
Co-Mutations and Possible Variation Tendency of the Spike RBD and Membrane Protein in SARS-CoV-2 by Machine Learning

 ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
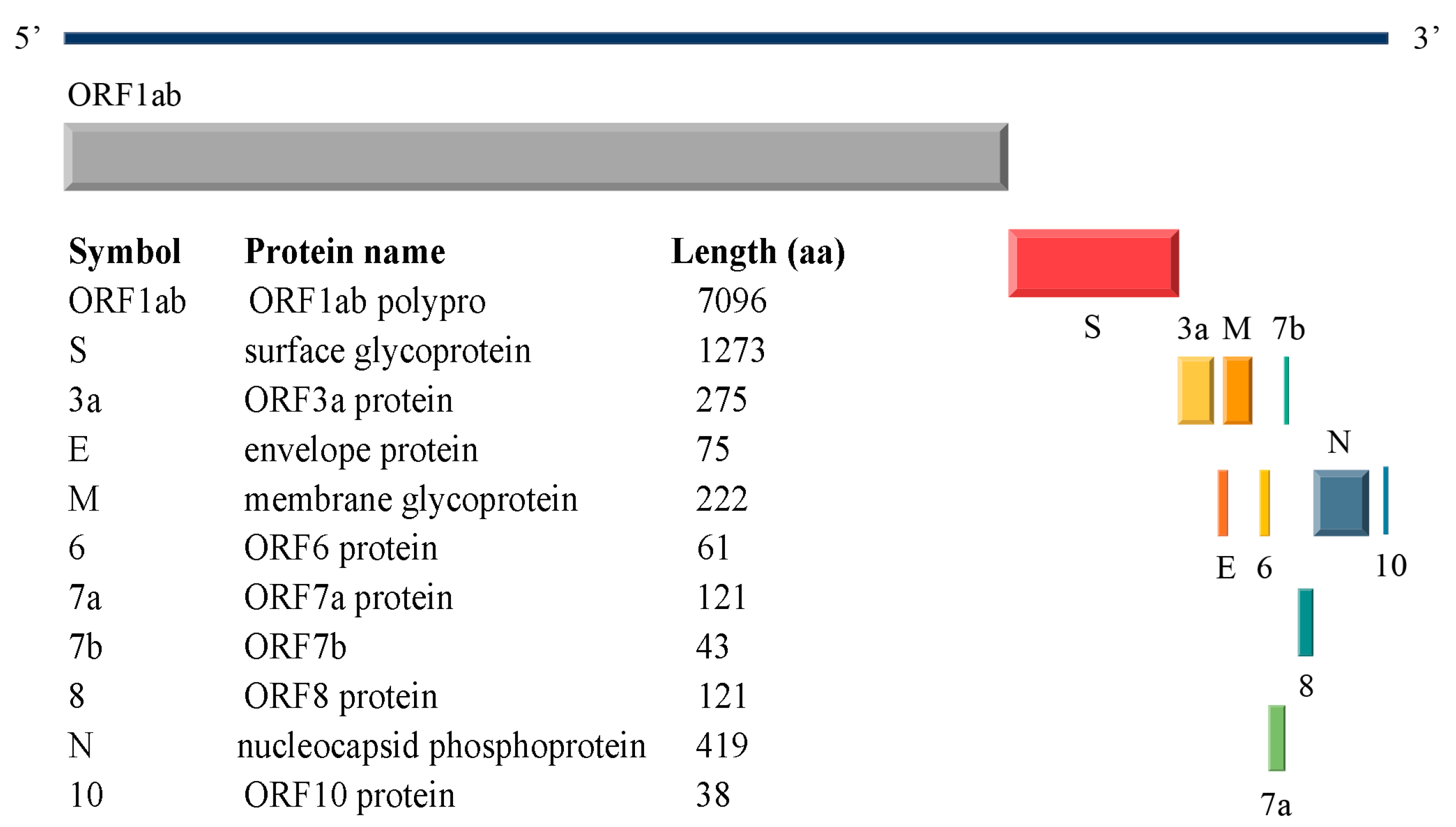
1.1. SARS-CoV-2 Structural Proteins
1.2. SARS-CoV-2 Variants
1.3. Mutational Correlations Analysis
2. Results
2.1. Variation Analyses at the Amino Acid Level
2.1.1. Results of Single Mutation Analysis
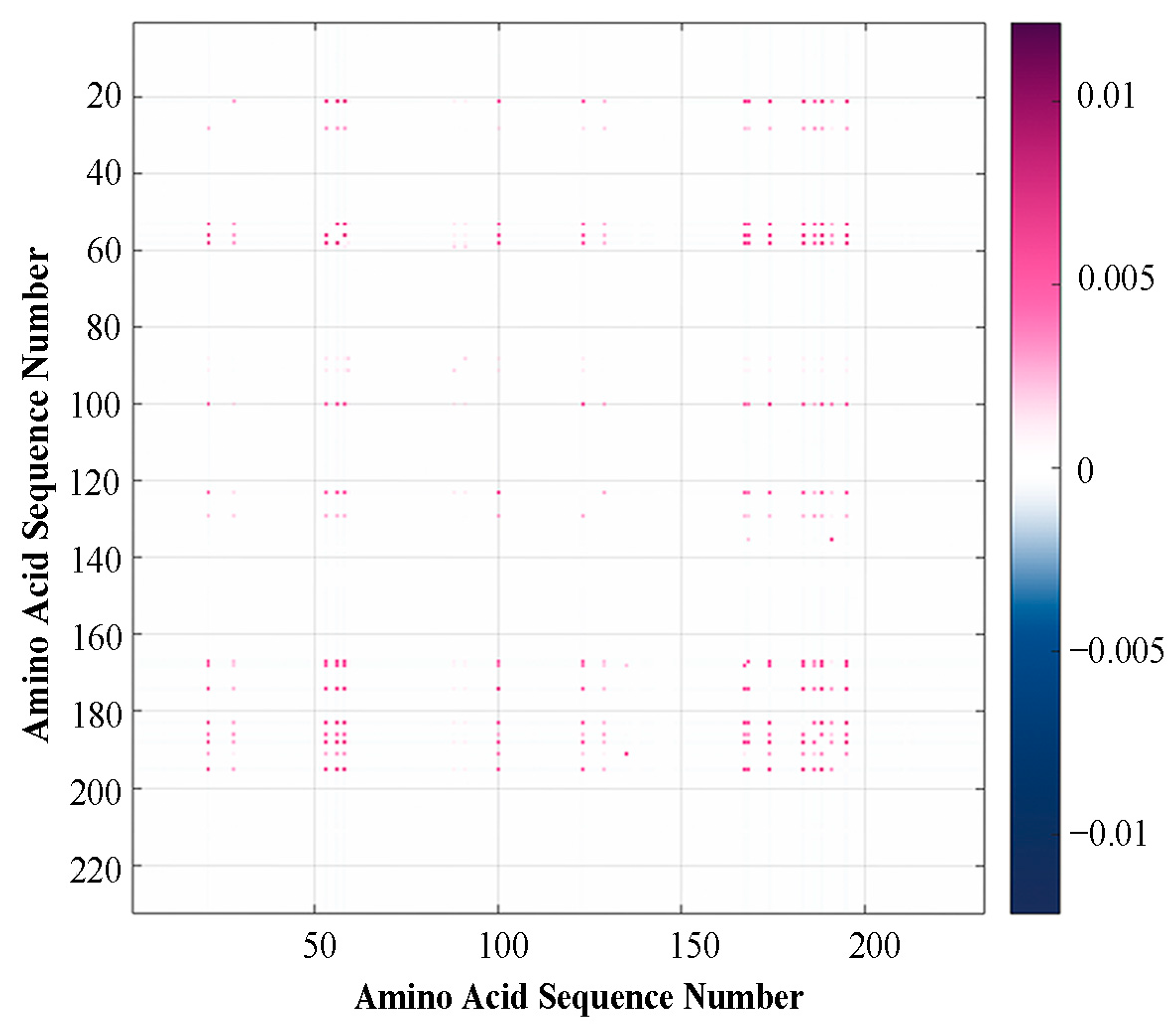
2.1.2. Results of Multiple Mutation Analysis
2.2. Possible Variation Tendency Identified by S2STM
3. Discussion
4. Materials and Methods
4.1. Sequence Retrieval and Preparation
4.2. Mutational Synergy Analysis
4.2.1. Single Mutations
4.2.2. Multiple Mutations
4.3. Sequence Translation
4.3.1. Sequence-to-Sequence Transformer Model
4.3.2. Model Parameters
4.3.3. Dataset Selection and Division
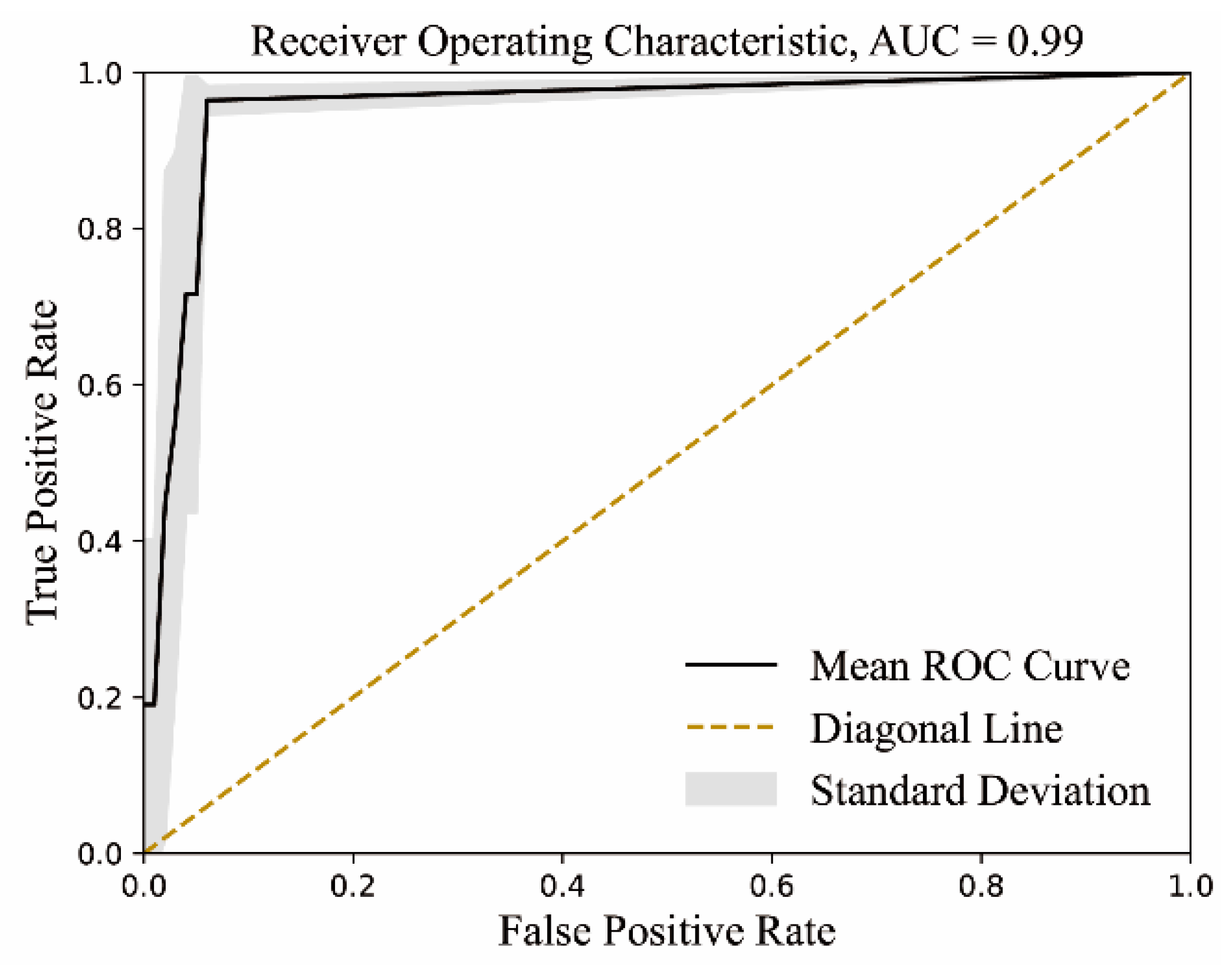
4.3.4. Model Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; COVID-19 Genomics UK Consortium; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Knisely, J.M.; Buyon, L.E.; Mandt, R.; Farkas, R.; Balasingam, S.; Bok, K.; Buchholz, U.J.; D’Souza, M.P.; Gordon, J.L.; King, D.F. Mucosal vaccines for SARS-CoV-2: Scientific gaps and opportunities—Workshop report. npj Vaccines 2023, 8, 53. [Google Scholar] [CrossRef]
- Menegale, F.; Manica, M.; Zardini, A.; Guzzetta, G.; Marziano, V.; d’Andrea, V.; Trentini, F.; Ajelli, M.; Poletti, P.; Merler, S. Evaluation of waning of SARS-CoV-2 vaccine–induced immunity: A systematic review and meta-analysis. JAMA Netw. Open 2023, 6, e2310650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Chalupka, A.; Richter, L.; Chakeri, A.; El-Khatib, Z.; Theiler-Schwetz, V.; Trummer, C.; Krause, R.; Willeit, P.; Benka, B.; Ioannidis, J.P. Effectiveness of a fourth SARS-CoV-2 vaccine dose in previously infected individuals from Austria. Eur. J. Clin. Investig. 2024, 54, e14136. [Google Scholar] [CrossRef] [PubMed]
- Polatoğlu, I.; Oncu-Oner, T.; Dalman, I.; Ozdogan, S. COVID-19 in early 2023: Structure, replication mechanism, variants of SARS-CoV-2, diagnostic tests, and vaccine & drug development studies. MedComm 2023, 4, e228. [Google Scholar]
- Sia, S.F.; Yan, L.-M.; Chin, A.W.H.; Fung, K.; Choy, K.-T.; Wong, A.Y.L.; Kaewpreedee, P.; Perera, R.A.P.M.; Poon, L.L.M.; Nicholls, J.M.; et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 2020, 583, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, S.; Wang, P.; Chen, X.; Bi, J.; Cheng, L.; Zhang, X. A comprehensive review of the analysis and integration of omics data for SARS-CoV-2 and COVID-19. Brief. Bioinform. 2021, 23, bbab446. [Google Scholar]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef]
- Munnink, B.B.O.; Sikkema, R.S.; Nieuwenhuijse, D.F.; Molenaar, R.J.; Munger, E.; Molenkamp, R.; van der Spek, A.; Tolsma, P.; Rietveld, A.; Brouwer, M.; et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 2021, 371, 172–177. [Google Scholar] [CrossRef]
- Satarker, S.; Nampoothiri, M. Structural Proteins in Severe Acute Respiratory Syndrome Coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Cano-Muñoz, M.; Jurado, S.; Morel, B.; Conejero-Lara, F. Conformational flexibility of the conserved hydrophobic pocket of HIV-1 gp41. Implications for the discovery of small-molecule fusion inhibitors. Int. J. Biol. Macromol. 2021, 192, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Cano-Muñoz, M.; Lucas, J.; Lin, L.-Y.; Cesaro, S.; Moog, C.; Conejero-Lara, F. Conformational stabilization of Gp41-mimetic miniproteins opens up new ways of inhibiting HIV-1 fusion. Int. J. Mol. Sci. 2022, 23, 2794. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Kratzel, A.; Barut, G.T.; Lang, R.M.; Aguiar Moreira, E.; Thomann, L.; Kelly, J.N.; Thiel, V. SARS-CoV-2 biology and host interactions. Nat. Rev. Microbiol. 2024, 22, 206–225. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zheng, T.; Xu, K.; Han, Y.; Xu, L.; Huang, E.; An, Y.; Cheng, Y.; Li, S.; Liu, M.; et al. A Universal Design of Betacoronavirus Vaccines against COVID-19, MERS, and SARS. Cell 2020, 182, 722–733. [Google Scholar] [CrossRef]
- Xu, K.; Gao, P.; Liu, S.; Lu, S.; Lei, W.; Zheng, T.; Liu, X.; Xie, Y.; Zhao, Z.; Guo, S.; et al. Protective prototype-Beta and Delta-Omicron chimeric RBD-dimer vaccines against SARS-CoV-2. Cell 2022, 185, 2265–2278. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- de Haan, C.A.; Rottier, P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar]
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J.; et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Du, N.; Lei, Y.; Dorje, S.; Qi, J.; Luo, T.; Gao, G.F.; Song, H. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. Embo J. 2020, 39, e105938. [Google Scholar] [CrossRef] [PubMed]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.; Matrosovich, M.; Klenk, H.D. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev. Med. Virol. 2002, 12, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhu, X.; Dwek, R.A.; Stevens, J.; Wilson, I.A. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 2008, 82, 10493–10501. [Google Scholar] [CrossRef] [PubMed]
- Katz, G.; Benkarroum, Y.; Wei, H.; Rice, W.J.; Bucher, D.; Alimova, A.; Katz, A.; Klukowska, J.; Herman, G.T.; Gottlieb, P. Morphology of influenza B/Lee/40 determined by cryo-electron microscopy. PLoS ONE 2014, 9, e88288. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef]
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Ze-Ze, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- Zhang, J.; Han, Z.B.; Liang, Y.; Zhang, X.F.; Jin, Y.Q.; Du, L.F.; Shao, S.; Wang, H.; Hou, J.W.; Xu, K.; et al. A mosaic-type trimeric RBD-based COVID-19 vaccine candidate induces potent neutralization against Omicron and other SARS-CoV-2 variants. Elife 2022, 11, e78633. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef] [PubMed]
- Makowski, E.K.; Schardt, J.S.; Smith, M.D.; Tessier, P.M. Mutational analysis of SARS-CoV-2 variants of concern reveals key tradeoffs between receptor affinity and antibody escape. PLoS Comput. Biol. 2022, 18, e1010160. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Lin, Y.; Gu, C.; Ding, L.; Lu, X.; Yang, Z.; Zhang, Y.; Ma, L.; Gu, W.; et al. Comprehensive mapping of binding hot spots of SARS-CoV-2 RBD-specific neutralizing antibodies for tracking immune escape variants. Genome Med. 2021, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gao, Y.; Li, T.; Li, T.; Lu, Y.; Zheng, L.; Liu, Y.; Yang, T.; Luo, F.; Song, S.; et al. Structures of Omicron spike complexes and implications for neutralizing antibody development. Cell Rep. 2022, 39, 110770. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Kmiec, D.; Koepke, L.; Zech, F.; Jacob, T.; Sparrer, K.M.J.; Kirchhoff, F. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol. 2022, 96, e02077. [Google Scholar] [CrossRef]
- Kannan, S.; Ali, P.S.S.; Sheeza, A. Omicron (B.1.1.529)—Variant of concern—Molecular profile and epidemiology: A mini review. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 8019–8022. [Google Scholar] [PubMed]
- Kupferschmidt, K. COVID-19 New mutations raise specter of ‘immune escape’. Science 2021, 371, 329–330. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, K.; Guarnaccia, T.; Carolan, L.A.; Maurer-Stroh, S.; Lee, R.T.C.; Job, E.; Reading, P.C.; Petrie, S.; McCaw, J.M.; McVernon, J.; et al. Antigenic Drift of the Pandemic 2009 A(H1N1) Influenza Virus in a Ferret Model. PLoS Pathog. 2013, 9, e1003354. [Google Scholar]
- Tewawong, N.; Prachayangprecha, S.; Vichiwattana, P.; Korkong, S.; Klinfueng, S.; Vongpunsawad, S.; Thongmee, T.; Theamboonlers, A.; Poovorawan, Y. Assessing Antigenic Drift of Seasonal Influenza A(H3N2) and A(H1N1)pdm09 Viruses. PLoS ONE 2015, 10, e0139958. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Martin, S.R.; Wharton, S.A.; McCauley, J.W. Biophysical Measurement of the Balance of Influenza A Hemagglutinin and Neuraminidase Activities. J. Biol. Chem. 2015, 290, 6516–6521. [Google Scholar] [CrossRef]
- Du, X.; Wang, Z.; Wu, A.; Song, L.; Cao, Y.; Hang, H.; Jiang, T. Networks of genomic co-occurrence capture characteristics of human influenza A (H3N2) evolution. Genome Res. 2008, 18, 178–187. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaverin, N.V.; Gambaryan, A.S.; Bovin, N.V.; Rudneva, I.A.; Shilov, A.A.; Khodova, O.M.; Varich, N.L.; Sinitsin, B.V.; Makarova, N.V.; Kropotkina, E.A. Postreassortment changes in influenza A virus hemagglutinin restoring HA-NA functional match. Virology 1998, 244, 315–321. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. S4), D49–D53. [Google Scholar] [CrossRef]
- Vincent, A.; Awada, L.; Brown, I.; Chen, H.; Claes, F.; Dauphin, G.; Donis, R.; Culhane, M.; Hamilton, K.; Lewis, N.; et al. Review of influenza A virus in swine worldwide: A call for increased surveillance and research. Zoonoses Public Health 2014, 61, 4–17. [Google Scholar] [CrossRef]
- Göbel, U.; Sander, C.; Schneider, R.; Valencia, A. Correlated Mutations and Residue Contacts in Proteins. Proteins: Struct. Funct. Bioinform. 1994, 18, 309–317. [Google Scholar] [CrossRef]
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108, E1293–E1301. [Google Scholar] [CrossRef]
- Hopf, T.A.; Ingraham, J.B.; Poelwijk, F.J.; Scharfe, C.P.; Springer, M.; Sander, C.; Marks, D.S. Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 2017, 35, 128–135. [Google Scholar] [CrossRef]
- Wang, H.; Zang, Y.; Zhao, Y.; Hao, D.; Kang, Y.; Zhang, J.; Zhang, Z.; Zhang, L.; Yang, Z.; Zhang, S. Sequence Matching between Hemagglutinin and Neuraminidase through Sequence Analysis Using Machine Learning. Viruses 2022, 14, 469. [Google Scholar] [CrossRef]
- Stigler, S.M. Francis Galton’s account of the invention of correlation. Stat. Sci. 1989, 4, 73–79. [Google Scholar] [CrossRef]
- Md Saad, R.; Ahmad, M.Z.; Abu, M.S.; Jusoh, M.S. Hamming distance method with subjective and objective weights for personnel selection. Sci. World J. 2014, 2014, 865495. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.P. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 1997, 30, 1145–1159. [Google Scholar] [CrossRef]
- Verma, S.; Patil, V.M.; Gupta, M.K. Mutation informatics: SARS-CoV-2 receptor-binding domain of the spike protein. Drug Discov. Today 2022, 27, 103312. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Hozumi, Y.; Yin, C.; Wei, G.-W. Emerging Vaccine-Breakthrough SARS-CoV-2 Variants. Acs Infect. Dis. 2022, 8, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Cano-Muñoz, M.; Polo-Megías, D.; Cámara-Artigas, A.; Gavira, J.A.; López-Rodríguez, M.J.; Laumond, G.; Schmidt, S.; Demiselle, J.; Bahram, S.; Moog, C. Novel chimeric proteins mimicking SARS-CoV-2 spike epitopes with broad inhibitory activity. Int. J. Biol. Macromol. 2022, 222, 2467–2478. [Google Scholar] [CrossRef]
- Braeye, T.; Catteau, L.; Brondeel, R.; van Loenhout, J.A.; Proesmans, K.; Cornelissen, L.; Van Oyen, H.; Stouten, V.; Hubin, P.; Billuart, M. Vaccine effectiveness against transmission of alpha, delta and omicron SARS-CoV-2-infection, Belgian contact tracing, 2021–2022. Vaccine 2023, 41, 3292–3300. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Okumura, K.; Padilla-Blanco, M.; Kosugi, Y.; Uriu, K.; Hinay, A.A.; Chen, L.; Plianchaisuk, A.; Kobiyama, K.; Ishii, K.J. Virological characteristics of the SARS-CoV-2 JN. 1 variant. Lancet Infect. Dis. 2024, 24, e82. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef]
- Tamura, T.; Mizuma, K.; Nasser, H.; Deguchi, S.; Padilla-Blanco, M.; Oda, Y.; Uriu, K.; Tolentino, J.E.; Tsujino, S.; Suzuki, R. Virological characteristics of the SARS-CoV-2 BA. 2.86 variant. Cell Host Microbe 2024, 32, 170–180.e112. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. In Proceedings of the 31st Annual Conference on Neural Information Processing Systems (NIPS), Long Beach, CA, USA, 4–9 December 2017; Association of Computing Machinery: Long Beach, CA, USA, 2017; pp. 6000–6010. [Google Scholar]
- Kingma, D.P.; Ba, J.L. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Savino, S.; Desmet, T.; Franceus, J. Insertions and deletions in protein evolution and engineering. Biotechnol. Adv. 2022, 60, 108010. [Google Scholar] [CrossRef]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A system for large-scale machine learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI), Savannah, GA, USA, 2–4 November 2016; Association of Computing Machinery: Savannah, GA, USA, 2016; pp. 265–283. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO Nomenclature or Designation | Pango Lineage | Spike RBD Mutations of Interest | M Mutations of Interest |
|---|---|---|---|
| Alpha | B.1.1.7 | N501Y | |
| Beta | B.1351 | K417N, N501Y, E484K | |
| Gamma | P.1 | K417T, N501Y, E484K | |
| Delta | B.1.617.2 | L452R, T478K | I82T |
| Omicron | B.1.1.529 | G339D, S371F/L, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S447N, T478K, E484A, Q493R, Q498R, N501Y, Y505H | D3G, Q19E, A63T, I82T |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Q.; Wang, H.; Xu, F.; Zhang, S.; Zhang, S.; Yang, Z.; Zhang, L. Co-Mutations and Possible Variation Tendency of the Spike RBD and Membrane Protein in SARS-CoV-2 by Machine Learning. Int. J. Mol. Sci. 2024, 25, 4662. https://doi.org/10.3390/ijms25094662
Ye Q, Wang H, Xu F, Zhang S, Zhang S, Yang Z, Zhang L. Co-Mutations and Possible Variation Tendency of the Spike RBD and Membrane Protein in SARS-CoV-2 by Machine Learning. International Journal of Molecular Sciences. 2024; 25(9):4662. https://doi.org/10.3390/ijms25094662
Chicago/Turabian StyleYe, Qiushi, He Wang, Fanding Xu, Sijia Zhang, Shengli Zhang, Zhiwei Yang, and Lei Zhang. 2024. "Co-Mutations and Possible Variation Tendency of the Spike RBD and Membrane Protein in SARS-CoV-2 by Machine Learning" International Journal of Molecular Sciences 25, no. 9: 4662. https://doi.org/10.3390/ijms25094662
APA StyleYe, Q., Wang, H., Xu, F., Zhang, S., Zhang, S., Yang, Z., & Zhang, L. (2024). Co-Mutations and Possible Variation Tendency of the Spike RBD and Membrane Protein in SARS-CoV-2 by Machine Learning. International Journal of Molecular Sciences, 25(9), 4662. https://doi.org/10.3390/ijms25094662