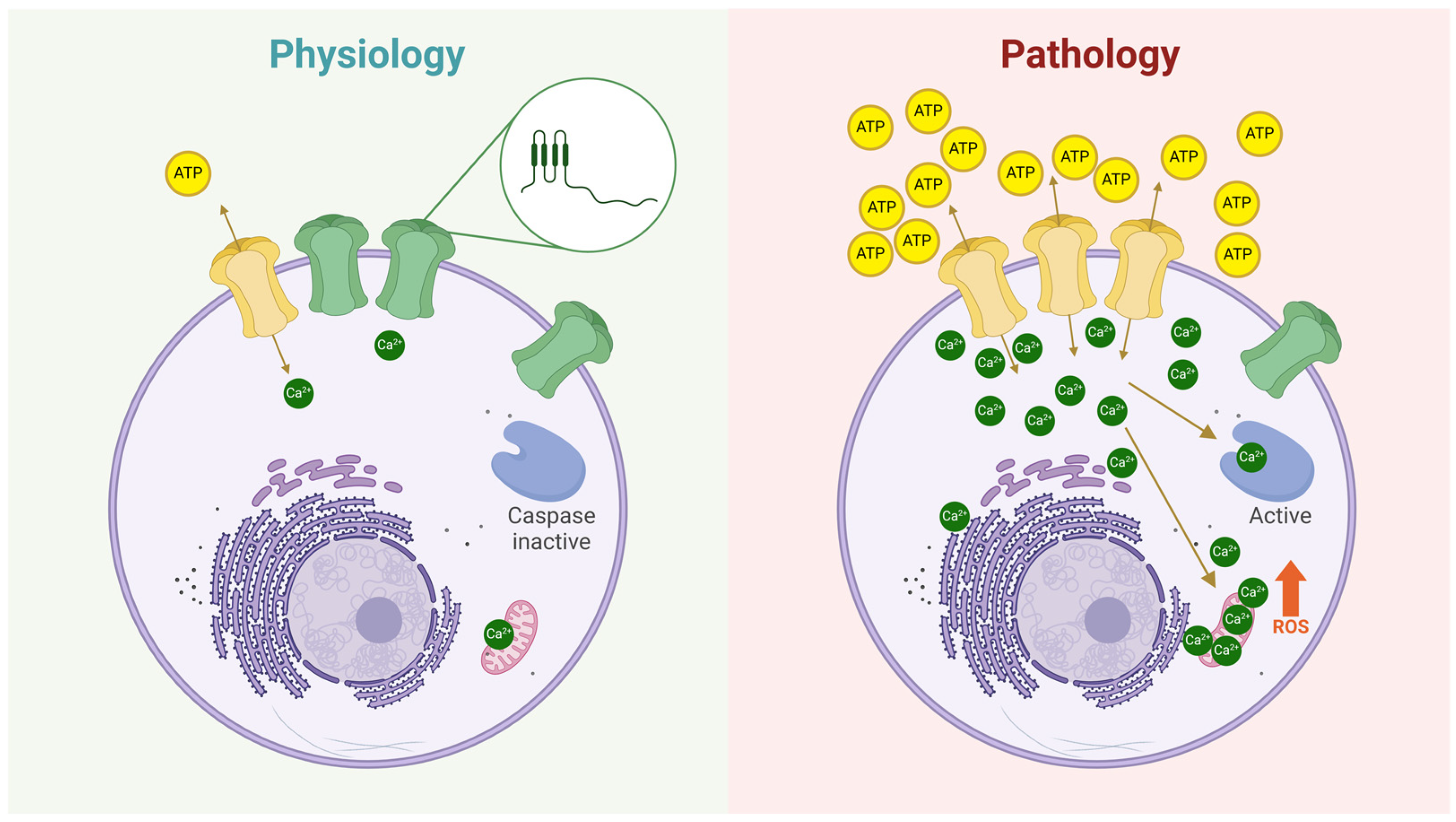
Hemichannels in Skeletal Muscle Atrophy
4. Targeting Hemichannels: A Novel Therapeutic Avenue for FSHD
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tawil, R.; McDermott, M.P.; Mendell, J.R.; Kissel, J.; Griggs, R.C.; FSH-DY Group. Facioscapulohumeral muscular dystrophy (FSHD): Design of natural history study and results of baseline testing. Neurology 1994, 44, 442. [Google Scholar] [CrossRef]
- Zampatti, S.; Colantoni, L.; Strafella, C.; Galota, R.M.; Caputo, V.; Campoli, G.; Pagliaroli, G.; Carboni, S.; Mela, J.; Peconi, C.; et al. Facioscapulohumeral muscular dystrophy (FSHD) molecular diagnosis: From traditional technology to the NGS era. Neurogenetics 2019, 20, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Salviati, L.; Desnuelle, C. Facioscapulohumeral muscular dystrophy. Biochim. Biophys. Acta 2015, 1852, 607–614. [Google Scholar] [CrossRef]
- Tawil, R.; Mah, J.K.; Baker, S.; Wagner, K.R.; Ryan, M.M.; Corbett, A.; McNamara, S.; van Engelen, B.; Rasko, J.; Raykar, V.; et al. Clinical practice considerations in facioscapulohumeral muscular dystrophy Sydney, Australia, 21 September 2015. Neuromuscul. Disord. 2016, 26, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Tawil, R.; McDermott, M.; Forrester, J.; Figlewicz, D.; Weiffenbach, B. Monozygotic twins with facioscapulohumeral dystrophy (FSHD): Implications for genotype/phenotype correlation. Muscle Nerve 1995, 18, S50–S55. [Google Scholar] [CrossRef]
- Tupler, R.; Barbierato, L.; Memmi, M.; Sewry, C.A.; De Grandis, D.; Maraschio, P.; Tiepolo, L.; Ferlini, A. Identical de novo mutation at the D4F104S1 locus in monozygotic male twins affected by facioscapulohumeral muscular dystrophy (FSHD) with different clinical expression. J. Med. Genet. 1998, 35, 778–783. [Google Scholar] [CrossRef]
- Banerji, C.R.S. PAX7 target gene repression associates with FSHD progression and pathology over 1 year. Hum. Mol. Genet. 2020, 29, 2124–2133. [Google Scholar] [CrossRef]
- Magdinier, F. Joining mainstream research on Facioscapulohumeral Dystophy: Disease prevalence in China. Lancet Reg. Health West. Pac. 2021, 18, 100328. [Google Scholar] [CrossRef]
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.W.; Mercier, J.; Coppée, F.; Belayew, A. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013, 17, 76–89. [Google Scholar] [CrossRef]
- Snider, L.; Geng, L.N.; Lemmers, R.J.L.F.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181. [Google Scholar] [CrossRef]
- Retamal, M.A.; Sáez, J.C. (Eds.) Hemichannels; From the Molecule to the Function; Frontiers Media SA: Lausanne, Switzerland, 2014; Volume 5. [Google Scholar] [CrossRef]
- Delvaeye, T.; Vandenabeele, P.; Bultynck, G.; Leybaert, L.; Krysko, D.V. Therapeutic Targeting of Connexin Channels: New Views and Challenges. Trends Mol. Med. 2018, 24, 1036–1053. [Google Scholar] [CrossRef]
- Sáez, J.C.; Retamal, M.A.; Basilio, D.; Bukauskas, F.F.; Bennett, M.V.L. Connexin-based gap junction hemichannels: Gating mechanisms. Biochim. Biophys. Acta-Biomembr. 2005, 1711, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Puebla, C.; Cisterna, B.A.; Escamilla, R.; Vargas, A.A.; Frank, M.; Martínez-Montero, P.; Prior, C.; Molano, J.; Esteban-Rodríguez, I.; et al. Fast skeletal myofibers of mdx mouse, model of Duchenne muscular dystrophy, express connexin hemichannels that lead to apoptosis. Cell. Mol. Life Sci. 2016, 73, 2583–2599. [Google Scholar] [CrossRef]
- Cea, L.A.; Bevilacqua, J.A.; Arriagada, C.; Cárdenas, A.M.; Bigot, A.; Mouly, V.; Sáez, J.C.; Caviedes, P. The absence of dysferlin induces the expression of functional connexin-based hemichannels in human myotubes. BMC Cell Biol. 2016, 17 (Suppl. 1), 127–136. [Google Scholar] [CrossRef] [PubMed]
- Duranti, E.; Villa, C. Influence of DUX4 Expression in Facioscapulohumeral Muscular Dystrophy and Possible Treatments. Int. J. Mol. Sci. 2023, 24, 9503. [Google Scholar] [CrossRef]
- Teveroni, E.; Pellegrino, M.; Sacconi, S.; Calandra, P.; Cascino, I.; Farioli-Vecchioli, S.; Puma, A.; Garibaldi, M.; Morosetti, R.; Tasca, G.; et al. Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J. Clin. Investig. 2017, 127, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Tonini, M.M.O.; Passos-Bueno, M.R.; Cerqueira, A.; Matioli, S.R.; Pavanello, R.; Zatz, M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul. Disord. 2004, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Schätzl, T.; Kaiser, L.; Deigner, H.P. Facioscapulohumeral muscular dystrophy: Genetics, gene activation and downstream signalling with regard to recent therapeutic approaches: An update. Orphanet J. Rare Dis. 2021, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.T.; Wljmenga, C.; Van Tlenhoven, E.A.E.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.B.; Hofker, M.H.; Fronts, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef]
- Lemmers, R.J.L.F.; Wohlgemuth, M.; Van Der Gaag, K.J.; Van Der Vliet, P.J.; Van Teijlingen, C.M.M.; De Knijff, P.; Padberg, G.W.; Frants, R.R.; Van Der Maarel, S.M. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2007, 81, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Gatica, L.V.; Rosa, A.L. A complex interplay of genetic and epigenetic events leads to abnormal expression of the DUX4 gene in facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2016, 26, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Van Overveld, P.G.M.; Lemmers, R.J.F.L.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; Van Ommen, G.J.B.; Frants, R.R.; Van Der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003, 35, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.M.; Garwick, S.E.; Mei, W.; Belayew, A.; Coppee, F.; Ladner, K.J.; Guttridge, D.; Yang, J.; Harper, S.Q. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 2011, 69, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Corona, E.D.; Jacquelin, D.; Gatica, L.; Rosa, A.L. Multiple protein domains contribute to nuclear import and cell toxicity of DUX4, a candidate pathogenic protein for facioscapulohumeral muscular dystrophy. PLoS ONE 2013, 8, e75614. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 729. [Google Scholar] [CrossRef]
- Zeng, W.; De Greef, J.C.; Chen, Y.Y.; Chien, R.; Kong, X.; Gregson, H.C.; Winokur, S.T.; Pyle, A.; Robertson, K.D.; Schmiesing, J.A.; et al. Specific Loss of Histone H3 Lysine 9 Trimethylation and HP1γ/Cohesin Binding at D4Z4 Repeats Is Associated with Facioscapulohumeral Dystrophy (FSHD). PLoS Genet. 2009, 5, e1000559. [Google Scholar] [CrossRef] [PubMed]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A Long ncRNA Links Copy Number Variation to a Polycomb/Trithorax Epigenetic Switch in FSHD Muscular Dystrophy. Cell 2012, 149, 819. [Google Scholar] [CrossRef]
- Vizoso, M.; Esteller, M. The activatory long non-coding RNA DBE-T reveals the epigenetic etiology of facioscapulohumeral muscular dystrophy. Cell Res. 2012, 22, 1413–1415. [Google Scholar] [CrossRef]
- Himeda, C.L.; Jones, T.I.; Virbasius, C.M.; Zhu, L.J.; Green, M.R.; Jones, P.L. Identification of Epigenetic Regulators of DUX4-fl for Targeted Therapy of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. 2018, 26, 1797. [Google Scholar] [CrossRef] [PubMed]
- Resnick, R.; Wong, C.J.; Hamm, D.C.; Bennett, S.R.; Skene, P.J.; Hake, S.B.; Henikoff, S.; van der Maarel, S.M.; Tapscott, S.J. DUX4-Induced Histone Variants H3.X and H3.Y Mark DUX4 Target Genes for Expression. Cell Rep. 2019, 29, 1812. [Google Scholar] [CrossRef] [PubMed]
- Kujirai, T.; Horikoshi, N.; Sato, K.; Maehara, K.; Machida, S.; Osakabe, A.; Kimura, H.; Ohkawa, Y.; Kurumizaka, H. Structure and function of human histone H3.Y nucleosome. Nucleic Acids Res. 2016, 44, 6127. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, S. The evolution of DUX4 gene regulation and its implication for facioscapulohumeral muscular dystrophy. Biochim. Biophys. Acta-Mol. Basis Dis. 2022, 1868, 166367. [Google Scholar] [CrossRef] [PubMed]
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017, 49, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Vuoristo, S.; Bhagat, S.; Hydén-Granskog, C.; Yoshihara, M.; Gawriyski, L.; Jouhilahti, E.M.; Ranga, V.; Tamirat, M.; Huhtala, M.; Kirjanov, I.; et al. DUX4 is a multifunctional factor priming human embryonic genome activation. iScience 2022, 25, 104137. [Google Scholar] [CrossRef]
- Banerji, C.R.S.; Zammit, P.S. PAX7 target gene repression is a superior FSHD biomarker than DUX4 target gene activation, associating with pathological severity and identifying FSHD at the single-cell level. Hum. Mol. Genet. 2019, 28, 2224–2236. [Google Scholar] [CrossRef] [PubMed]
- Sasaki-Honda, M.; Jonouchi, T.; Arai, M.; Hotta, A.; Mitsuhashi, S.; Nishino, I.; Matsuda, R.; Sakurai, H. A patient-derived iPSC model revealed oxidative stress increases facioscapulohumeral muscular dystrophy-causative DUX4. Hum. Mol. Genet. 2018, 27, 4024–4035. [Google Scholar] [CrossRef] [PubMed]
- Kowaljow, V.; Marcowycz, A.; Ansseau, E.; Conde, C.B.; Sauvage, S.; Mattéotti, C.; Arias, C.; Corona, E.D.; Nuñez, N.G.; Leo, O.; et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007, 17, 611–623. [Google Scholar] [CrossRef]
- Banerji, C.R.S.; Zammit, P.S. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: Roles of DUX4 and PAX7. EMBO Mol. Med. 2021, 13, e13695. [Google Scholar] [CrossRef] [PubMed]
- Lassche, S.; Ottenheijm, C.A.C.; Voermans, N.C.; Westeneng, H.J.; Janssen, B.H.; van der Maarel, S.M.; Hopman, M.T.; Padberg, G.W.; Stienen, G.J.M.; van Engelen, B.G.M. Determining the role of sarcomeric proteins in facioscapulohumeral muscular dystrophy: A study protocol. BMC Neurol. 2013, 13, 144. [Google Scholar] [CrossRef]
- Zernov, N.; Skoblov, M. Genotype-phenotype correlations in FSHD. BMC Med. Genom. 2019, 12, 43. [Google Scholar] [CrossRef]
- Banerji, C.R.S.; Panamarova, M.; Pruller, J.; Figeac, N.; Hebaishi, H.; Fidanis, E.; Saxena, A.; Contet, J.; Sacconi, S.; Severini, S.; et al. Dynamic transcriptomic analysis reveals suppression of PGC1α/ERRα drives perturbed myogenesis in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2019, 28, 1244–1259. [Google Scholar] [CrossRef] [PubMed]
- Hiramuki, Y.; Kure, Y.; Saito, Y.; Ogawa, M.; Ishikawa, K.; Mori-Yoshimura, M.; Oya, Y.; Takahashi, Y.; Kim, D.S.; Arai, N.; et al. Simultaneous measurement of the size and methylation of chromosome 4qA-D4Z4 repeats in facioscapulohumeral muscular dystrophy by long-read sequencing. J. Transl. Med. 2022, 20, 517. [Google Scholar] [CrossRef]
- Gould, T.; Jones, T.I.; Jones, P.L. Precise Epigenetic Analysis Using Targeted Bisulfite Genomic Sequencing Distinguishes FSHD1, FSHD2, and Healthy Subjects. Diagnostics 2021, 11, 1469. [Google Scholar] [CrossRef] [PubMed]
- Balog, J.; Thijssen, P.E.; Shadle, S.; Straasheijm, K.R.; van der Vliet, P.J.; Krom, Y.D.; van den Boogaard, M.L.; de Jong, A.; F Lemmers, R.J.L.; Tawil, R.; et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics 2015, 10, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Nykänen, S.; Vuoristo, S. DUX4, the rockstar of embryonic genome activation? Int. J. Dev. Biol. 2024, in press. [Google Scholar] [CrossRef] [PubMed]
- Ferreboeuf, M.; Mariot, V.; Furling, D.; Butler-Browne, G.; Mouly, V.; Dumonceaux, J. Nuclear protein spreading: Implication for pathophysiology of neuromuscular diseases. Hum. Mol. Genet. 2014, 23, 4125–4133. [Google Scholar] [CrossRef]
- Rickard, A.M.; Petek, L.M.; Miller, D.G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015, 24, 5901–5914. [Google Scholar] [CrossRef]
- Hangul, C.; Celik, E.G.; Kaya, H.; Eroglu, O.; Uysal, H.; Karauzum, S.B. Estradiol differentially regulates DUX4, β-catenin and PAX3/PAX7 in primary myoblasts of facioscapulohumeral muscular dystrophy patients. Turk. J. Biochem. 2021, 46, 435–444. [Google Scholar] [CrossRef]
- Hangül, C.; Bozkurt, S.; Bilge, U.; Özdem, S.; Altunbaş, H.; Uysal, H.; Koç, F.; Karaüzüm, S.B. The ratios of estradiol and progesterone to testosterone influence the severity of facioscapulohumeral muscular dystrophy. Neurol. Sci. Neurophysiol. 2020, 37, 190–196. [Google Scholar] [CrossRef]
- Banerji, C.R.S.; Panamarova, M.; Zammit, P.S. DUX4 expressing immortalized FSHD lymphoblastoid cells express genes elevated in FSHD muscle biopsies, correlating with the early stages of inflammation. Hum. Mol. Genet. 2020, 29, 2285–2299. [Google Scholar] [CrossRef]
- Hauerslev, S.; Ørngreen, M.C.; Hertz, J.M.; Vissing, J.; Krag, T.O. Muscle regeneration and inflammation in patients with facioscapulohumeral muscular dystrophy. Acta Neurol. Scand. 2013, 128, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Dahlqvist, J.R.; Andersen, G.; Khawajazada, T.; Vissing, C.; Thomsen, C.; Vissing, J. Relationship between muscle inflammation and fat replacement assessed by MRI in facioscapulohumeral muscular dystrophy. J. Neurol. 2019, 266, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Arahata, K.; Ishihara, T.; Fukunaga, H.; Orimo, S.; Lee, J.H.; Goto, K.; Nonaka, I. Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): Immunocytochemical and genetic analyses. Muscle Nerve 1995, 18, S56–S66. [Google Scholar] [CrossRef]
- van den Heuvel, A.; Lassche, S.; Mul, K.; Greco, A.; San León Granado, D.; Heerschap, A.; Küsters, B.; Tapscott, S.J.; Voermans, N.C.; van Engelen, B.G.M.; et al. Facioscapulohumeral dystrophy transcriptome signatures correlate with different stages of disease and are marked by different MRI biomarkers. Sci. Rep. 2022, 12, 1426. [Google Scholar] [CrossRef]
- Friedman, S.D.; Poliachik, S.L.; Carter, G.T.; Budech, C.B.; Bird, T.D.; Shaw, D.W.W. The magnetic resonance imaging spectrum of facioscapulohumeral muscular dystrophy. Muscle Nerve 2012, 45, 500–506. [Google Scholar] [CrossRef]
- Dahlqvist, J.R.; Poulsen, N.S.; Østergaard, S.T.; Fornander, F.; De Stricker Borch, J.; Danielsen, E.R.; Thomsen, C.; Vissing, J. Evaluation of inflammatory lesions over 2 years in facioscapulohumeral muscular dystrophy. Neurology 2020, 95, E1211–E1221. [Google Scholar] [CrossRef] [PubMed]
- Frisullo, G.; Frusciante, R.; Nociti, V.; Tasca, G.; Renna, R.; Iorio, R.; Patanella, A.K.; Iannaccone, E.; Marti, A.; Rossi, M.; et al. CD8+ T cells in facioscapulohumeral muscular dystrophy patients with inflammatory features at muscle MRI. J. Clin. Immunol. 2011, 31, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Nunes, A.M.; Daoudlarian, D.; Pini, J.; Martinuzzi, E.; Barbosa, S.; Ramirez, M.; Puma, A.; Villa, L.; Cavalli, M.; et al. Identification of Serum Interleukin 6 Levels as a Disease Severity Biomarker in Facioscapulohumeral Muscular Dystrophy. J. Neuromuscul. Dis. 2022, 9, 83–93. [Google Scholar] [CrossRef]
- Wosiski-Kuhn, M.; Caress, J.B.; Cartwright, M.S.; Hawkins, G.A.; Milligan, C. Interleukin 6 (IL6) level is a biomarker for functional disease progression within IL6R358Ala variant groups in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, P.; Bou Saada, Y.; Dib, C.; Ansseau, E.; Barat, A.; Hamade, A.; Dessen, P.; Robert, T.; Lazar, V.; Louzada, R.A.N.; et al. DUX4-induced constitutive DNA damage and oxidative stress contribute to aberrant differentiation of myoblasts from FSHD patients. Free Radic. Biol. Med. 2016, 99, 244–258. [Google Scholar] [CrossRef]
- Passerieux, E.; Hayot, M.; Jaussent, A.; Carnac, G.; Gouzi, F.; Pillard, F.; Picot, M.C.; Böcker, K.; Hugon, G.; Pincemail, J.; et al. Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: A double-blind randomized controlled clinical trial. Free Radic. Biol. Med. 2015, 81, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Söhl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 2009, 458, 597–602. [Google Scholar] [CrossRef]
- Koval, M. Pathways and control of connexin oligomerization. Trends Cell Biol. 2006, 16, 159–166. [Google Scholar] [CrossRef]
- Thomas, T.; Jordan, K.; Simek, J.; Shao, Q.; Jedeszko, C.; Walton, P.; Laird, D.W. Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J. Cell Sci. 2005, 118, 4451–4462. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.-J.; Liu, X.-Z.; Tu, L.; Sun, Y. Cytomembrane Trafficking Pathways of Connexin 26, 30, and 43. Int. J. Mol. Sci. 2023, 24, 10349. [Google Scholar] [CrossRef]
- Li, F.; Sugishita, K.; Su, Z.; Ueda, I.; Barry, W.H. Activation of connexin-43 hemichannels can elevate [Ca2+]i and [Na+]i in rabbit ventricular myocytes during metabolic inhibition. J. Mol. Cell. Cardiol. 2001, 33, 2145–2155. [Google Scholar] [CrossRef]
- Schalper, K.A.; Sánchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Sáez, J.C. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.C.; Figueroa, V.; Zoghbi, M.E.; Saéz, J.C.; Reuss, L.; Altenberg, G.A. Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem. 2012, 287, 40826–40834. [Google Scholar] [CrossRef] [PubMed]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.G.; Charles, A.C. Intercellular Calcium Signaling in Astrocytes via ATP Release through Connexin Hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Riquelme, M.A.; Gu, S.; Jiang, J.X. Connexin hemichannels mediate glutathione transport and protect lens fiber cells from oxidative stress. J. Cell Sci. 2018, 131, jcs212506. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.-C.; Wyeth, M.S.; Baltan-Tekkok, S.; Ransom, B.R. Functional hemichannels in astrocytes: A novel mechanism of glutamate release. J. Neurosci. 2003, 23, 3588–3596. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 Hemichannels and Gap Junction Channels in Astrocytes Are Regulated Oppositely by Proinflammatory Cytokines Released from Activated Microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.E.; Saez, J.C.; Bukauskas, F.F.; Bennett, M.V.L. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad. Sci. USA 2003, 100, 11388–11393. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.T.; Poudyal, N.; Verdugo, D.A.; Peña, F.; Stehberg, J.; Retamal, M.A. KI04 an Aminoglycosides-Derived Molecule Acts as an Inhibitor of Human Connexin46 Hemichannels Expressed in HeLa Cells. Biomolecules 2023, 13, 411. [Google Scholar] [CrossRef]
- Gaete, P.S.; Kumar, D.; Fernandez, C.I.; Valdez Capuccino, J.M.; Bhatt, A.; Jiang, W.; Lin, Y.-C.; Liu, Y.; Harris, A.L.; Luo, Y.L.; et al. Large-pore connexin hemichannels function like molecule transporters independent of ion conduction. Proc. Natl. Acad. Sci. USA 2024, 121, e2403903121. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Reyes, E.P.; García, I.E.; Pinto, B.; Martínez, A.D.; González, C. Diseases associated with leaky hemichannels. Front. Cell. Neurosci. 2015, 9, 267. [Google Scholar] [CrossRef]
- Liang, G.S.L.L.; De Miguel, M.; Gómez-Hernández, J.M.; Glass, J.D.; Scherer, S.S.; Mintz, M.; Barrio, L.C.; Fischbeck, K.H. Severe neuropathy with leaky connexin32 hemichannels. Ann. Neurol. 2005, 57, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, H.A.; Meşe, G.; Srinivas, M.; White, T.W.; Verselis, V.K. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J. Gen. Physiol. 2010, 136, 47–62. [Google Scholar] [CrossRef]
- Saez, J.; Berthoud, V.; Branes, M.; Martinez, A.; Beyer, E. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sinovas, A.; Sánchez, J.A.; Valls-Lacalle, L.; Consegal, M.; Ferreira-González, I. Connexins in the Heart: Regulation, Function and Involvement in Cardiac Disease. Int. J. Mol. Sci. 2021, 22, 4413. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Rohrbach, S.; Weissmann, N.; Schulz, R. Importance of Cx43 for Right Ventricular Function. Int. J. Mol. Sci. 2021, 22, 987. [Google Scholar] [CrossRef] [PubMed]
- Lucero, C.M.; Andrade, D.C.; Toledo, C.; Díaz, H.S.; Pereyra, K.V.; Diaz-Jara, E.; Schwarz, K.G.; Marcus, N.J.; Retamal, M.A.; Quintanilla, R.A.; et al. Cardiac remodeling and arrhythmogenesis are ameliorated by administration of Cx43 mimetic peptide Gap27 in heart failure rats. Sci. Rep. 2020, 10, 6878. [Google Scholar] [CrossRef]
- Andrade, D.C.; Iturriaga, R.; Toledo, C.; Lucero, C.M.; Díaz, H.S.; Arce-Álvarez, A.; Retamal, M.A.; Marcus, N.J.; Alcayaga, J.; Del Rio, R. Topical Application of Connexin43 Hemichannel Blocker Reduces Carotid Body-Mediated Chemoreflex Drive in Rats. Adv. Exp. Med. Biol. 2018, 1071, 61–68. [Google Scholar] [CrossRef]
- Leybaert, L.; De Smet, M.A.J.; Lissoni, A.; Allewaert, R.; Roderick, H.L.; Bultynck, G.; Delmar, M.; Sipido, K.R.; Witschas, K. Connexin hemichannels as candidate targets for cardioprotective and anti-arrhythmic treatments. J. Clin. Investig. 2023, 133, 6. [Google Scholar] [CrossRef] [PubMed]
- Araya, R.; Eckardt, D.; Riquelme, M.A.; Willecke, K.; Sáez, J.C. Presence and importance of connexin43 during myogenesis. Cell Commun. Adhes. 2003, 10, 451–456. [Google Scholar] [CrossRef]
- Merrifield, P.A.; Laird, D.W. Connexins in skeletal muscle development and disease. Semin. Cell Dev. Biol. 2016, 50, 67–73. [Google Scholar] [CrossRef]
- Cea, L.A.; Riquelme, M.A.; Cisterna, B.A.; Puebla, C.; Vega, J.L.; Rovegno, M.; Sáez, J.C. Connexin- and pannexin-based channels in normal skeletal muscles and their possible role in muscle atrophy. J. Membr. Biol. 2012, 245, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Sáez, J.C.; Cisterna, B.A.; Vargas, A.; Cardozo, C.P. Regulation of pannexin and connexin channels and their functional role in skeletal muscles. Cell. Mol. Life Sci. 2015, 72, 2929–2935. [Google Scholar] [CrossRef] [PubMed]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Balboa, E.; Puebla, C.; Vargas, A.A.; Cisterna, B.A.; Escamilla, R.; Regueira, T.; Sáez, J.C. Dexamethasone-induced muscular atrophy is mediated by functional expression of connexin-based hemichannels. Biochim. Biophys. Acta 2016, 1862, 1891–1899. [Google Scholar] [CrossRef]
- Cisterna, B.A.; Vargas, A.A.; Puebla, C.; Sáez, J.C. Connexin hemichannels explain the ionic imbalance and lead to atrophy in denervated skeletal muscles. Biochim. Biophys. Acta 2016, 1862, 2168–2176. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Cisterna, B.A.; Puebla, C.; Frank, M.; Figueroa, X.F.; Cardozo, C.; Willecke, K.; Latorre, R.; Sáez, J.C. De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 16229–16234. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Balboa, E.; Vargas, A.A.; Puebla, C.; Brañes, M.C.; Escamilla, R.; Regueira, T.; Sáez, J.C. De novo expression of functional connexins 43 and 45 hemichannels increases sarcolemmal permeability of skeletal myofibers during endotoxemia. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 2765–2773. [Google Scholar] [CrossRef] [PubMed]
- Balboa, E.; Saavedra, F.; Cea, L.A.; Ramírez, V.; Escamilla, R.; Vargas, A.A.; Regueira, T.; Sáez, J.C. Vitamin E Blocks Connexin Hemichannels and Prevents Deleterious Effects of Glucocorticoid Treatment on Skeletal Muscles. Int. J. Mol. Sci. 2020, 21, 4094. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Crespo Yanguas, S.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; Kwak, B.R.; Green, C.R.; Cogliati, B.; Vinken, M. Connexins and their channels in inflammation. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, M.; Wang, H.Q.; Zhang, N.N.; Li, X.M.; Yang, X.Y.; Chen, A.P.; Yan, X.; Zhang, Z.; Chu, S.F.; et al. Inflammation and Connexin 43 profiles in the prefrontal cortex are relevant to stress susceptibility and resilience in mice. Pharmacol. Biochem. Behav. 2024, 239, 173757. [Google Scholar] [CrossRef]
- Sáez, J.C.; Contreras-Duarte, S.; Gómez, G.I.; Labra, V.C.; Santibañez, C.A.; Gajardo-Gómez, R.; Avendaño, B.C.; Díaz, E.F.; Montero, T.D.; Velarde, V.; et al. Connexin 43 Hemichannel Activity Promoted by Pro-Inflammatory Cytokines and High Glucose Alters Endothelial Cell Function. Front. Immunol. 2018, 9, 1899. [Google Scholar] [CrossRef]
- Chanson, M.; Berclaz, P.-Y.; Scerri, I.; Dudez, T.; Wernke-Dollries, K.; Pizurki, L.; Pavirani, A.; Fiedler, M.A.; Suter, S. Regulation of Gap Junctional Communication by a Pro-Inflammatory Cytokine in Cystic Fibrosis Transmembrane Conductance Regulator-Expressing but Not Cystic Fibrosis Airway Cells. Am. J. Pathol. 2001, 158, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Cortés, C.J.; Reuss, L.; Bennett, M.V.L.; Sáez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480. [Google Scholar] [CrossRef] [PubMed]
- Lucero, C.M.; Prieto-Villalobos, J.; Marambio-Ruiz, L.; Balmazabal, J.; Alvear, T.F.; Vega, M.; Barra, P.; Retamal, M.A.; Orellana, J.A.; Gómez, G.I. Hypertensive Nephropathy: Unveiling the Possible Involvement of Hemichannels and Pannexons. Int. J. Mol. Sci. 2022, 23, 15936. [Google Scholar] [CrossRef] [PubMed]
- León-Paravic, C.G.; Figueroa, V.A.; Guzmán, D.J.; Valderrama, C.F.; Vallejos, A.A.; Fiori, M.C.; Altenberg, G.A.; Reuss, L.; Retamal, M.A. Carbon monoxide (CO) is a novel inhibitor of connexin hemichannels. J. Biol. Chem. 2014, 289, 36150–36157. [Google Scholar] [CrossRef] [PubMed]
- Sirago, G.; Candia, J.; Franchi, M.V.; Sarto, F.; Monti, E.; Toniolo, L.; Reggiani, C.; Giacomello, E.; Zampieri, S.; Hartnell, L.M.; et al. Upregulation of Sarcolemmal Hemichannels and Inflammatory Transcripts with Neuromuscular Junction Instability during Lower Limb Unloading in Humans. Biology 2023, 12, 431. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Jones, P.L. FSHD Therapeutic Strategies: What Will It Take to Get to Clinic? J. Pers. Med. 2022, 12, 865. [Google Scholar] [CrossRef]
- Wang, L.H.; Tawil, R. Current Therapeutic Approaches in FSHD. J. Neuromuscul. Dis. 2021, 8, 441–451. [Google Scholar] [CrossRef]
- Chen, T.H.; Wu, Y.Z.; Tseng, Y.H. Early-Onset Infantile Facioscapulohumeral Muscular Dystrophy: A Timely Review. Int. J. Mol. Sci. 2020, 21, 7783. [Google Scholar] [CrossRef]
- DeSimone, A.M.; Cohen, J.; Lek, M.; Lek, A. Cellular and animal models for facioscapulohumeral muscular dystrophy. Dis. Model. Mech. 2020, 13, dmm046904. [Google Scholar] [CrossRef]
- Statland, J.; Donlin-Smith, C.M.; Tapscott, S.J.; Van Der Maarel, S.M.; Tawil, R. Multiplex Screen of Serum Biomarkers in Facioscapulohumeral Muscular Dystrophy. J. Neuromuscul. Dis. 2014, 1, 181–190. [Google Scholar] [CrossRef]
- Fernández, G.; Arias-Bravo, G.; Bevilacqua, J.A.; Castillo-Ruiz, M.; Caviedes, P.; Sáez, J.C.; Cea, L.A. Myofibers deficient in connexins 43 and 45 expression protect mice from skeletal muscle and systemic dysfunction promoted by a dysferlin mutation. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165800. [Google Scholar] [CrossRef]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine prevents renal alterations in diabetic rats. J. Diabetes Res. 2013, 2013, 165800. [Google Scholar] [CrossRef]
- Yi, C.; Ezan, P.; Fernández, P.; Schmitt, J.; Sáez, J.C.; Giaume, C.; Koulakoff, A. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017, 65, 1607–1625. [Google Scholar] [CrossRef]
- Sáez, J.C.; Burrell, J.C.; Cahill, C.M.; Cullen, D.K.; Devi, L.A.; Gilbert, R.J.; Graham, Z.A.; Gurvich, V.J.; Havton, L.A.; Iyengar, R.; et al. Pharmacology of boldine: Summary of the field and update on recent advances. Front. Pharmacol. 2024, 15, 1427147. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Fernández, G.; Arias-Bravo, G.; Castillo-Ruiz, M.; Escamilla, R.; Brañes, M.C.; Sáez, J.C. Blockade of Hemichannels Normalizes the Differentiation Fate of Myoblasts and Features of Skeletal Muscles from Dysferlin-Deficient Mice. Int. J. Mol. Sci. 2020, 21, 6025. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Vásquez, W.; Hernández-Salinas, R.; Vielma, A.Z.; Castillo-Ruiz, M.; Velarde, V.; Salgado, M.; Sáez, J.C. Skeletal Muscle Atrophy Induced by Diabetes Is Mediated by Non-Selective Channels and Prevented by Boldine. Biomolecules 2023, 13, 708. [Google Scholar] [CrossRef]
- Burrell, J.C.; Vu, P.T.; Alcott, O.J.B.; Toro, C.A.; Cardozo, C.; Cullen, D.K. Orally administered boldine reduces muscle atrophy and promotes neuromuscular recovery in a rodent model of delayed nerve repair. Front. Cell. Neurosci. 2023, 17, 1240916. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.A.; Toro, C.A.; Harlow, L.; Lavin, K.M.; Cardozo, C.P.; Wende, A.R.; Graham, Z.A. Assessing the impact of boldine on the gastrocnemius using multiomics profiling at 7 and 28 days post-complete spinal cord injury in young male mice. Physiol. Genom. 2023, 55, 297–313. [Google Scholar] [CrossRef]
- Riquelme, M.A.; Kar, R.; Gu, S.; Jiang, J.X. Antibodies targeting extracellular domain of connexins for studies of hemichannels. Neuropharmacology 2013, 75, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Walrave, L.; Pierre, A.; Albertini, G.; Aourz, N.; De Bundel, D.; Van Eeckhaut, A.; Vinken, M.; Giaume, C.; Leybaert, L.; Smolders, I. Inhibition of astroglial connexin43 hemichannels with TAT-Gap19 exerts anticonvulsant effects in rodents. Glia 2018, 66, 1788–1804. [Google Scholar] [CrossRef] [PubMed]
- Linsambarth, S.; Carvajal, F.J.; Moraga-Amaro, R.; Mendez, L.; Tamburini, G.; Jimenez, I.; Verdugo, D.A.; Gómez, G.I.; Jury, N.; Martínez, P.; et al. Astroglial gliotransmitters released via Cx43 hemichannels regulate NMDAR-dependent transmission and short-term fear memory in the basolateral amygdala. FASEB J. 2022, 36, e22134. [Google Scholar] [CrossRef]
- Chen, C.-H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin 43-based therapeutics for dermal wound healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Ghatnekar, G.; Gourdie, R.G.; Potts, J.D. Impact of the controlled release of a connexin 43 peptide on corneal wound closure in an STZ model of type I diabetes. PLoS ONE 2014, 9, e86570. [Google Scholar] [CrossRef]
- Che, J.; DePalma, T.J.; Sivakumar, H.; Mezache, L.S.; Tallman, M.M.; Venere, M.; Swindle-Reilly, K.; Veeraraghavan, R.; Skardal, A. αCT1 peptide sensitizes glioma cells to temozolomide in a glioblastoma organoid platform. Biotechnol. Bioeng. 2023, 120, 1108–1119. [Google Scholar] [CrossRef]
- Tardif, J.C. Antioxidants: The good, the bad and the ugly. Can. J. Cardiol. 2006, 22 (Suppl. B), 61B–65B. [Google Scholar] [CrossRef] [PubMed]
- Tyuryaeva, I.; Lyublinskaya, O. Expected and Unexpected Effects of Pharmacological Antioxidants. Int. J. Mol. Sci. 2023, 24, 9303. [Google Scholar] [CrossRef]
- Lamers, C. Overcoming the shortcomings of peptide-based therapeutics. Future Drug Discov. 2022, 4, FDD75. [Google Scholar] [CrossRef]
- Gemel, J.; Kilkus, J.; Dawson, G.; Beyer, E.C. Connecting Exosomes and Connexins. Cancers 2019, 11, 476. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.; Catoe, H.; Werner, R. MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006, 34, 5863–5871. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.M.; Ramirez, M.; Jones, T.I.; Jones, P.L. Identification of candidate miRNA biomarkers for facioscapulohumeral muscular dystrophy using DUX4-based mouse models. Dis. Model. Mech. 2021, 14, dmm049016. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Ubilla, M.; Retamal, M.A. The Unexplored Role of Connexin Hemichannels in Promoting Facioscapulohumeral Muscular Dystrophy Progression. Int. J. Mol. Sci. 2025, 26, 373. https://doi.org/10.3390/ijms26010373
Díaz-Ubilla M, Retamal MA. The Unexplored Role of Connexin Hemichannels in Promoting Facioscapulohumeral Muscular Dystrophy Progression. International Journal of Molecular Sciences. 2025; 26(1):373. https://doi.org/10.3390/ijms26010373
Chicago/Turabian StyleDíaz-Ubilla, Macarena, and Mauricio A. Retamal. 2025. "The Unexplored Role of Connexin Hemichannels in Promoting Facioscapulohumeral Muscular Dystrophy Progression" International Journal of Molecular Sciences 26, no. 1: 373. https://doi.org/10.3390/ijms26010373
APA StyleDíaz-Ubilla, M., & Retamal, M. A. (2025). The Unexplored Role of Connexin Hemichannels in Promoting Facioscapulohumeral Muscular Dystrophy Progression. International Journal of Molecular Sciences, 26(1), 373. https://doi.org/10.3390/ijms26010373