
Inhibition of the JAK-STAT Pathway in the Treatment of Psoriasis: A Review of the Literature





Abstract
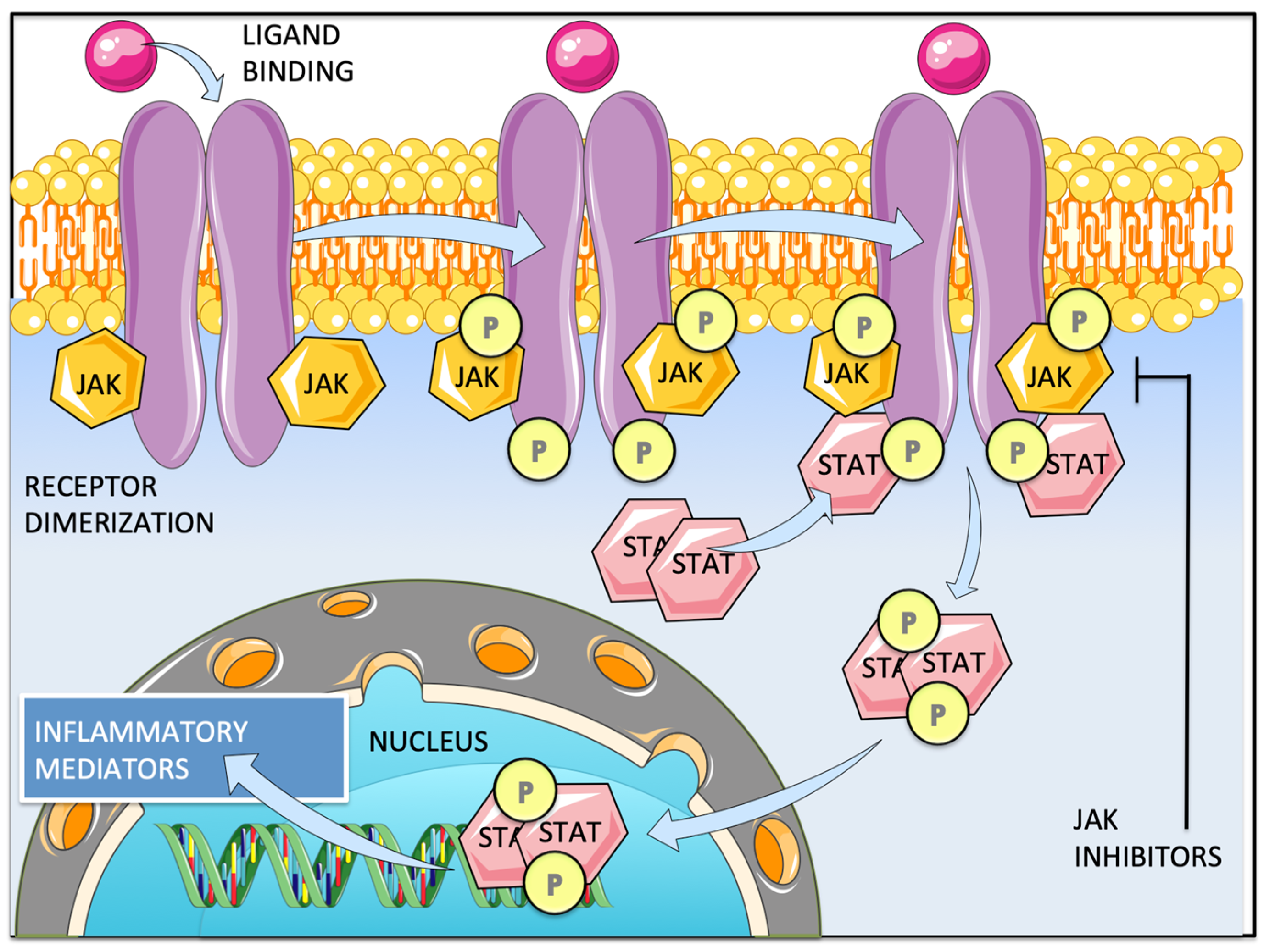
1. Introduction
2. Methods
3. Results
3.1. Efficacy
3.1.1. Tofacitinib
- Moderate-to-Severe Plaque Psoriasis
- Psoriatic Arthritis
3.1.2. Deucravacitinib
- Moderate-to-Severe Plaque Psoriasis
- Psoriatic Arthritis
3.1.3. Upadacitinib
- Psoriatic Arthritis
3.1.4. Other oral JAKis
3.1.5. Topical JAKis
3.2. Safety
3.2.1. Tofacitinib and Upadacitinib
3.2.2. Deucravacitinib
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Michalek, I.M.; Loring, B.; John, S.M.; World Health Organization. Global Report on Psoriasis; World Health Organization: Geneva, Switzerland, 2016; pp. 5–6. ISBN 9789241565189. [Google Scholar]
- Parisi, R.; Iskandar, I.Y.K.; Kontopantelis, E.; Augustin, M.; Griffiths, C.E.M.; Ashcroft, D.M. National, regional, and worldwide epidemiology of psoriasis: Systematic analysis and modelling study. BMJ 2020, 369, m1590. [Google Scholar] [CrossRef] [PubMed]
- Prevalence Heat Map. Global Psoriasis Atlas. Available online: https://www.globalpsoriasisatlas.org/en/explore/prevalence-heatmap (accessed on 3 March 2024).
- Meyer, A.; Kirveskoski, H.; Melancia, J.; Koukopoulou, T.; Sutton, P.; van de Kerkhof, P.C. Briefing Book: IFPA Forum 2022; International Federation of Psoriatic Disease Associations: Bromma, Sweden, 2022; pp. 7–12. [Google Scholar]
- Hernandez-Nicols, B.F.; Robledo-Pulido, J.J.; Alvarado-Navarro, A. Etiopathogenesis of Psoriasis: Integration of Proposed Theories. Immunol. Investig. 2024, 53, 348–415. [Google Scholar] [CrossRef] [PubMed]
- Ogdie, A.; Langan, S.; Love, T.; Haynes, K.; Shin, D.; Seminara, N.; Mehta, N.N.; Troxel, A.; Choi, H.; Gelfand, J.M. Prevalence and treatment patterns of psoriatic arthritis in the UK. Rheumatology 2013, 52, 568–575. [Google Scholar] [CrossRef]
- Yang, Q.; Qu, L.; Tian, H.; Hu, Y.; Peng, J.; Yu, X.; Yu, C.; Pei, Z.; Wang, G.; Shi, B.; et al. Prevalence and characteristics of psoriatic arthritis in Chinese patients with psoriasis. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1409–1414. [Google Scholar] [CrossRef]
- Carneiro, J.N.; de Paula, A.P.; Martins, G.A. Psoriatic arthritis in patients with psoriasis: Evaluation of clinical and epidemiological features in 133 patients followed at the University Hospital of Brasília. An. Bras. Dermatol. 2012, 87, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, G.; Waxman, R.; Helliwell, P.S. The prevalence of psoriatic arthritis in people with psoriasis. Arthritis Care Res. 2009, 61, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Gladman, D.D.; Helliwell, P.; Khraishi, M.M.; Fuiman, J.; Bananis, E.; Alvarez, D. Comparative performance of psoriatic arthritis screening tools in patients with psoriasis in European/North American dermatology clinics. J. Am. Acad. Dermatol. 2014, 71, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Alinaghi, F.; Calov, M.; Kristensen, L.E.; Gladman, D.D.; Coates, L.C.; Jullien, D.; Gottlieb, A.B.; Gisondi, P.; Wu, J.J.; Thyssen, J.P.; et al. Prevalence of psoriatic arthritis in patients with psoriasis: A systematic review and meta-analysis of observational and clinical studies. J. Am. Acad. Dermatol. 2019, 80, 251–265.e19. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Zhang, X.; Du, Y.; Dai, S.-M. Global and regional epidemiology of psoriatic arthritis in patients with psoriasis: A comprehensive systematic analysis and modelling study. J. Autoimmun. 2024, 145, 103202. [Google Scholar] [CrossRef]
- Gladman, D.D.; Brockbank, J. Psoriatic arthritis. Expert Opin. Investig. Drugs 2000, 9, 1511–1522. [Google Scholar] [CrossRef]
- Ahlehoff, O.; Gislason, G.H.; Jørgensen, C.H.; Lindhardsen, J.; Charlot, M.; Olesen, J.B.; Abildstrøm, S.Z.; Skov, L.; Torp-Pedersen, C.; Hansen, P.R. Psoriasis and risk of atrial fibrillation and ischaemic stroke: A Danish Nationwide Cohort Study. Eur. Heart J. 2012, 33, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Harskamp, C.T.; Ledo, L.; Rogers, J.H.; Armstrong, E.J. Coronary Artery Disease in Patients with Psoriasis Referred for Coronary Angiography. Am. J. Cardiol. 2012, 109, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Hillary, T.M.; Vanhoutvin, T.; Peeters, M.; Imbrechts, M.; Vanassche, T.; Garmyn, M.; Vermeire, S. A Prospective, Monocentric Case-Control Study on Uncontrolled Psoriasis as Independent Risk Factor for a Hypercoagulable State. Dermatol. Ther. 2024, 14, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Solberg, S.M. Psoriasis in Norway: A prescription-based registry study of psoriasis-associated comorbidities. Eur. J. Dermatol. 2023, 33, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B. Risk of Myocardial Infarction in Patients with Psoriasis. JAMA 2006, 296, 1735. [Google Scholar] [CrossRef] [PubMed]
- Davidovici, B.B.; Sattar, N.; Jörg, P.C.; Puig, L.; Emery, P.; Barker, J.N.; Van De Kerkhof, P.; Ståhle, M.; Nestle, F.O.; Girolomoni, G.; et al. Psoriasis and Systemic Inflammatory Diseases: Potential Mechanistic Links between Skin Disease and Co-Morbid Conditions. J. Investig. Dermatol. 2010, 130, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clinical Reviews in Allergy and Immunology; Humana Press Inc.: Totowa, NJ, USA, 2018; pp. 379–390. [Google Scholar] [CrossRef]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.J.; Fearon, U. The pathogenesis of psoriatic arthritis. Lancet 2018, 391, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.M.; Armstrong, A.W.; Gudjonsson, J.E.; Barker, J.N.W.N. Psoriasis. Lancet 2021, 397, 1301–1315. [Google Scholar] [CrossRef]
- Robustillo-Villarino, M.; Alegre-Sancho, J.J.; de los Ángeles Martínez-Ferrer, M. Evaluation of the sexual sphere in patients with psoriatic arthritis. Reumatol. Clínica 2023, 19, 249–254. [Google Scholar] [CrossRef]
- Chu, M.; Shen, S.; Zhu, Z.; Li, Z.; Bai, Y.; Ma, J.; Hao, J.; Wang, L.; Fu, M.; Dang, E.; et al. Association of psoriasis with depression, anxiety, and suicidality: A bidirectional two-sample Mendelian randomization study. J. Dermatol. 2023, 50, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Svedbom, A.; Ståhle, M. Itch Is More Important than Clinical Disease Severity for Depression in Psoriasis: A Prospective Register Study. J. Investig. Dermatol. 2023, 143, 2529–2532.e1. [Google Scholar] [CrossRef] [PubMed]
- Watts, S.; Nagib, L.G.; Bamford, S.; Sheraz, A.; Tahir, H. A Patient Survey Exploring the Burden of Inflammatory Back Pain in Patients with Known Psoriasis. Cureus 2024, 16, e51968. [Google Scholar] [CrossRef]
- Nymand, L.; Kristensen, L.E.; Thomsen, S.F.; Thyssen, J.P.; Egeberg, A. Characteristics and drivers of fatigue in patients with psoriasis and psoriatic arthritis: A cross sectional study. J. Am. Acad. Dermatol. 2024, in press. [Google Scholar] [CrossRef]
- Gossec, L.; Humphries, B.; Rutherford, M.; Taieb, V.; Willems, D.; Tillett, W. Improvement in work productivity among psoriatic arthritis patients treated with biologic or targeted synthetic drugs: A systematic literature review and meta-analysis. Arthritis Res. Ther. 2024, 26, 50. [Google Scholar] [CrossRef]
- Gudu, T.; Gossec, L. Quality of life in psoriatic arthritis. Expert Rev. Clin. Immunol. 2018, 14, 405–417. [Google Scholar] [CrossRef]
- Coates, L.C.; Soriano, E.R.; Corp, N.; Bertheussen, H.; Callis Duffin, K.; Campanholo, C.B.; Chau, J.; Eder, L.; Fernández-Ávila, D.G.; FitzGerald, O.; et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA): Updated treatment recommendations for psoriatic arthritis 2021. Nat. Rev. Rheumatol. 2022, 18, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Gossec, L.; Baraliakos, X.; Kerschbaumer, A.; de Wit, M.; McInnes, I.; Dougados, M.; Primdahl, J.; McGonagle, D.G.; Aletaha, D.; Balanescu, A.; et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann. Rheum. Dis. 2020, 79, 700–712. [Google Scholar] [CrossRef]
- Pathirana, D.; Ormerod, A.D.; Saiag, P.; Smith, C.; Spuls, P.I.; Nast, A.; Barker, J.; Bos, J.D.; Burmester, G.R.; Chimenti, S.; et al. European S3-Guidelines on the systemic treatment of psoriasis vulgaris. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1–70. [Google Scholar] [CrossRef]
- Koren, J.; Lambert, J.L.; Thomsen, S.F.; McAteer, H.; Fabbrocini, G.; Corazza, V.; Jullien, D.; Augustin, M.; Warren, R.B.; de Rie, M.A.; et al. Elevating the Standard of Care for Patients with Psoriasis: ‘Calls to Action’ from Epicensus, a Multistakeholder Pan-European Initiative. Dermatol. Ther. 2023, 13, 245–268. [Google Scholar] [CrossRef]
- Gonzalez-Cantero, A.; Boehncke, W.H.; De Sutter, J.; Zamorano, J.L.; Lambert, J.; Puig, L. Statins and psoriasis: Position statement by the Psoriasis Task Force of the European Academy of Dermatology and Venerology. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 1697–1705. [Google Scholar] [CrossRef]
- Moreland, L.W.; Baumgartner, S.W.; Schiff, M.H.; Tindall, E.A.; Fleischmann, R.M.; Weaver, A.L.; Ettlinger, R.E.; Cohen, S.; Koopman, W.J.; Mohler, K.; et al. Treatment of Rheumatoid Arthritis with a Recombinant Human Tumor Necrosis Factor Receptor (p75)–Fc Fusion Protein. N. Engl. J. Med. 1997, 337, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Kamata, M.; Tada, Y. Efficacy and Safety of Biologics for Psoriasis and Psoriatic Arthritis and Their Impact on Comorbidities: A Literature Review. Int. J. Mol. Sci. 2020, 21, 1690. [Google Scholar] [CrossRef] [PubMed]
- Bolognia, J.L.; Jorizzo, J.J.; Schaffer, J.V.; Callen, J.P.; Cerroni, L.; Heymann, W.R.; Hruza, G.J.; Mancini, A.J.; McGrath, J.; Patterson, J.W.; et al. (Eds.) Dermatology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 2, pp. 2244–2253. ISBN 978-0-7020-6275-9. [Google Scholar]
- Tsai, Y.-C.; Tsai, T.-F. Switching biologics in psoriasis—Practical guidance and evidence to support. Expert Rev. Clin. Pharmacol. 2020, 13, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Florek, A.G.; Wang, C.J.; Armstrong, A.W. Treatment preferences and treatment satisfaction among psoriasis patients: A systematic review. Arch. Dermatol. Res. 2018, 310, 271–319. [Google Scholar] [CrossRef]
- Eliasson, L.; Bewley, A.P.; Mughal, F.; Johnston, K.M.; Kuznik, A.; Patel, C.; Lloyd, A.J. Evaluation of psoriasis patients’ attitudes toward benefit-risk and therapeutic trade-offs in their choice of treatments. Patient Prefer. Adherence 2017, 11, 353–362. [Google Scholar] [CrossRef]
- Yadav, V.; Varum, F.; Bravo, R.; Furrer, E.; Basit, A.W. Gastrointestinal stability of therapeutic anti-TNF α IgG1 monoclonal antibodies. Int. J. Pharm. 2016, 502, 181–187. [Google Scholar] [CrossRef]
- Carmona-Rocha, E.; Rusiñol, L.; Puig, L. New and Emerging Oral/Topical Small-Molecule Treatments for Psoriasis. Pharmaceutics 2024, 16, 239. [Google Scholar] [CrossRef]
- Bonelli, M.; Kerschbaumer, A.; Kastrati, K.; Ghoreschi, K.; Gadina, M.; Heinz, L.X.; Smolen, J.S.; Aletaha, D.; O’Shea, J.; Laurence, A. Selectivity, efficacy and safety of JAKinibs: New evidence for a still evolving story. Ann. Rheum. Dis. 2024, 83, 139–160. [Google Scholar] [CrossRef]
- Nash, P.; Kerschbaumer, A.; Dörner, T.; Dougados, M.; Fleischmann, R.M.; Geissler, K.; McInnes, I.; Pope, J.E.; Van Der Heijde, D.; Stoffer-Marx, M.; et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: A consensus statement. Ann. Rheum. Dis. 2021, 80, 71–87. [Google Scholar] [CrossRef]
- Miot, H.A.; Criado, P.R.; de Castro, C.C.S.; Ianhez, M.; Talhari, C.; Ramos, P.M. JAK-STAT pathway inhibitors in dermatology. An. Bras. Dermatol. 2023, 98, 656–677. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862, Erratum in Nat. Rev. Drug Discov. 2018, 17, 78. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.; Sharma, A.; Mehta, H.; Sarkar, R. Emerging role of topical Janus kinase inhibitors in dermatological disorders: A review. Clin. Exp. Dermatol. 2023, 48, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Rafael, A.; Torres, T. Topical therapy for psoriasis: A promising future. Focus on JAK and phosphodiesterase-4 inhibitors. Eur. J. Dermatol. 2016, 26, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Słuczanowska-Głąbowska, S.; Ziegler-Krawczyk, A.; Szumilas, K.; Pawlik, A. Role of Janus Kinase Inhibitors in Therapy of Psoriasis. J. Clin. Med. 2021, 10, 4307. [Google Scholar] [CrossRef]
- Bacon, C.M.; McVicar, D.W.; Ortaldo, J.R.; Rees, R.C.; O’Shea, J.J.; Johnston, J.A. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: Differential use of Janus family tyrosine kinases by IL-2 and IL-12. J. Exp. Med. 1995, 181, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Strober, B.; Buonanno, M.; Clark, J.D.; Kawabata, T.; Tan, H.; Wolk, R.; Valdez, H.; Langley, R.G.; Harness, J.; Menter, A.; et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br. J. Dermatol. 2013, 169, 992–999. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of Innate and Adaptive Immune Responses by Tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Kurtovic, N.; Halilovic, E. Serum Concentrations of Interferon Gamma (IFN- and #947;) in Patients with Psoriasis: Correlation with Clinical Type and Severity of the Disease. Med. Arch. 2018, 72, 410. [Google Scholar] [CrossRef] [PubMed]
- Chimalakonda, A.; Burke, J.; Cheng, L.; Catlett, I.; Tagen, M.; Zhao, Q.; Patel, A.; Shen, J.; Girgis, I.G.; Banerjee, S.; et al. Selectivity Profile of the Tyrosine Kinase 2 Inhibitor Deucravacitinib Compared with Janus Kinase 1/2/3 Inhibitors. Dermatol. Ther. 2021, 11, 1763–1776. [Google Scholar] [CrossRef] [PubMed]
- Dowty, M.E.; Lin, T.H.; Jesson, M.I.; Hegen, M.; Martin, D.A.; Katkade, V.; Menon, S.; Telliez, J.B. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol. Res. Perspect. 2019, 7, e00537. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 Increases the Innate Immunity of Tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, H.; Lin, W.; Lu, L.; Su, J.; Chen, X. Signaling pathways and targeted therapies for psoriasis. Signal Transduct. Target. Ther. 2023, 8, 437. [Google Scholar] [CrossRef]
- Burke, J.R.; Cheng, L.; Gillooly, K.M.; Strnad, J.; Zupa-Fernandez, A.; Catlett, I.M.; Zhang, Y.; Heimrich, E.M.; McIntyre, K.W.; Cunningham, M.D.; et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 2019, 11, eaaw1736. [Google Scholar] [CrossRef]
- O’Shea, J.J. Targeting the Jak/STAT pathway for immunosuppression. Ann. Rheum. Dis. 2004, 63, ii67–ii71. [Google Scholar] [CrossRef]
- Girolomoni, G.; Strohal, R.; Puig, L.; Bachelez, H.; Barker, J.; Boehncke, W.H.; Prinz, J.C. The role of IL-23 and the IL-23/Th17 immune axis in the pathogenesis and treatment of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1616–1626. [Google Scholar] [CrossRef]
- Jacobson, N.G.; Szabo, S.J.; Weber-Nordt, R.M.; Zhong, Z.; Schreiber, R.D.; Darnell, J.E., Jr.; Murphy, K.M. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J. Exp. Med. 1995, 181, 1755–1762. [Google Scholar] [CrossRef]
- Watford, W.T.; Hissong, B.D.; Bream, J.H.; Kanno, Y.; Muul, L.; O’Shea, J.J. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol. Rev. 2004, 202, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Scheidegger, D.; Palmer-Lehmann, K.; Lane, P.; Lanzavecchia, A.; Alber, G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J. Exp. Med. 1996, 184, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.M.; Way, S.S. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology 2009, 126, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A. T-Helper 17 Cells in Psoriatic Plaques and Additional Genetic Links between IL-23 and Psoriasis. J. Investig. Dermatol. 2008, 128, 1064–1067. [Google Scholar] [CrossRef] [PubMed]
- Gisondi, P.; Bellinato, F.; Maurelli, M.; Geat, D.; Zabotti, A.; McGonagle, D.; Girolomoni, G. Reducing the Risk of Developing Psoriatic Arthritis in Patients with Psoriasis. Psoriasis Targets Ther. 2022, 12, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Podder, I.; De, A.; Das, S. Psoriatic Arthritis: A Comprehensive Update for Dermatologists with Review of Literature. Indian J. Dermatol. 2022, 67, 381–386. [Google Scholar] [PubMed]
- Papp, K.A.; Menter, A.; Strober, B.; Langley, R.G.; Buonanno, M.; Wolk, R.; Gupta, P.; Krishnaswami, S.; Tan, H.; Harness, J.A. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: A Phase 2b randomized placebo-controlled dose-ranging study. Br. J. Dermatol. 2012, 167, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.M.; Jesson, M.I.; Li, X.; Elrick, M.M.; Funckes-Shippy, C.L.; Warner, J.D.; Gross, C.J.; Dowty, M.E.; Ramaiah, S.K.; Hirsch, J.L.; et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J. Inflamm. 2010, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Luo, Y.; O’Shea, J.J.; Nakayamada, S. Janus kinase-targeting therapies in rheumatology: A mechanisms-based approach. Nat. Rev. Rheumatol. 2022, 18, 133–145. [Google Scholar] [CrossRef]
- Bissonnette, R.; Iversen, L.; Sofen, H.; Griffiths, C.E.; Foley, P.; Romiti, R.; Bachinsky, M.; Rottinghaus, S.T.; Tan, H.; Proulx, J.; et al. Tofacitinib withdrawal and retreatment in moderate-to-severe chronic plaque psoriasis: A randomized controlled trial. Br. J. Dermatol. 2015, 172, 1395–1406. [Google Scholar] [CrossRef]
- Bachelez, H.; Van De Kerkhof, P.C.; Strohal, R.; Kubanov, A.; Valenzuela, F.; Lee, J.H.; Yakusevich, V.; Chimenti, S.; Papacharalambous, J.; Proulx, J.; et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: A phase 3 randomised non-inferiority trial. Lancet 2015, 386, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Menter, M.A.; Abe, M.; Elewski, B.; Feldman, S.R.; Gottlieb, A.B.; Langley, R.; Luger, T.; Thaci, D.; Buonanno, M.; et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: Results from two randomized, placebo-controlled, phase III trials. Br. J. Dermatol. 2015, 173, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tsai, T.F.; Lee, M.G.; Zheng, M.; Wang, G.; Jin, H.; Gu, J.; Li, R.; Liu, Q.; Chen, J.; et al. The efficacy and safety of tofacitinib in Asian patients with moderate to severe chronic plaque psoriasis: A Phase 3, randomized, double-blind, placebo-controlled study. J. Dermatol. Sci. 2017, 88, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.; Rigby, W.; Azevedo, V.F.; Behrens, F.; Blanco, R.; Kaszuba, A.; Kudlacz, E.; Wang, C.; Menon, S.; Hendrikx, T.; et al. Tofacitinib for Psoriatic Arthritis in Patients with an Inadequate Response to TNF Inhibitors. N. Engl. J. Med. 2017, 377, 1525–1536. [Google Scholar] [CrossRef]
- Mease, P.; Hall, S.; FitzGerald, O.; van der Heijde, D.; Merola, J.F.; Avila-Zapata, F.; Cieślak, D.; Graham, D.; Wang, C.; Menon, S.; et al. Tofacitinib or Adalimumab versus Placebo for Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef]
- Leng, X.; Lin, W.; Liu, S.; Kanik, K.; Wang, C.; Wan, W.; Jiang, Z.; Liu, Y.; Liu, S.; Zhang, Z.; et al. Efficacy and safety of tofacitinib in Chinese patients with active psoriatic arthritis: A phase 3, randomised, double-blind, placebo-controlled study. RMD Open 2023, 9, e002559. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.; Clark, J.D.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Cueto, I.; Wang, C.Q.; Tan, H.; Wolk, R.; Rottinghaus, S.T.; Whitley, M.Z.; et al. Tofacitinib attenuates pathologic immune pathways in patients with psoriasis: A randomized phase 2 study. J. Allergy Clin. Immunol. 2016, 137, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Papp, K.A.; Tan, H.; Tyring, S.; Wolk, R.; Buonanno, M. Efficacy of tofacitinib, an oral janus kinase inhibitor, on clinical signs of moderate-to- severe plaque psoriasis in different body regions. J. Drugs Dermatol. 2014, 13, 252–256. [Google Scholar] [PubMed]
- Valenzuela, F.; Papp, K.A.; Pariser, D.; Tyring, S.K.; Wolk, R.; Buonanno, M.; Wang, J.; Tan, H.; Valdez, H. Effects of tofacitinib on lymphocyte sub-populations, CMV and EBV viral load in patients with plaque psoriasis. BMC Dermatol. 2015, 15, 8. [Google Scholar] [CrossRef]
- Schmieder, G.J.; Draelos, Z.D.; Pariser, D.M.; Banfield, C.; Cox, L.; Hodge, M.; Kieras, E.; Parsons-Rich, D.; Menon, S.; Salganik, M.; et al. Efficacy and safety of the Janus kinase 1 inhibitor PF-04965842 in patients with moderate-to-severe psoriasis: Phase II, randomized, double-blind, placebo-controlled study. Br. J. Dermatol. 2018, 179, 54–62. [Google Scholar] [CrossRef]
- Mamolo, C.; Harness, J.; Tan, H.; Menter, A. Tofacitinib (CP-690,550), an oral Janus kinase inhibitor, improves patient-reported outcomes in a phase 2b, randomized, double-blind, placebo-controlled study in patients with moderate-to-severe psoriasis. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 192–203. [Google Scholar] [CrossRef]
- Bushmakin, A.G.; Mamolo, C.; Cappelleri, J.C.; Stewart, M. The relationship between pruritus and the clinical signs of psoriasis in patients receiving tofacitinib. J. Dermatol. Treat. 2015, 26, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, C.E.; Vender, R.; Sofen, H.; Kircik, L.; Tan, H.; Rottinghaus, S.T.; Bachinsky, M.; Mallbris, L.; Mamolo, C. Effect of tofacitinib withdrawal and re-treatment on patient-reported outcomes: Results from a Phase 3 study in patients with moderate to severe chronic plaque psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 323–332. [Google Scholar] [CrossRef]
- Valenzuela Ahumada, F.; Paul, C.; Mallbris, L.; Tan, H.; Papacharalambous, J.; Valdez, H.; Mamolo, C. Tofacitinib versus etanercept or placebo in patients with moderate to severe chronic plaque psoriasis: Patient-reported outcomes from a Phase 3 study. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tomalin, L.; Lee, J.; Fitz, L.J.; Berstein, G.; Correa-da Rosa, J.; Garcet, S.; Lowes, M.A.; Valdez, H.; Wolk, R.; et al. Reduction of Inflammatory and Cardiovascular Proteins in the Blood of Patients with Psoriasis: Differential Responses between Tofacitinib and Etanercept after 4 Weeks of Treatment. J. Investig. Dermatol. 2018, 138, 273–281. [Google Scholar] [CrossRef]
- Menter, M.A.; Papp, K.A.; Cather, J.; Leonardi, C.; Pariser, D.M.; Krueger, J.G.; Wohlrab, J.; Amaya-Guerra, M.; Kaszuba, A.; Nadashkevich, O.; et al. Efficacy of Tofacitinib for the Treatment of Moderate-to-Severe Chronic Plaque Psoriasis in Patient Subgroups from Two Randomised Phase 3 Trials. J. Drugs Dermatol. 2016, 15, 568–580. [Google Scholar] [PubMed]
- Tan, H.; Valdez, H.; Griffiths, C.E.; Mrowietz, U.; Tallman, A.; Wolk, R.; Gordon, K. Early clinical response to tofacitinib treatment as a predictor of subsequent efficacy: Results from two phase 3 studies of patients with moderate-to-severe plaque psoriasis. J. Dermatol. Treat. 2017, 28, 3–7. [Google Scholar] [CrossRef]
- Merola, J.F.; Elewski, B.; Tatulych, S.; Lan, S.; Tallman, A.; Kaur, M. Efficacy of tofacitinib for the treatment of nail psoriasis: Two 52-week, randomized, controlled phase 3 studies in patients with moderate-to-severe plaque psoriasis. J. Am. Acad. Dermatol. 2017, 77, 79–87.e1. [Google Scholar] [CrossRef]
- Abe, M.; Nishigori, C.; Torii, H.; Ihn, H.; Ito, K.; Nagaoka, M.; Isogawa, N.; Kawaguchi, I.; Tomochika, Y.; Kobayashi, M.; et al. Tofacitinib for the treatment of moderate to severe chronic plaque psoriasis in Japanese patients: Subgroup analyses from a randomized, placebo-controlled phase 3 trial. J. Dermatol. 2017, 44, 1228–1237. [Google Scholar] [CrossRef]
- Asahina, A.; Etoh, T.; Igarashi, A.; Imafuku, S.; Saeki, H.; Shibasaki, Y.; Tomochika, Y.; Toyoizumi, S.; Nagaoka, M.; Ohtsuki, M. Oral tofacitinib efficacy, safety and tolerability in Japanese patients with moderate to severe plaque psoriasis and psoriatic arthritis: A randomized, double-blind, phase 3 study. J. Dermatol. 2016, 43, 869–880. [Google Scholar] [CrossRef]
- Strand, V.; de Vlam, K.; Covarrubias-Cobos, J.A.; Mease, P.J.; Gladman, D.D.; Graham, D.; Wang, C.; Cappelleri, J.C.; Hendrikx, T.; Hsu, M.A. Tofacitinib or adalimumab versus placebo: Patient-reported outcomes from OPAL Broaden—A phase III study of active psoriatic arthritis in patients with an inadequate response to conventional synthetic disease-modifying antirheumatic drugs. RMD Open 2019, 5, e000806. [Google Scholar] [CrossRef] [PubMed]
- van der Heijde, D.; Gladman, D.D.; FitzGerald, O.; Kavanaugh, A.; Graham, D.; Wang, C.; Fallon, L. Radiographic progression according to baseline C-reactive protein levels and other risk factors in psoriatic arthritis treated with tofacitinib or adalimumab. J. Rheumatol. 2019, 46, 1089–1096. [Google Scholar] [CrossRef]
- Strand, V.; de Vlam, K.; Covarrubias-Cobos, J.A.; Mease, P.J.; Gladman, D.D.; Chen, L.; Kudlacz, E.; Wu, J.; Cappelleri, J.C.; Hendrikx, T.; et al. Effect of tofacitinib on patient-reported outcomes in patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors in the phase III, randomised controlled trial: OPAL beyond. RMD Open 2019, 5, e000808. [Google Scholar] [CrossRef]
- Giles, J.T.; Ogdie, A.; Gomez-Reino, J.J.; Helliwell, P.S.; Germino, R.; Stockert, L.; Young, P.; Joseph, W.; Mundayat, R.; Graham, D.; et al. Impact of baseline body mass index on the efficacy and safety of tofacitinib in patients with psoriatic arthritis. RMD Open 2021, 7, e001486. [Google Scholar] [CrossRef]
- Hoy, S.M. Deucravacitinib: First Approval. Drugs 2022, 82, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Catlett, I.M.; Aras, U.; Hansen, L.; Liu, Y.; Bei, D.; Girgis, I.G.; Murthy, B. First-in-human study of deucravacitinib: A selective, potent, allosteric small-molecule inhibitor of tyrosine kinase 2. Clin. Transl. Sci. 2023, 16, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Gordon, K.; Thaçi, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Gooderham, M.; Warren, R.B.; Papp, K.A.; Strober, B.; Thaçi, D.; Morita, A.; Szepietowski, J.C.; Imafuku, S.; Colston, E.; et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: Efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J. Am. Acad. Dermatol. 2023, 88, 29–39. [Google Scholar] [CrossRef]
- Strober, B.; Thaçi, D.; Sofen, H.; Kircik, L.; Gordon, K.B.; Foley, P.; Rich, P.; Paul, C.; Bagel, J.; Colston, E.; et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: Efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 Program fOr Evaluation of TYK2 inhibitor psoriasis second trial. J. Am. Acad. Dermatol. 2023, 88, 40–51. [Google Scholar] [CrossRef]
- Mease, P.J.; Deodhar, A.A.; Van Der Heijde, D.; Behrens, F.; Kivitz, A.J.; Neal, J.; Kim, J.; Singhal, S.; Nowak, M.; Banerjee, S. Efficacy and safety of selective TYK2 inhibitor, deucravacitinib, in a phase II trial in psoriatic arthritis. Ann. Rheum. Dis. 2022, 81, 815–822. [Google Scholar] [CrossRef]
- Lebwohl, M.; Warren, R.B.; Sofen, H.; Imafuku, S.; Paul, C.; Szepietowski, J.C.; Spelman, L.; Passeron, T.; Vritzali, E.; Napoli, A.; et al. Deucravacitinib in plaque psoriasis: 2-year safety and efficacy results from the phase III POETYK trials. Br. J. Dermatol. 2024, 190, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Bhatnagar, S.; Parmentier, J.M.; Nakasato, P.; Wung, P. Upadacitinib: Mechanism of action, clinical, and translational science. Clin. Transl. Sci. 2024, 17, e13688. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Kato, K.; Magrey, M.; Merola, J.F.; Kishimoto, M.; Pacheco-Tena, C.; Haaland, D.; Chen, L.; Duan, Y.; Zueger, P.; et al. Upadacitinib in patients with psoriatic arthritis and an inadequate response to non-biological therapy: 56-week data from the phase 3 SELECT-PsA 1 study. RMD Open 2021, 7, e001838. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Lertratanakul, A.; Anderson, J.K.; Papp, K.; Van den Bosch, F.; Tsuji, S.; Dokoupilova, E.; Keiserman, M.; Wang, X.; Zhong, S.; et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann. Rheum. Dis. 2021, 80, 312–320. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Kato, K.; Magrey, M.; Merola, J.F.; Kishimoto, M.; Haaland, D.; Chen, L.; Duan, Y.; Liu, J.; Lippe, R.; et al. Efficacy and Safety of Upadacitinib in Patients with Psoriatic Arthritis: 2-Year Results from the Phase 3 SELECT-PsA 1 Study. Rheumatol. Ther. 2023, 10, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Lertratanakul, A.; Papp, K.A.; van den Bosch, F.E.; Tsuji, S.; Dokoupilova, E.; Keiserman, M.W.; Bu, X.; Chen, L.; McCaskill, R.M.; et al. Upadacitinib in Patients with Psoriatic Arthritis and Inadequate Response to Biologics: 56-Week Data from the Randomized Controlled Phase 3 SELECT-PsA 2 Study. Rheumatol. Ther. 2021, 8, 903–919. [Google Scholar] [CrossRef]
- Baraliakos, X.; Ranza, R.; Östör, A.; Ciccia, F.; Coates, L.C.; Rednic, S.; Walsh, J.A.; Douglas, K.; Gao, T.; Kato, K.; et al. Efficacy and safety of upadacitinib in patients with active psoriatic arthritis and axial involvement: Results from two phase 3 studies. Arthritis Res. Ther. 2023, 25, 56. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Setty, A.; Papp, K.; Van den Bosch, F.; Tsuji, S.; Keiserman, M.; Carter, K.; Li, Y.; McCaskill, R.; McDearmon-Blondell, E.; et al. Upadacitinib in patients with psoriatic arthritis and inadequate response to biologics: 3-year results from the open-label extension of the randomised controlled phase 3 SELECT-PsA 2 study. Clin. Exp. Rheumatol. 2023, 41, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Menter, M.A.; Raman, M.; Disch, D.; Schlichting, D.E.; Gaich, C.; Macias, W.; Zhang, X.; Janes, J.M. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br. J. Dermatol. 2016, 174, 1266–1276. [Google Scholar] [CrossRef]
- Forman, S.B.; Pariser, D.M.; Poulin, Y.; Vincent, M.S.; Gilbert, S.A.; Kieras, E.M.; Qiu, R.; Yu, D.; Papacharalambous, J.; Tehlirian, C.; et al. TYK2/JAK1 Inhibitor PF-06700841 in Patients with Plaque Psoriasis: Phase IIa, Randomized, Double-Blind, Placebo-Controlled Trial. J. Investig. Dermatol. 2020, 140, 2359–2370.e5. [Google Scholar] [CrossRef]
- Mease, P.; Helliwell, P.; Silwinska-Stanczyk, P.; Miakisz, M.; Ostor, A.; Peeva, E.; Vincent, M.S.; Sun, Q.; Sikirica, V.; Winnette, R.; et al. Efficacy and Safety of the TYK2/JAK1 Inhibitor Brepocitinib for Active Psoriatic Arthritis: A Phase IIb Randomized Controlled Trial. Arthritis Rheumatol. 2023, 75, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Coates, L.C.; Helliwell, P.S.; Stanislavchuk, M.; Rychlewska-Hanczewska, A.; Dudek, A.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Harrison, P.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Socié, G.; Schroeder, M.A.; Abhyankar, S.; Vaz, C.P.; Kwon, M.; Clausen, J.; Volodin, L.; Giebel, S.; Chacon, M.J.; et al. Efficacy and safety of itacitinib versus placebo in combination with corticosteroids for initial treatment of acute graft-versus-host disease (GRAVITAS-301): A randomised, multicentre, double-blind, phase 3 trial. Lancet Haematol. 2022, 9, e14–e25. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Luchi, M.; Fidelus-Gort, R.; Jackson, S.; Zhang, H.; Flores, R.; Newton, R.; Scherle, P.; Yeleswaram, S.; Chen, X.; et al. A randomized, double-blind, placebo-controlled, dose-escalation study of the safety and efficacy of INCB039110, an oral janus kinase 1 inhibitor, in patients with stable, chronic plaque psoriasis. J. Dermatol. Treat. 2016, 27, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Takeuchi, T.; Morita, Y.; Kaneko, Y.; Terada, W. Safety and effectiveness of peficitinib 100 mg/day in patients achieving clinical remission from a long-term open-label extension study in Japan, Korea, and Taiwan (RAJ2). Mod. Rheumatol. 2023, road110. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Pariser, D.; Catlin, M.; Wierz, G.; Ball, G.; Akinlade, B.; Zeiher, B.; Krueger, J.G. A phase 2a randomized, double-blind, placebo-controlled, sequential dose-escalation study to evaluate the efficacy and safety of ASP015K, a novel Janus kinase inhibitor, in patients with moderate-to-severe psoriasis. Br. J. Dermatol. 2015, 173, 767–776. [Google Scholar] [CrossRef]
- Tehlirian, C.; Singh, R.S.; Pradhan, V.; Roberts, E.S.; Tarabar, S.; Peeva, E.; Vincent, M.S.; Gale, J.D. Oral tyrosine kinase 2 inhibitor PF-06826647 demonstrates efficacy and an acceptable safety profile in participants with moderate-to-severe plaque psoriasis in a phase 2b, randomized, double-blind, placebo-controlled study. J. Am. Acad. Dermatol. 2022, 87, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Ludbrook, V.J.; Hicks, K.J.; Hanrott, K.E.; Patel, J.S.; Binks, M.H.; Wyres, M.R.; Watson, J.; Wilson, P.; Simeoni, M.; Schifano, L.A.; et al. Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase IIa study. Br. J. Dermatol. 2016, 174, 985–995. [Google Scholar] [CrossRef]
- Landis, M.N.; Smith, S.R.; Berstein, G.; Fetterly, G.; Ghosh, P.; Feng, G.; Pradhan, V.; Aggarwal, S.; Banfield, C.; Peeva, E.; et al. Efficacy and safety of topical brepocitinib cream for mild-to-moderate chronic plaque psoriasis: A phase IIb randomized double-blind vehicle-controlled parallel-group study. Br. J. Dermatol. 2023, 189, 33–41. [Google Scholar] [CrossRef]
- Punwani, N.; Scherle, P.; Flores, R.; Shi, J.; Liang, J.; Yeleswaram, S.; Levy, R.; Williams, W.; Gottlieb, A. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J. Am. Acad. Dermatol. 2012, 67, 658–664. [Google Scholar] [CrossRef]
- Punwani, N.; Burn, T.; Scherle, P.; Flores, R.; Shi, J.; Collier, P.; Hertel, D.; Haley, P.; Lo, Y.; Waeltz, P.; et al. Downmodulation of key inflammatory cell markers with a topical Janus kinase 1/2 inhibitor. Br. J. Dermatol. 2015, 173, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Ports, W.C.; Khan, S.; Lan, S.; Lamba, M.; Bolduc, C.; Bissonnette, R.; Papp, K. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br. J. Dermatol. 2013, 169, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Bissonnette, R.; Gooderham, M.; Feldman, S.R.; Iversen, L.; Soung, J.; Draelos, Z.; Mamolo, C.; Purohit, V.; Wang, C.; et al. Treatment of plaque psoriasis with an ointment formulation of the Janus kinase inhibitor, tofacitinib: A Phase 2b randomized clinical trial. BMC Dermatol. 2016, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, F.; Korman, N.J.; Bissonnette, R.; Bakos, N.; Tsai, T.F.; Harper, M.K.; Ports, W.C.; Tan, H.; Tallman, A.; Valdez, H.; et al. Tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: Long-term safety and efficacy in an open-label extension study. Br. J. Dermatol. 2018, 179, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Nash, P.; Coates, L.C.; Fleishaker, D.; Kivitz, A.J.; Mease, P.J.; Gladman, D.D.; FitzGerald, O.; Wang, C.; Wu, J.; Hsu, M.A.; et al. Safety and efficacy of tofacitinib up to 48 months in patients with active psoriatic arthritis: Final analysis of the OPAL Balance long-term extension study. Lancet Rheumatol. 2021, 3, e270–e283. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Charles-Schoeman, C.; Cohen, S.; Fallon, L.; Woolcott, J.; Yun, H.; Kremer, J.; Greenberg, J.; Malley, W.; Onofrei, A.; et al. Incidence of venous and arterial thromboembolic events reported in the tofacitinib rheumatoid arthritis, psoriasis and psoriatic arthritis development programmes and from real-world data. Ann. Rheum. Dis. 2020, 79, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.E.; Strober, B.; Poddubnyy, D.; Leung, Y.Y.; Jo, H.; Kwok, K.; Vranic, I.; Fleishaker, D.L.; Fallon, L.; Yndestad, A.; et al. Association between baseline cardiovascular risk and incidence rates of major adverse cardiovascular events and malignancies in patients with psoriatic arthritis and psoriasis receiving tofacitinib. Ther. Adv. Musculoskelet. Dis. 2023, 15. [Google Scholar] [CrossRef]
- Kristensen, L.E.; Danese, S.; Yndestad, A.; Wang, C.; Nagy, E.; Modesto, I.; Rivas, J.; Benda, B. Identification of two tofacitinib subpopulations with different relative risk versus TNF inhibitors: An analysis of the open label, randomised controlled study ORAL Surveillance. Ann. Rheum. Dis. 2023, 82, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Charles-Schoeman, C.; McInnes, I.B.; Veale, D.J.; Thiers, B.; Nurmohamed, M.; Graham, D.; Wang, C.; Jones, T.; Wolk, R.; et al. Changes in Lipid Levels and Incidence of Cardiovascular Events Following Tofacitinib Treatment in Patients with Psoriatic Arthritis: A Pooled Analysis Across Phase III and Long-Term Extension Studies. Arthritis Care Res. 2019, 71, 1387–1395. [Google Scholar] [CrossRef]
- Catlett, I.M.; Hu, Y.; Gao, L.; Banerjee, S.; Gordon, K.; Krueger, J.G. Molecular and clinical effects of selective tyrosine kinase 2 inhibition with deucravacitinib in psoriasis. J. Allergy Clin. Immunol. 2022, 149, 2010–2020.e8. [Google Scholar] [CrossRef]
- McInnes, I.B.; Anderson, J.K.; Magrey, M.; Merola, J.F.; Liu, Y.; Kishimoto, M.; Jeka, S.; Pacheco-Tena, C.; Wang, X.; Chen, L.; et al. Trial of Upadacitinib and Adalimumab for Psoriatic Arthritis. N. Engl. J. Med. 2021, 384, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarska, A.; Kwiatkowska, D.; Skrzypek, K.K.; Kowalewski, Z.T.; Jaworecka, K.; Reich, A. Pathomechanism of Pruritus in Psoriasis and Atopic Dermatitis: Novel Approaches, Similarities and Differences. Int. J. Mol. Sci. 2023, 24, 14734. [Google Scholar] [CrossRef] [PubMed]
- Pfizer Receives Complete Response Letter from FDA for Oral XELJANZ® (Tofacitinib Citrate) Supplemental New Drug Application for Moderate to Severe Chronic Plaque Psoriasis|Pfizer. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer_receives_complete_response_letter_from_fda_for_oral_xeljanz_tofacitinib_citrate_supplemental_new_drug_application_for_moderate_to_severe_chronic_plaque_psoriasis (accessed on 18 April 2024).
- European Medicine Agency. EMA Confirms Measures to Minimise Risk of Serious Side Effects with Janus Kinase Inhibitors for Chronic Inflammatory Disorders. EMA/27681/2023. 27 January 2023. Available online: https://www.ema.europa.eu/en/medicines/human/referrals/janus-kinase-inhibitors-jaki (accessed on 18 April 2024).
- Ytterberg, S.R.; Bhatt, D.L.; Mikuls, T.R.; Koch, G.G.; Fleischmann, R.; Rivas, J.L.; Germino, R.; Menon, S.; Sun, Y.; Wang, C.; et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N. Engl. J. Med. 2022, 386, 316–326. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cytokines | Signaling Pathway | Effect upon Inhibition with JAKis | |
|---|---|---|---|
| IL-12, -23 | JAK2/ TYK2 | STAT3, STAT4 | inhibition if the IL-23/IL-17 axis, inhibition of Th1 differentiation, decreased IFN-γ production |
| IL-2, -4, -7, -9, -15, -21 (common γ-chain) | JAK1/ JAK3 | STAT1, STAT3, STAT5, STAT6 | inhibition of Th, cT, and NK-cell activity |
| IL-22 | JAK1/TYK2 | STAT3 | normalization of keratinocyte differentiation and barrier function, reduction of antimicrobial peptide secretion by keratinocytes |
| IFN-γ | JAK1/ JAK2 | STAT1 | inhibition of Th1 activity, inhibition of T cell migration, inhibition of keratinocyte proliferation |
| IFN-α, -β | JAK1/ TYK2 | STAT1, STAT2, STAT3 | inhibition of T-cell mediated inflammation |
| EPO. TPO, GM-CSF | JAK2/ JAK2 | STAT5 | decrease in hemoglobin, lymphocyte, and neutrophil count |
| Tofacitinib | ||||||||
|---|---|---|---|---|---|---|---|---|
| Trial | Population—Diagnosis and No. | Type of Study | Trial Arms | Primary Endpoint | Results | Any Adverse Event | Serious Adverse Events | |
| NCT00678210 Papp et al., 2012 [72] | Pso 197 | phase 2 | T2 T5 T10 placebo | %PASI75 at week 12 | 25% 40.8% 66.7% 2% | 551% 57.1% 61.2% 60% | 4.1% 2% 0 0 | |
| NCT01186744 Bissonnette et al., 2015 [75] | Pso 666 | phase 3 | Treatment period: | |||||
| T5 T10 | %PASI75 and PGA0/1 at week 24 | 33.5% 55.2% | 61.9% 69.9% | 1.8% 3.3% | ||||
| Treatment-withdrawal period: | ||||||||
| T5 Placebo for T5 T10 Placebo for T10 | % maintaining PASI75 at week 40 | 56.2% 23.3% 62.3% 26.1% | 54.8% 50% 62.2% 42.9% | |||||
| % maintaining PGA0/1 at week 40 | 49.9% 22.9% 63.9% 18% | |||||||
| Re-treatment period: | ||||||||
| T5 T5 from placebo T10 T10 from placebo | % who relapsed and regained PASI75 at week 56 | T5 from placebo | 36.8% | 29.6% 507% 40.5% 53.3% | 3.7% 4% 2.4% 1.7% | |||
| T10 from placebo | 61% | |||||||
| % who relapsed and regained PGA0/1 at week 56 | T5 from placebo | 44.8% | ||||||
| T10 from placebo | 57.1% | |||||||
| NCT01241591 (OPT Compare) Bachelez et al., 2015 [76] | Pso 1101 | phase 3 | T5 T10 Etanercept Placebo | %PASI75 | 39.5% 63.6% 58.8% 5.6% | 55% 60% 57% 51% | 2% 2% 2% 2% | |
| %PGA score of 0 or 1 at week 12 | 47.1% 68.2% 66.3% 15% | |||||||
| NCT01276639 (OPT Pivotal 1) Papp et al., 2015 [77] | Pso 901 | phase 3 | T5 T10 placebo | %PASI75 at week 16 | 39.9% 59.2% 6.2% | 51% 61.1% 50.3% | 2.2% 2.8% 2.8% | |
| %PGA 0/1 at week 16 | 41.9% 59.2% 9% | |||||||
| NCT01309737 (OPT Pivotal 2) Papp et al., 2015 [77] | Pso 960 | phase 3 | T5 T10 placebo | %PASI75 at week 16 | 46% 59.6% 11.4% | 55.8% 55.6% 47.4% | 2.9% 1.3% 1% | |
| %PGA 0/1 at week 16 | 46% 59.1% 10.9% | |||||||
| NCT01815424 Zhang et al., 2017 [78] | Pso 266 | phase 3 | T5 T10 Placebo | %PASI75 at week 16 | 54.6% 81.1% 12.5% | 64.8% 67.8% 48.9% | 2.3% 0 0 | |
| % PGA 0/1 at week 16 | 52.3% 75.6% 19.3% | |||||||
| NCT01882439 (OPAL Beyond) Gladman et al., 2017 [79] | PsA 395 | phase 3 | T5 T10 placebo | %ACR20 at 3 months | 50% 47% 24% | 55% 53% 44% | 1% 2% 2% | |
| change in HAQ-DI at 3 months | −0.39 −0.35 −0.14 | |||||||
| NCT01877668 (OPAL Broaden) Mease et al., 2017 [80] | PsA 422 | phase 3 | T5 T10 adalimumab placebo | %ACR20 at 3 months | 50% 61% 52% 33% | 39% 45% 46% 35% | 3% 1% 1% 1% | |
| change in HAQ-DI at 3 months | −0.35 −0.40 −0.38 −0.18 | |||||||
| NCT03486457 Leng et al., 2022 [81] | PsA 204 | phase 3 | T5 placebo | %ACR50 at 3 months | 38.2% 5.9% | 68.4% 75% | 0 4.4% | |
| Deucravacitinib | |||||||
|---|---|---|---|---|---|---|---|
| Trial | Population—Diagnosis and No. | Type of Study | Trial Arms | Primary Endpoint | Results | Any Adverse Event | Serious Adverse Events |
| NCT02931838 Papp et al., 2018 [102] | Pso 267 | Phase 2 | deucravacitinib 3 mg/2 d deucravacitinib 3 mg/d deucravacitinib 3 mg × 2/d deucravacitinib 6 mg × 2/d deucravacitinib 12 mg/d placebo | %PASI75 at week 12 | 9% 39% 69% 67% 75% 7% | 59% 55% 64% 80% 77% 51% | 2% 2% 2% 0 0 2% |
| NCT03624127 (POETYK PSO-1) Armstrong et al., 2023 [103] | Pso 666 | phase 3 | deucravacitinib 6 mg apremilast 30 mg placebo | %PASI75 at week 16 for deucravacitinib vs. placebo | 58.4% vs. 12.7% | 53% 55.4% 42.4% | 2.1% 2.4% 5.5% |
| %PGA 0/1 at week 16 for deucravacitinib vs. placebo | 53.6% vs. 7.2% | ||||||
| NCT03611751 (POETYK PSO-2) Strober et al., 2022 [104] | Pso 885 | Phase 3 | deucravacitinib 6 mg apremilast 30 mg placebo | %PASI75 at week 16 for deucravacitinib vs. placebo | 53.0% vs. 9.4% | 57.5% 59.1% 54.3% | 1.6% 0.4% 1.2% |
| %PGA 0/1 at week 16 for deucravacitinib vs. placebo | 49.5% vs. 8.6% | ||||||
| NCT03881059 Mease et al., 2022 [105] | PsA 203 | Phase 2 | deucravacitinib 6 mg deucravacitinib 12 mg placebo | % ACR20 at Week 16 | 62.7% 52.9% 31.8% | 65.7% 65.7% 42.4% | 0 0 1.5% |
| Upadacitinib | |||||||
|---|---|---|---|---|---|---|---|
| Trial | Population—Diagnosis and No. | Type of Study | Trial Arms | Primary Endpoint | Results | Any Adverse Event | Serious Adverse Events |
| NCT03104400 (SELECT-PsA 1) McInnes et al., 2021 [108] | PsA 1704 | phase 3 | U15 U30 Placebo Adalimumab | %ACR20 at week 12 | 70.6% 78.5% 36.2% 65.0% | 66.9% 72.3% 59.6% 64.8% | 3.3% 6.1% 3.1% 3.7% |
| NCT03104374 (SELECT-PsA 2) Mease et al., 2020 [109] | PsA | phase 3 | U15 U30 Placebo Adalimumab | %ACR20 at week 12 | 56.9% 63.8% 24.1% | 64% 78% 65.6% | 5.7% 8.3% 1.9% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furtunescu, A.R.; Georgescu, S.R.; Tampa, M.; Matei, C. Inhibition of the JAK-STAT Pathway in the Treatment of Psoriasis: A Review of the Literature. Int. J. Mol. Sci. 2024, 25, 4681. https://doi.org/10.3390/ijms25094681
Furtunescu AR, Georgescu SR, Tampa M, Matei C. Inhibition of the JAK-STAT Pathway in the Treatment of Psoriasis: A Review of the Literature. International Journal of Molecular Sciences. 2024; 25(9):4681. https://doi.org/10.3390/ijms25094681
Chicago/Turabian StyleFurtunescu, Andreea Roxana, Simona Roxana Georgescu, Mircea Tampa, and Clara Matei. 2024. "Inhibition of the JAK-STAT Pathway in the Treatment of Psoriasis: A Review of the Literature" International Journal of Molecular Sciences 25, no. 9: 4681. https://doi.org/10.3390/ijms25094681
APA StyleFurtunescu, A. R., Georgescu, S. R., Tampa, M., & Matei, C. (2024). Inhibition of the JAK-STAT Pathway in the Treatment of Psoriasis: A Review of the Literature. International Journal of Molecular Sciences, 25(9), 4681. https://doi.org/10.3390/ijms25094681