

Fetal Hypoglycemia Induced by Placental SLC2A3-RNA Interference Alters Fetal Pancreas Development and Transcriptome at Mid-Gestation
 ,
, 
 ,
,
Abstract
1. Introduction
2. Results
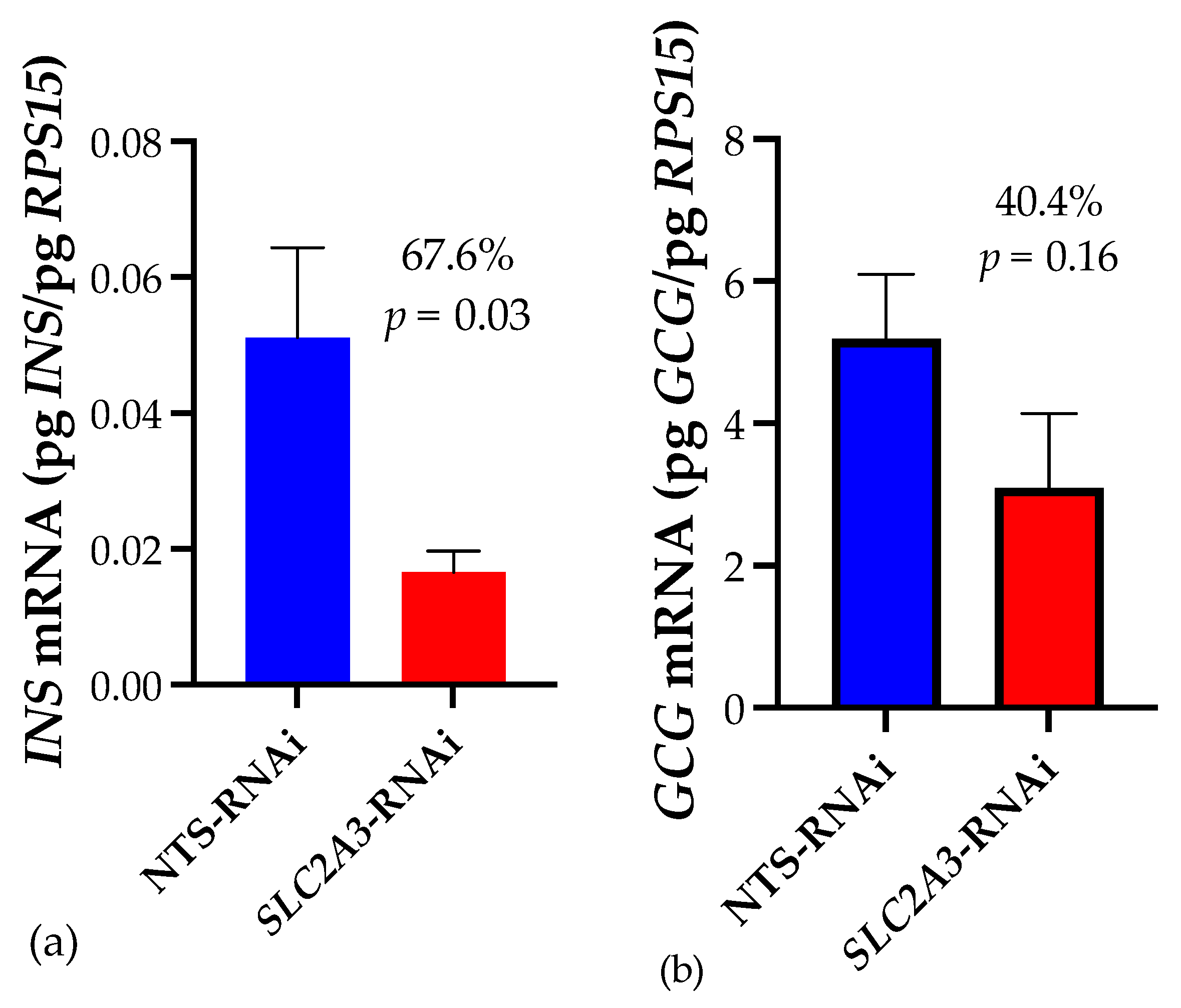
2.1. Quantitative Real-Time PCR
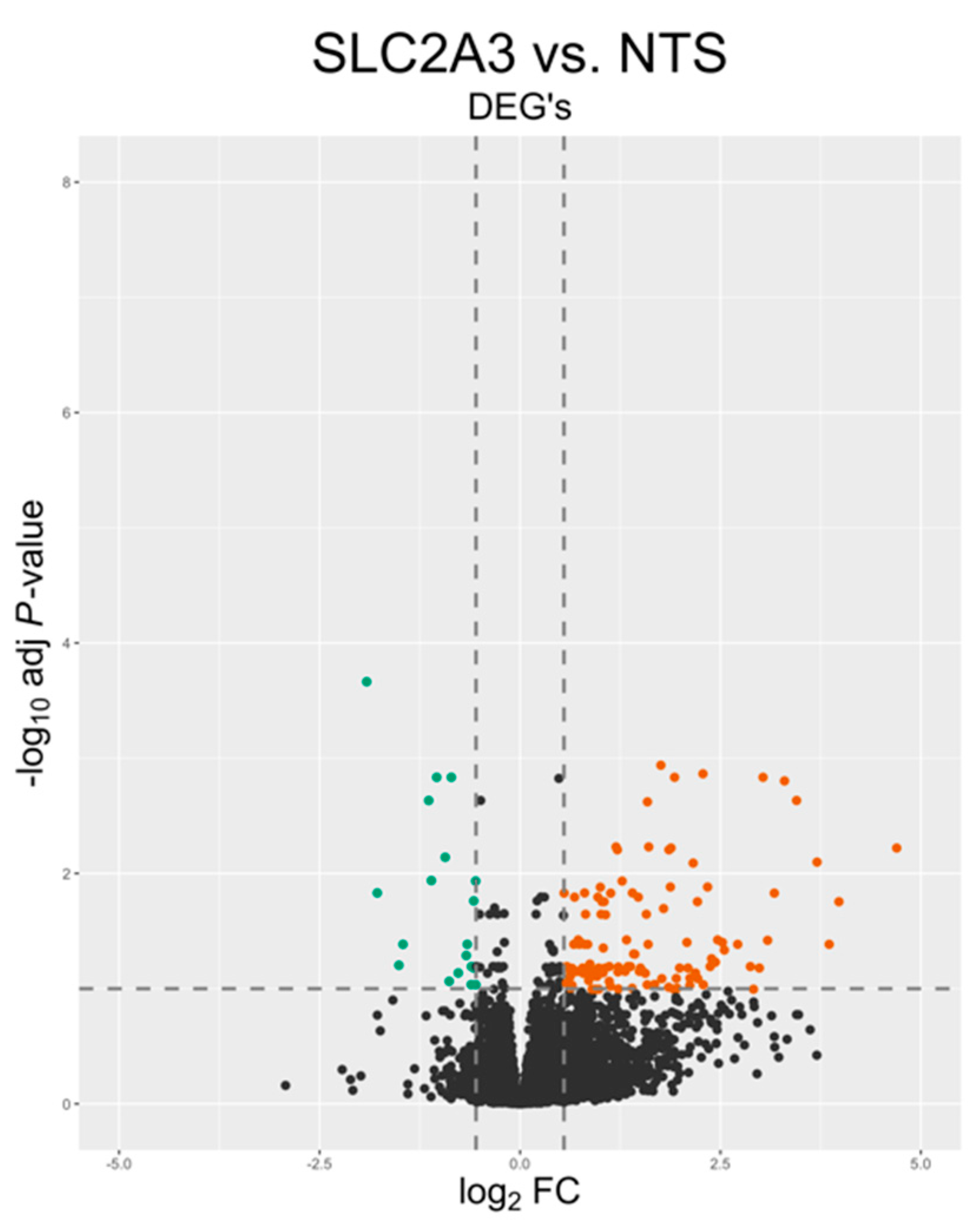
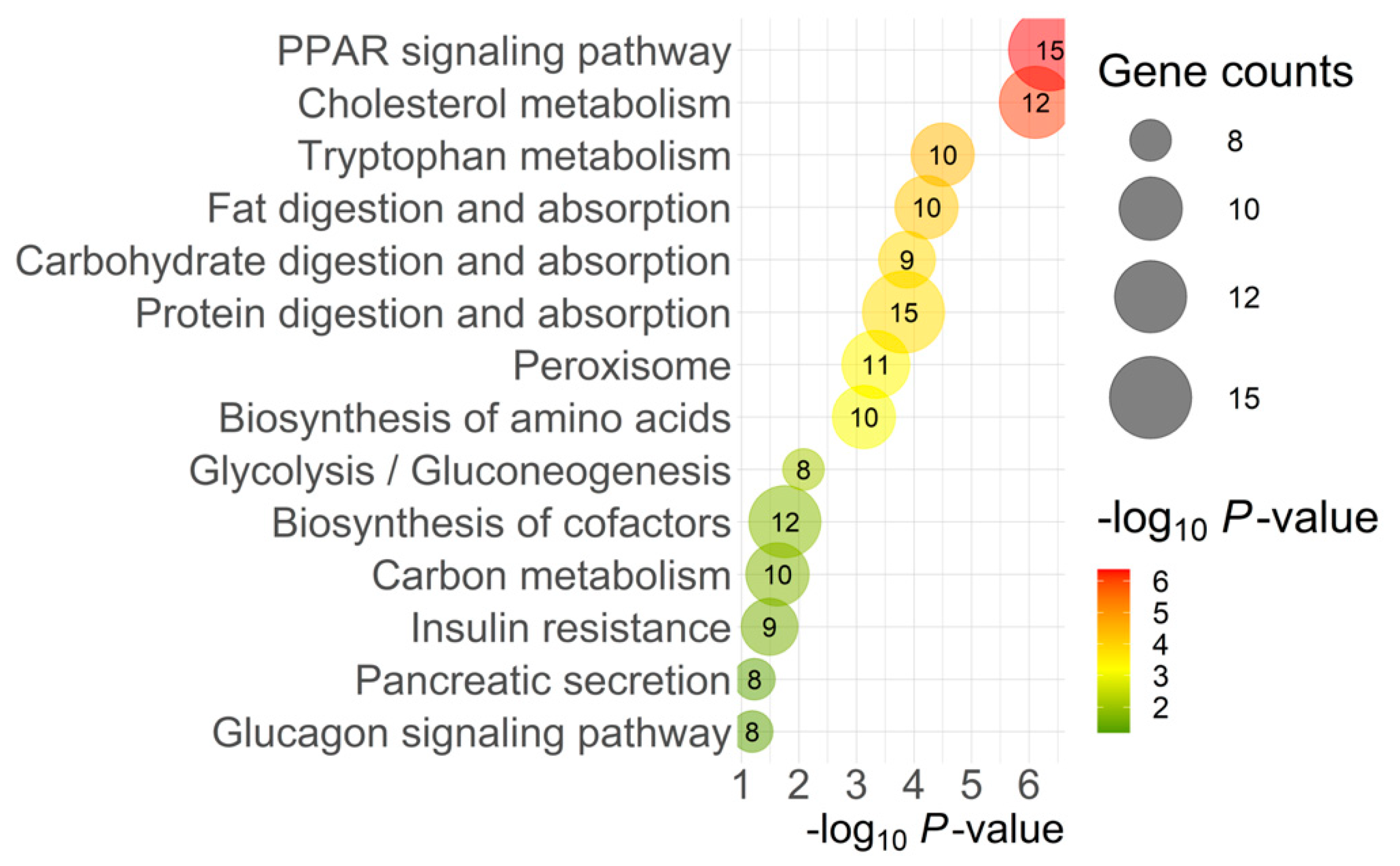
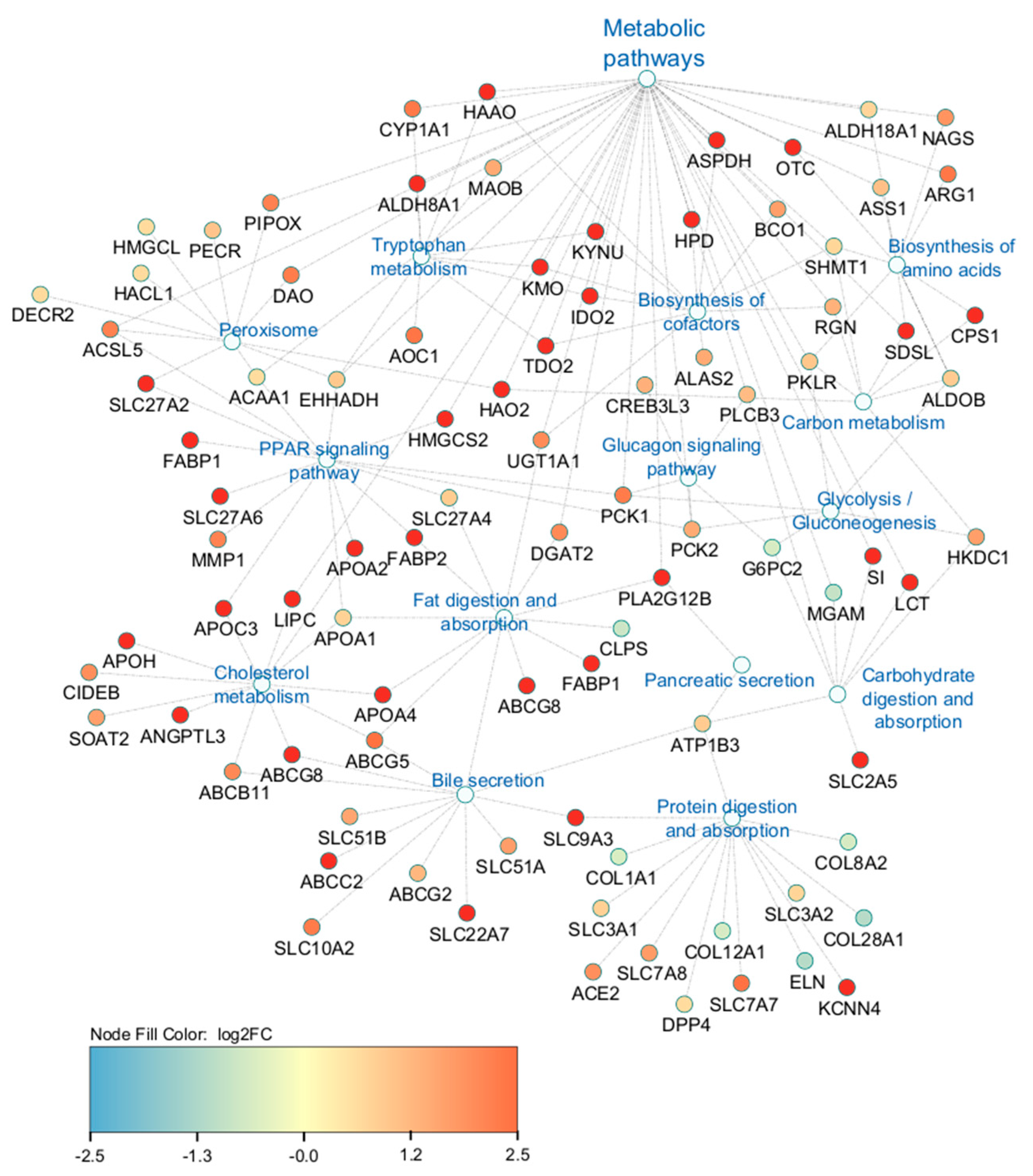
2.2. Transcriptomic Analysis
3. Discussion
4. Materials and Methods
4.1. Generation of Lentivirus and SLC2A3-RNAi Pregnancies
4.2. RNA Isolation
4.3. cDNA Synthesis and Quantitative Real-Time PCR
4.4. RNA Sequencing, Alignments, and Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marconi, A.M.; Paolini, C.L. Nutrient transport across the intrauterine growth-restricted placenta. Semin. Perinatol. 2008, 32, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Limesand, S.W.; Rozance, P.J.; Zerbe, G.O.; Hutton, J.C.; Hay, W.W., Jr. Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology 2006, 147, 1488–1497. [Google Scholar] [CrossRef]
- Limesand, S.W.; Rozance, P.J.; Smith, D.; Hay, W.W., Jr. Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1716–E1725. [Google Scholar] [CrossRef]
- Tanner, A.R.; Lynch, C.S.; Kennedy, V.C.; Ali, A.; Winger, Q.A.; Rozance, P.J.; Anthony, R.V. CSH RNA interference reduces global nutrient uptake and umbilical blood flow resulting in intrauterine growth restriction. Int. J. Mol. Sci. 2021, 22, 8150. [Google Scholar] [CrossRef]
- Wooding, F.B.P.; Fowden, A.L.; Bell, A.W.; Ehrhardt, R.A.; Limesand, S.W.; Hay, W.W., Jr. Localisation of glucose transport in the ruminant placenta: Implications for sequential use of transporter isoforms. Placenta 2005, 26, 626–640. [Google Scholar] [CrossRef]
- Limesand, S.W.; Hay, W.W., Jr. Adaptation of ovine fetal pancreatic insulin secretion to chronic hypoglycaemia and euglycaemic correction. J. Physiol. 2003, 547, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hay, W.W., Jr. Placental-fetal glucose exchange and fetal glucose metabolism. Trans. Am. Clin. Climatol. Assoc. 2006, 117, 321–340. [Google Scholar]
- Jimenez-Chillaron, J.C.; Hernandez-Valencia, M.; Reamer, C.; Fisher, S.; Joszi, A.; Hirshman, M.; Oge, A.; Walrond, S.; Przybyla, R.; Boozer, C.; et al. Beta-cell secretory dysfunction in the pathogenesis of low birth weight associated diabetes: A murine model. Diabetes 2005, 54, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, F.A.; De Prins, F.; Aerts, L.; Verjans, M. The endocrine pancreas in small-for-dates infants. Br. J. Obstet. Gynaecol. 1977, 84, 751–753. [Google Scholar] [CrossRef]
- Lynch, C.S.; Kennedy, V.C.; Tanner, A.R.; Ali, A.; Winger, Q.A.; Rozance, P.J.; Anthony, R.V. Impact of placental SLC2A3 deficiency during the first-half of gestation. Int. J. Mol. Sci. 2022, 23, 12530. [Google Scholar] [CrossRef]
- Economides, D.L.; Nicolaides, K.H. Blood glucose and oxygen tension levels in small-for-gestational-age fetuses. Am. J. Obstet. Gynecol. 1989, 160, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, B.H.; Limesand, S.W.; Rozance, P.J. The impact of IUGR on pancreatic islet development and β-cell function. J. Endocrinol. 2017, 235, R73–R76. [Google Scholar] [CrossRef] [PubMed]
- Dassaye, R.; Naidoo, S.; Cerf, M.E. Transcription factor regulation of pancreatic organogenesis, differentiation and maturation. Islets 2016, 8, 13–34. [Google Scholar] [CrossRef]
- Gittes, G.K. Developmental biology of the pancreas: A comprehensive review. Dev. Biol. 2009, 326, 4–35. [Google Scholar] [CrossRef]
- Hebrok, M.; Kim, S.K.; St Jacques, B.; McMahon, A.P.; Melton, D.A. Regulation of pancreas development by hedgehog signaling. Development 2000, 127, 4905–4913. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; Baumann, D.; Alejandro, E.U. Fetal undernutrition, placental insufficiency, and pancreatic β-cell development programming in utero. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R867–R878. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.P.; Wang, A.; Sander, M. Pancreas organogenesis: From lineage determination to morphogenesis. Annu. Rev. Cell Dev. Biol. 2019, 29, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Odom, D.T.; Zizlsperger, N.; Gordon, D.B.; Bell, G.W.; Rinaldi, N.J.; Murray, H.L.; Volkert, T.L.; Schreiber, J.; Rolfe, P.A.; Gifford, D.K.; et al. Control of pancreas and liver gene expression by HNF transcription factors. Science 2004, 303, 1378–1381. [Google Scholar] [CrossRef]
- Chen, W.S.; Manova, K.; Weinstein, D.C.; Duncan, S.A.; Plump, A.S.; Prezioso, V.R.; Bachvarova, R.F.; Darnell, J.E., Jr. Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev. 1994, 8, 2466–2477. [Google Scholar] [CrossRef]
- Maestro, M.A.; Cardalda, C.; Boj, S.F.; Luco, R.F.; Servitja, J.M.; Ferrer, J. Distinct roles of HNF1beta, HNF1alpha, and HNF4alpha in regulating pancreas development, beta-cell function and growth. Endocr. Dev. 2007, 12, 33–45. [Google Scholar]
- Peterson, M.; Gauvin, M.; Pillai, S.; Jones, A.; McFadden, K.; Cameron, K.; Reed, S.; Zinn, S.; Govoni, K. Maternal under- and over-nutrition during gestation causes islet hypertrophy and sex-specific changes to pancreas DNA methylation in fetal sheep. Animals 2021, 11, 2531. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A. Cholecystokinin (CCK) regulation of pancreatic acinar cells: Physiological actions and signal transduction mechanisms. Compr. Physiol. 2019, 9, 535–564. [Google Scholar] [PubMed]
- Liu, G.; Pakala, S.; Gu, D.; Krahl, T.; Mocnik, L.; Sarvetnick, N. Cholecystokinin expression in the developing and regenerating pancreas and intestine. J. Endocrinol. 2001, 169, 233–240. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saillan-Barreau, C.; Dufresne, M.; Clerc, P.; Sanchez, D.; Corominola, H.; Moriscot, C.; Guy-Crotte, O.; Escrieut, C.; Vaysse, N.; Gomis, R.; et al. Evidence for a functional role of the cholecystokinin-B/gastrin receptor in the human fetal and adult pancreas. Diabetes 1999, 48, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- Venezia, O.; Islam, S.; Cho, C.; Timme-Laragy, A.R.; Sant, K.E. Modulation of PPAR signaling disrupts pancreas development in the zebrafish, Danio rerio. Toxicol. Appl. Pharmacol. 2021, 426, 115653. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Barg, S.; Vallois, D.; Lahiri, S.; Roger, C.; Yessoufou, A.; Pradevand, S.; McDonald, A.; Bonal, C.; Reimann, F.; et al. PPARβ/δ affects pancreatic β cell mass and insulin secretion in mice. J. Clin. Investig. 2012, 122, 4105–4117. [Google Scholar] [CrossRef]
- Zhao, Y.F. Free fatty acid receptors in the endocrine regulation of glucose metabolism: Insight from gastrointestinal-pancreatic adipose interactions. Front. Endocrinol. 2022, 13, 956277. [Google Scholar] [CrossRef] [PubMed]
- Cen, J.; Sargsyan, E.; Bergsten, P. Fatty acids stimulate insulin secretion from human pancreatic islets at fasting glucose concentrations via mitochondria-dependent and -independent mechanisms. Nutr. Metab. 2016, 13, 59. [Google Scholar] [CrossRef]
- Ježek, P.; Jabůrek, M.; Holendová, B.; Plecitá-Hlavatá, L. Fatty acid-stimulated insulin secretion vs. lipotoxicity. Molecules 2018, 23, 1483. [Google Scholar] [CrossRef]
- Haber, E.P.; Ximenes, H.M.; Procópio, J.; Carvalho, C.R.; Curi, R.; Carpinelli, A.R. Pleiotropic effects of fatty acids on pancreatic beta-cells. J Cell. Physiol. 2002, 194, 1–12. [Google Scholar] [CrossRef]
- Acharya, R.; Shetty, S.S.; Kumari, S.N. Fatty acid transport proteins (FATPs) in cancer. Chem. Phys. Lipids 2023, 250, 105269. [Google Scholar] [CrossRef]
- Best, L.; Jarman, E.; Brown, P.D. A dual action of saturated fatty acids on electrical activity in rat pancreatic β-cells. Role of volume-regulated anion channel and KATP channel currents. J. Physiol. 2011, 589, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Chueire, V.B.; Muscelli, E. Effect of free fatty acids on insulin secretion, insulin sensitivity and incretin effect—A narrative review. Arch. Endocrinol. Metab. 2021, 65, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Wen, H.; Xu, P.; Chen, J.; Xue, M.; Li, J.; Wang, M.; Song, C.; Li, H. PPAR signaling maintains metabolic homeostasis under hypothermia in freshwater drum (Aplodinotus grunniens). Metabolites 2023, 13, 102. [Google Scholar] [CrossRef]
- Henry, C.; Close, A.-F.; Buteau, J. A critical role for the neural zinc factor ST18 in pancreatic β-cell apoptosis. J. Biol. Chem. 2014, 289, 8413–8419. [Google Scholar] [CrossRef]
- Wesolowski, S.R.; Hay, W.W., Jr. Role of placental insufficiency and intrauterine growth restriction on the activation of fetal hepatic glucose production. Mol. Cell. Endocrinol. 2016, 435, 61–68. [Google Scholar] [CrossRef]
- Bosma, K.J.; Rahim, M.; Oeser, J.K.; McGuinness, O.P.; Young, J.D.; O’Brien, R.M. G6PC2 confers protection against hypoglycemia upon ketogenic diet feeding and prolonged fasting. Mol. Metab. 2020, 41, 101043. [Google Scholar] [CrossRef] [PubMed]
- Rahim, M.; Nakhe, A.Y.; Banerjee, D.R.; Overway, E.M.; Bosma, K.J.; Rosch, J.C.; Oeser, J.K.; Wang, B.; Lippmann, E.S.; Jacobson, D.A.; et al. Glucose-6-phosphatase catalytic subunit 2 negatively regulates glucose oxidation and insulin secretion in pancreatic β-cells. J. Biol. Chem. 2022, 298, 101729. [Google Scholar] [CrossRef]
- Baker, C.M.; Goetzmann, L.N.; Cantlon, J.D.; Jeckel, K.M.; Winger, Q.A.; Anthony, R.V. Development of ovine chorionic somato-mammotropin hormone-deficient pregnancies. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R837–R846. [Google Scholar] [CrossRef]
- National Research Council. Nutrient Requirements of Small Ruminants: Sheep, Goats, Cervids, and New World Camelids; The National Academies Press: Washington, DC, USA, 2007. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Jeckel, K.M.; Boyarko, A.C.; Bouma, G.J.; Winger, Q.A.; Anthony, R.V. Chorionic somatomammotropin impacts early fetal growth and placental gene expression. J. Endocrinol. 2018, 237, 301–310. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sample I.D. | Raw Reads | Quality Control (QC) Reads | Mapping Rate (%) |
|---|---|---|---|---|
| SLC2A3-RNAi | 2_472 | 201,264,985.3 | 185,063,154 | 98.26 |
| 3_472 | 175,336,416.6 | 160,783,494 | 98.06 | |
| 5_472 | 190,077,663.5 | 175,232,598 | 98.24 | |
| 6_472 | 192,195,658 | 176,704,688 | 98.21 | |
| NTS-RNAi | 7_SC | 169,767,017.4 | 155,846,122 | 98.27 |
| 8_SC | 179,660,289.9 | 164,874,248 | 98.11 | |
| 11_SC | 129,489,997.8 | 118,975,410 | 98.22 | |
| 12_SC | 162,587,201.6 | 149,141,240 | 98.19 |
| Gene | Name | Category | Log2FC | FDR |
|---|---|---|---|---|
| CLPS | Colipase | Fat digestion and absorption | −0.80984 | 0.000412 |
| CCK | Cholecystokinin | Pancreatic secretion | 2.85173 | 2.48 × 10−13 |
| SHH | Sonic Hedgehog Signaling Molecule | Cellular development/growth and proliferation; tissue development | 1.39917 | 0.023658 |
| PLCB3 | Phospholipase C Beta 3 | IP3-DAG signaling | 1.17916 | 4.17 × 10−12 |
| SLC26A3 | Solute Carrier Family 26 Member 3 | Ion transport; pancreatic secretion | 2.57893 | 4.02 × 10−9 |
| SLC27A2 | Solute Carrier Family 27 | PPAR signaling; fatty acid transport | 2.60986 | 3.77 × 10−10 |
| SLC27A4 | Solute Carrier Family 27 | PPAR signaling; fatty acid transport | 0.8981 | 0.00025893 |
| TDO2 | Tryptophan 2,3-dioxygenase | Tryptophan metabolism | 4.23593 | 0.0088385 |
| HAA0 | Hydroxyacid Oxidase | Tryptophan metabolism; biosynthesis of cofactors | 3.13823 | 7.24 × 10−13 |
| KYNU | Kynureninase | Tryptophan metabolism; biosynthesis of cofactors | 2.62505 | 1.02 × 10−7 |
| HNF4A | Hepatocyte Nuclear Factor 4 Alpha | Negative regulation of cell proliferation | 0.58768 | 0.00751424 |
| ST18 | ST18 C2H2C-type zinc finger transcription factor | Negative regulation of cell proliferation; apoptosis | −0.59897 | 0.00458748 |
| HS3ST1 | Heparan sulfate-glucosamine 3-sulfotransferase 1 | Cell signaling; growth factor binding; tissue development | 0.88156 | 0.07424441 |
| CREB3L3 | cAMP Responsive Element Binding Protein 3 Like 3 | Glucagon signaling; insulin resistance; pancreatic secretion | 1.3806 | 8.09 × 10−12 |
| FBP1 | Fructose-bisphosphatase 1 | Glucagon signaling; carbon metabolism; glycolysis/gluconeogenesis | 0.6707 | 0.0013089 |
| LDHA | Lactate dehydrogenase A | Glucagon signaling; glycolysis/gluconeogenesis | 0.73 | 0.00382388 |
| PCK1; PCK2 | Phosphoenolpyruvate carboxykinase 1; 2 | Glucagon signaling; insulin resistance; glycolysis/gluconeogenesis; PPAR signaling | 2.2652; 1.4892 | 1.5541 × 10−7; 2.31 × 10−18 |
| G6PC2 | Islet specific glucose-6-phosphatase catalytic subunit 2 | Glucagon signaling; insulin resistance; glycolysis/gluconeogenesis; carbohydrate digestion | −0.6281 | 0.02096731 |
| Term | Count | p-Value | Genes | Fold Enrichment | FDR |
|---|---|---|---|---|---|
| Carbohydrate metabolic process | 10 | 0.00353 | LDHA, MGAM, GNPDA1, SI, SLC3A1, SLC3A2, NPL, RENBP, FBP1, GLB1L3 | 3.26251 | 0.44330 |
| Cholesterol homeostasis | 10 | 0.00003 | EHD1, ABCG8, ABCG5, DGAT2, LIPC, SOAT2, HNF4A, APOA2, ANGPTL3, ABCB11 | 6.24377 | 0.03353 |
| Negative regulation of cell proliferation | 10 | 0.08406 | SLC9A3R1, HNF4A, CLDN19, PTK2B, PTH1R, PODN, ST18, SKAP2, ENPP7, BMP5 | 1.88614 | 0.99922 |
| Cell surface receptor signaling pathway | 6 | 0.05824 | VIPR1, ADGRG7, EDN3, GLP2R, PTH1R, F2 | 2.85899 | 0.99922 |
| Lipid catabolic process | 6 | 0.05054 | PLCB3, LIPC, PLA2G12B, CLPS, NEU1, PLBD1 | 2.97648 | 0.99922 |
| Cholesterol metabolic process | 5 | 0.01663 | CUBN, LIPC, SOAT2, APOA2, ANGPTL3 | 5.02971 | 0.97370 |
| Fatty acid metabolic process | 5 | 0.01510 | NAAA, CYP1A1, ANGPTL3, HACL1, ABCB11 | 5.17341 | 0.92632 |
| Gluconeogenesis | 4 | 0.00888 | G6PC2, PCK1, FBP1, PCK2 | 9.05347 | 0.71451 |
| Phospholipid homeostasis | 4 | 0.00379 | HNF4A, ANGPTL3, ITGB6, ABCB11 | 12.07130 | 0.44330 |
| Cell–cell signaling | 3 | 0.08660 | SHH, GJB4, IHH | 6.03565 | 0.99922 |
| Digestion | 3 | 0.04159 | CLPS, CYM, AKR1D1 | 9.05347 | 0.99922 |
| Fatty acid transport | 3 | 0.01449 | SLC27A6, SLC27A2, SLC27A4 | 15.52024 | 0.92632 |
| cDNA | Forward Primer (5′ → 3′) | Reverse Primer (5′ → 3′) | Product, bp |
|---|---|---|---|
| RPS15 | ATCATTCTGCCCGAGATGGTG | TGCTTGACGGGCTTGTAGGTG | 134 |
| INS | GAGAGCGCGGCTTCTTCTAC | CGGGGCAGGTCTAGTTACAG | 198 |
| GCG | ACTCACAGGGCACATTCACC | CGGCGGAGTTCTTCAACGAT | 274 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, V.C.; Lynch, C.S.; Tanner, A.R.; Winger, Q.A.; Gad, A.; Rozance, P.J.; Anthony, R.V. Fetal Hypoglycemia Induced by Placental SLC2A3-RNA Interference Alters Fetal Pancreas Development and Transcriptome at Mid-Gestation. Int. J. Mol. Sci. 2024, 25, 4780. https://doi.org/10.3390/ijms25094780
Kennedy VC, Lynch CS, Tanner AR, Winger QA, Gad A, Rozance PJ, Anthony RV. Fetal Hypoglycemia Induced by Placental SLC2A3-RNA Interference Alters Fetal Pancreas Development and Transcriptome at Mid-Gestation. International Journal of Molecular Sciences. 2024; 25(9):4780. https://doi.org/10.3390/ijms25094780
Chicago/Turabian StyleKennedy, Victoria C., Cameron S. Lynch, Amelia R. Tanner, Quinton A. Winger, Ahmed Gad, Paul J. Rozance, and Russell V. Anthony. 2024. "Fetal Hypoglycemia Induced by Placental SLC2A3-RNA Interference Alters Fetal Pancreas Development and Transcriptome at Mid-Gestation" International Journal of Molecular Sciences 25, no. 9: 4780. https://doi.org/10.3390/ijms25094780
APA StyleKennedy, V. C., Lynch, C. S., Tanner, A. R., Winger, Q. A., Gad, A., Rozance, P. J., & Anthony, R. V. (2024). Fetal Hypoglycemia Induced by Placental SLC2A3-RNA Interference Alters Fetal Pancreas Development and Transcriptome at Mid-Gestation. International Journal of Molecular Sciences, 25(9), 4780. https://doi.org/10.3390/ijms25094780